Abstract
Using various versions of quantum-chemical calculation, namely four versions of density functional theory (DFT), (DFT B3PW91/TZVP, DFT M06/TZVP, DFT B3PW91/Def2TZVP, and DFT M06/Def2TZVP) and two versions of the MP method (MP2/TZVP and MP3/TZVP), the existence possibility of the carbon-nitrogen-containing compound having an unusual M: nitrogen ratio of 1:20, unknown for these elements at present, was shown. Structural parameters data are presented; it was noted that, as may be expected, CN4 grouping has practically a tetrahedral structure, and the chemical bond lengths formed by nitrogen atoms and a carbon atom in the frameworks of each of the calculation methods indicated above are equal to each other. Thermodynamical parameters, NBO analysis data, and HOMO/LUMO images for this compound are also presented. A good agreement between the calculated data obtained using the above three quantum-chemical methods was noticed, too.
1. Introduction
As is well known, carbon and nitrogen together form a very significant number of chemical compounds—neutral as well as cations and anions. At present, as a minimum, several dozen neutral compounds are known to contain atoms of only these two elements; the simplest (and most known) of these are the cyanogens C2N2 (cyanogen N≡C–C≡N, isocyanogen C=N–C≡N, and diisocyanogen C=N–N=C) and tricarbon tetranitride or, simply carbon nitride C3N4, first described more than 100 years ago and considered in more detail in a number of works, in particular [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. The carbon:nitrogen ratio in known (C,N)-binuclear compounds containing atoms of only those two elements varies from 69:1 in diheterofullerene (C69N)2 [16] to 1:12 in tetraazidomethane C(N3)4 [17] and (dodecaazadodecatetraene-1,4, 7,10)carbon(IV) C(N12) [18] with structural formulas I and II, respectively.

The existence of compound I was confirmed experimentally [17], while the existence of compound II was predicted based on the results of quantum chemical calculations carried out using DFT B3PW91/TZVP, MP2/TZVP, and MP3/TZVP methods [18]. Accordingly, these two C(N12) isomers are the most nitrogen-rich compounds, containing only C and N atoms. However, the specified ratio C:N = 1:12 is not the maximum possible, and one can in principle assume the possibility of the existence of carbon-nitrogen-containing chemical compounds with an even greater number of nitrogen atoms per carbon atom. An example of such a compound is tetra(pentaazolato)carbon(IV) C(N5)4, having structural formula III in which the C:N ratio is equal to 1:20.

Such binuclear compounds, containing only nitrogen and carbon atoms, where the C:N ratio is less than 1:10, are of considerable interest as potential high-energetic substances [17,19,20]. As far as is known, compound III has not yet been considered in the literature even theoretically; in this connection, the given communication is devoted to the consideration of the question of the possibility of its existence and, in the case of a positive conclusion, to the determination of the parameters of its molecular structure and thermodynamic characteristics using modern quantum-chemical methods of calculation, namely the various versions of density functional theory (DFT) and Möller–Plesset perturbation theory (MP).
2. Results and Discussion
According to the data of each of the above six methods of quantum chemical calculation, the chemical compound of the composition with the gross formula C(N20) is capable of independent existence as an isolated molecule, at least in the gas phase. The lengths of chemical bonds and bond angles between atoms in this chemical compound, calculated by these methods, are presented in Table 1. As it is easy to see when comparing the data presented in it, the values of the above key parameters of the molecular structure are very close to each other, and therefore it seems appropriate to discuss them together.

Table 1.
Bond lengths and bond angles in the C(N20) [C(N5)4] calculated by and various versions of DFT and MP methods.
Taking into account the fact that the carbon atom in the C(N20) compound under consideration with the above structural formula III is bonded to four nitrogen atoms, we can expect that these N atoms should be located at the vertices of a regular tetrahedron or a polyhedron close to it. Indeed, the calculation of the molecular structure of C(N20) using each of the six quantum chemical methods we used indicates that a grouping of four nitrogen atoms bonded to a carbon atom forms an almost regular tetrahedron. Wherein, firstly, the C–N bond lengths are equal to each other (although they somewhat depend on the calculation method); secondly, the angles (NCN) differ little from each other, and their values are close to the values of similar angles in a regular tetrahedron (109.5°) (Table 1). In this regard, it is worth noting that the non-bonding angles (NNN) formed by those nitrogen atoms that are bonded to the C atom are close to 60° in each of the six methods (i.e., to the angles of a regular triangle). The pentazole fragments are strictly coplanar since the sum of the internal bond angles in each of them is the same and coincides with the sum of the internal angles in a flat pentagon (540°).
As should be expected, within the framework of each of these methods, they are completely identical to each other, although the sets of these angles differ somewhat from each other; the lengths of the N–N chemical bonds in them are equal in pairs (Table 1). That is characteristic, the C–N bond lines are in the same plane as the planes of the corresponding pentazole fragments. In general, in qualitative terms (in appearance), the molecular structures of this compound obtained by these six calculation methods show almost complete similarity with each other; an example of an image of such a structure is shown in Figure 1. The values of the electrical dipole moments of this compound obtained by each of the DFT B3PW91/TZVP, MP2/TZVP, and MP3/TZVP methods practically do not differ from 0.00 Debye units, which, taking into account the quasi-tetrahedral molecular structure of C(N20) (point group of symmetry D2) obtained by each of these methods, seems to be quite expected.
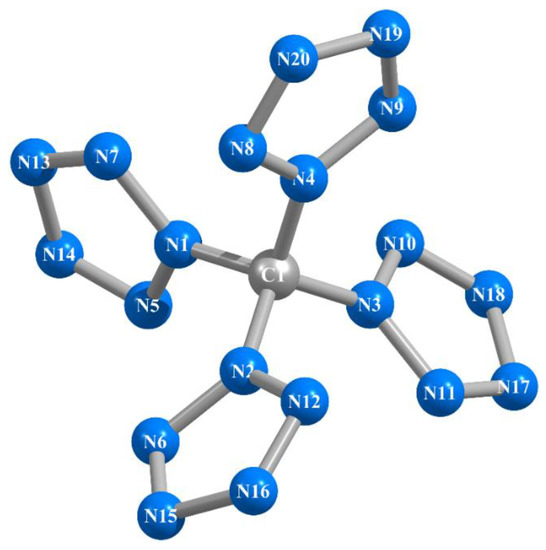
Figure 1.
Molecular structure of the C(N20) compound obtained as a result of MP3/TZVP quantum-chemical calculation.
The key data of the NBO analysis, namely the values of the effective charges on the carbon and nitrogen atoms in the test compound, obtained by the DFT, MP2, and MP3 methods, are presented in Table 2. The selected data of the NBO analysis of the C(N20) compound under examination are presented in the Supplementary Materials. As can be seen from these data, the effective charges on individual atoms calculated by the above methods with an accuracy of 0.01 are quite close to each other. Be that as it may, these values, judging by their absolute values, are much less than +4.00 ē (for the C1 atom) and –1.00 ē (for the N1–N4 atoms bonded to the C1 atom by chemical bonds), which would be the case if all chemical bonds between C and N atoms were ionic. In our opinion, this fact indicates the presence of a high degree of electron density delocalization in this compound. The ground state of this compound in the framework of each of these methods is a spin singlet (MS = 1), and the values of the operator of the square of the intrinsic angular momentum of the total spin of the system <S**2> are equal to 0. In addition, according to the calculation data in the framework of any of the above six methods, the nearest excited state with a different MS value, namely, the spin triplet (MS = 3), is significantly higher in energy than the ground state (for example, according to the B3PW91/TZVP method, by 403 kJ/mol). Testing the wave functions of the ground state for stability using the standard procedure STABLE = OPT showed that the wave function of the ground state at MS = 1 is stable with respect to the perturbations under consideration.
Table 2.
NBO analysis data for the C(N20) calculated by DFT B3PW91/TZVP, DFT B3PW91/Def2TZVP, DFT M06/TZVP, DFT M06/Def2TZVP, MP2/TZVP, and MP3/TZVP methods.
Images of the highest occupied and lowest vacant (unoccupied) molecular orbitals (HOMO and LUMO, respectively) obtained by using DFT B3PW91/TZVP, DFT B3PW91/Def2TZVP, DFT M06/TZVP, and DFT M06/Def2TZVP quantum-chemical methods are presented in Figure 2. As can be seen from this, the LUMO shapes obtained by each of these three methods are quite close to each other. As for HOMO, the above similarity is observed only for orbitals obtained by DFT methods.
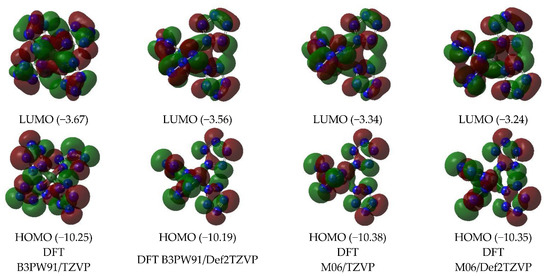
Figure 2.
The images of highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals in the C(N20) (ground state—spin singlet, MS = 1) according to DFT B3PW91/TZVP, DFT B3PW91/Def2TZVP, DFT M06/TZVP, and DFT M06/Def2TZVP methods. The energies values of these molecular orbitals (in brackets) are given in eV.
B3PW91/Def2TZVP, M06/TZVP, and M06/Def2TZVP, while HOMO obtained by the B3PW91/TZVP method has a different shape (Figure 2). The energies of these HOMO and LUMO obtained by DFT methods, as can be seen from Figure 2, are quite close to each other. However, they differ significantly from the energies of similar MO obtained using MP methods, which, taking into account the above, also seem quite natural and predictable.
The standard thermodynamic parameters of formation (∆fH0, S0, and ∆fG0) for the chemical compound under study are given in Table 3. As may be seen from it, all these parameters are positive, and, therefore, this compound, as should be expected, cannot be obtained from the most thermodynamically stable simple substances formed by carbon and nitrogen (i.e., graphite and molecular nitrogen N2). In connection with the foregoing, it is of particular interest to consider the reaction of the interaction of C(N20) with molecular oxygen, which proceeds in the gas phase according to Reaction (1)
and the decomposition reactions of this compound according to any of the Reactions (2)–(4)
C(N20) (gas) + O2 (gas) → CO2 (gas) + 10N2 (gas)
C(N20) (gas) → C (gas) + 10N2 (gas)
C(N20) (gas) → C (graphite) + 10N2 (gas)
C(N20) (gas) → C (diamond) + 10N2 (gas)
Table 3.
Standard thermodynamic parameters of formation at T = 298.15 K and P = 101,325 Pa (∆fH0, S0 and ∆fG0) for C(N20) calculated by B3PW91/TZVP and G4 methods.
The thermodynamic parameters of all these reactions are presented in Table 4. As can be seen from the data indicated in it, the reaction of the interaction of the compound C(N20) under consideration with molecular oxygen (1) is strongly exothermic, since its enthalpy ∆rH0 < 0, and the numerical value of this thermodynamic parameter according to its absolute value is very high (more than 2000 kJ). However, since the value of its entropy ∆rS0 > 0, then according to the well-known Gibbs–Helmholtz Equation (5),
at any temperature T, the relation ∆rG(T) < 0 will take place. Therefore, the process described by Equation (1) is irreversible. In this regard, it is interesting to note that a similar situation occurs for Reactions (2)–(4) (Table 4), the last of which, in principle, can be used even to obtain an allotropic modification of carbon—diamond within the isobaric process.
∆rG(T) = ∆rH0 − T∆rS0
Table 4.
Standard enthalpies (∆rH0) and standard entropies (∆rS0) of the Reactions (1)–(4) at T = 298.15 K and P = 101,325 Pa according to data of B3PW91/TZVP and G4 methods.
3. Method
To carry out the quantum-chemical calculation, in this work, we used the density functional theory (DFT), which combines the standard extended split valence basis set TZVP and the hybrid functional B3PW91, described in detail in Refs [22,23,24] and used by us, in particular, in [25,26,27]. The use of the B3PW91/TZVP method, in this case, is because, according to [22,23,24], it allows one to obtain, as a rule, the most accurate (i.e., close to experimental) values of the geometric parameters of molecular structures, as well as much more accurate values of thermodynamic and other physical-chemical parameters in comparison with other variants of the DFT method. For comparison, other, later versions of the DFT method were also used, namely the M06 functional [28] and the newer redefinitions of the TZVP basis set—Def2TZVP [29]. The calculations were carried out using the Gaussian09 program package [30]. As in our previous articles, in which this method of calculation was used [25,26,27], the correspondence of the found stationary points to the energy minima in all cases was proved by calculating the second derivatives of the energy to the coordinates of the atoms; wherein, all equilibrium structures corresponding to the minimum points on the potential energy surfaces had only real (and, moreover, always positive) frequency values. Of the optimized structures for further consideration, the one with the lowest total energy was selected. Since the structure of C(N20) may be a priori nontrivial, in order not to encounter a probable overestimation of the stability of such a structure when calculating by DFT methods, we decided to use an “honest” ab initio method for calculation along with DFT methods, as far as our capabilities allow. As it is well known, the DFT methods when calculating structures with a nontrivial distribution of electron density can, in some cases, lead to an incorrect order of orbitals and even an incorrect molecular structure. To exclude such a possibility, we decided to carry out the calculation using some ab initio quantum chemical methods. However, unfortunately, it was not possible to complete the calculation with such methods with a stricter account of electronic correlation as CCSD and QCISD due to high computational costs. That is why, in addition to the calculation by the DFT B3PW91/TZVP, DFT B3PW91/Def2TZVP, DFT M06/TZVP, and DFT M06/Def2TZVP methods, as an alternative, we used Möller-Plesset perturbation theory methods [31]. Unfortunately, we could only use the methods of second and third-order perturbation theory, namely MP2 [32] and MP3 [33], in combination with the extended split valence TZVP basis set, each of which is noticeably less computationally intensive than the CCSD and QCISD methods. (At the moment, it was also not possible to complete the calculation using the MP4 method, since in terms of computational complexity it is comparable to the CCSD method). We could not use other ab initio methods for the calculation since the technical and time resources at our disposal did not allow us to do this.
Natural bond orbital (NBO) analysis was carried out, using NBO version 3.1, integrated with the Gaussian09 program package [30], according to the methodology described in detail [34]. NBO methods are well known for their excellent numerical stability and convergence with respect to basis set expansion that is sensibly proportionate to the convergence of energy and other calculated wavefunction properties (unlike Mulliken analysis and related overlap-dependent methods in this case). The standard thermodynamic parameters of formation (∆fH0, S0, and ∆fG0) for the C(N20) compound under examination were calculated using the G4 method described in [35].
4. Conclusions
As follows from the above, the presented results of quantum chemical calculations performed using six different methods, namely four DFT methods (B3PW91/TZVP, B3PW91/Def2TZVP, M06/TZVP, and M06/Def2TZVP) and two MP methods (MP2/TZVP, MP3/TZVP), unambiguously testify in favor of the possibility of the existence of a new (and not yet experimentally discovered) chemical compound of carbon and nitrogen with the composition C(N20) [C(N5)4]. This substance is characterized by an unusual (and so far the highest among all currently known binary compounds of carbon and nitrogen) ratio between the number of N atoms and the number of C atoms (20:1) and has a quasi-tetrahedral molecular structure. At the same time, the use of DFT methods with B3PW91 and M06 potentials both with the TZVP basis and with the more advanced Def2TZVP basis leads to practically the same results (although the Def2TZVP set contains a much larger number of basis functions—651 versus 399—and its use requires much more energy—time costs). A similar situation occurs when comparing the results of the calculations of C(N20) by the above variants of the DFT method with the results of the calculations by the MP2 and MP3 methods, the use of which is even more expensive. Thus, there is every reason to state that the DFT methods generally correctly describe the molecular structure of the compound under study, at least in a qualitative sense.
Judging by the very high value of ∆fG0 (>2500 kJ/mol), the C(N20) [C(N5)4] compound under consideration is indeed a high-energy substance; if it is obtained experimentally, it will undoubtedly find some practical application, at least in the above capacity. Predicting the possibility of the existence of such an exotic chemical and modeling its molecular and electronic structures using modern quantum chemical calculations can serve as a very useful tool in solving the problems associated with this synthesis. On the other hand, its synthesis may be of great importance for the further development of physical chemistry and chemical technology of both its constituent chemical elements, which, as is known, play an extremely important role in nature.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24065172/s1.
Author Contributions
Conceptualization, O.V.M.; methodology, O.V.M. and D.V.C.; software, D.V.C.; validation, O.V.M. and D.V.C.; formal analysis, O.V.M. and D.V.C.; investigation, O.V.M. and D.V.C.; resources, D.V.C.; data curation, D.V.C.; writing—original draft preparation, O.V.M. and D.V.C.; writing—review and editing, O.V.M.; visualization, O.V.M. and D.V.C.; supervision, O.V.M.; project administration, O.V.M.; funding acquisition, D.V.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study did not require institutional approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
No unpublished data was created or analyzed in this article.
Acknowledgments
All quantum-chemical calculations were performed at the Joint Supercomputer Center of Russian Academy of Sciences—Branch of Federal Scientific Center “Scientific Research Institute for System Analysis of the RAS”, which is acknowledgement for technical support. Contribution of author Denis V. Chachkov was funded by the state assignment to the Federal Scientific Center “Scientific Research Institute for System Analysis of the RAS” for scientific research. Moreover, this study was carried out using the equipment of the Center for Collective Use “Nanomaterials and Nanotechnology” of the Kazan National Research Technological University with the financial support of the Ministry of Science and Higher Education of the Russian Federation under agreement No. 075-15-2021-699.
Conflicts of Interest
The authors declare that they have no conflict of interest, financial or otherwise.
References
- Ringer, A.L.; Sherrill, C.D.; King, R.A.; Crawford, T.D. Low-lying singlet excited states of isocyanogen. Int. J. Quantum Chem. 2008, 106, 1137–1140. [Google Scholar] [CrossRef]
- Brotherton, T.K.; Lynn, J.W. The Synthesis and Chemistry of Cyanogen. Chem. Revs. 1959, 59, 841–883. [Google Scholar] [CrossRef]
- Bircumshaw, L.L.; Tayler, F.M.; Whiffen, D.H. Paracyanogen: Its formation and properties. Part I. J. Chem. Soc. 1954, 931–935. [Google Scholar] [CrossRef]
- Franklin, E.C. The Ammono Carbonic Acids. J. Am. Chem. Soc. 1922, 44, 486–509. [Google Scholar] [CrossRef]
- Liu, A.; Cohen, M.L. Prediction of new low compressibility solids. Science 1989, 245, 841–843. [Google Scholar] [CrossRef]
- Pozdnyakov, O.F.; Blinov, L.N.; Arif, M.; Pozdnyakov, A.O.; Filippov, S.N.; Semencha, A.V. Mass spectrometry of carbon nitride C3N4. Tech. Phys. Lett. 2005, 31, 1001–1003. [Google Scholar] [CrossRef]
- Arif, M.; Blinov, L.N.; Lappalainen, R.; Filippov, S.N. Preparation of powdered carbon nitride C3N4. Glass Phys. Chem. 2004, 30, 573–575. [Google Scholar] [CrossRef]
- Cao, C.-B.; Lv, Q.; Zhu, H.-S. Carbon nitride prepared by solvothermal method. Diam. Relat. Mater. 2003, 12, 1070–1074. [Google Scholar] [CrossRef]
- Holst, J.R.; Gillan, E.G. From Triazines to Heptazines: Deciphering the Local Structure of Amorphous Nitrogen-Rich Carbon Nitride Materials. J. Am. Chem. Soc. 2008, 130, 7373–7379. [Google Scholar] [CrossRef] [PubMed]
- Mansor, N.; Belen Jorge, A.; Corà, F.; Gibbs, C.; Jervis, R.; McMillan, P.F.; Wang, X.; Brett, D.J.L. Graphitic Carbon Nitride Supported Catalysts for Polymer Electrolyte Fuel Cells. J. Phys. Chem. C 2014, 118, 6831–6838. [Google Scholar] [CrossRef]
- Fina, F.; Callear, S.K.; Carins, G.M.; Irvine, J.T.S. Structural Investigation of Graphitic Carbon Nitride via XRD and Neutron Diffraction. Chem. Mater. 2015, 27, 2612–2618. [Google Scholar] [CrossRef]
- Korsunskii, B.L.; Pepekin, V.I. On the way to carbon nitride. Russ. Chem. Rev. 1997, 66, 901–912. [Google Scholar] [CrossRef]
- Thomas, A.; Fischer, A.; Goettmann, F.; Antonietti, M.; Müller, J.-O.; Schlögl, R.; Carlsson, J.M. Graphitic carbon nitride materials: Variation of structure and morphology and their use as metal-free catalysts. J. Mater. Chem. 2008, 18, 4893–4908. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, J.; Li, B.; Sun, H.; Wang, S. Engineered Graphitic Carbon Nitride-Based Photocatalysts for Visible-Light-Driven Water Splitting: A Review. Energy Fuels 2021, 35, 6504–6526. [Google Scholar] [CrossRef]
- Nuber, B.; Hirsch, A. A new route to nitrogen heterofullerenes and the first synthesis of (C69N)2. Chem. Commun. 1996, 1421–1422. [Google Scholar] [CrossRef]
- Banert, K.; Joo, Y.-H.; Ruffer, T.; Walfort, B.; Lang, H. The Exciting Chemistry of Tetraazidomethane. Angew. Chem. Int. Ed. 2007, 46, 1168–1171. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. A new chemical compound with an unusual ratio of number of carbon and nitrogen atoms–C(N12): Quantum-chemical modelling. RSC Adv. 2021, 11, 35974–35981. [Google Scholar] [CrossRef]
- Forquet, V.; Miró Sabaté, C.; Chermette, H.; Jacob, G.; Labarthe, É.; Delalu, H.; Darwich, C. Energetic Properties of Rocket Propellants Evaluated through the Computational Determination of Heats of Formation of Nitrogen-Rich Compounds. Chem. Asian J. 2016, 11, 730–744. [Google Scholar] [CrossRef]
- Dhenain, A.; Darwich, C.; Miró Sabaté, C.; Le, D.-M.; Bougrine, A.-J.; Delalu, H.; Lacôte, E.; Payen, L.; Guitton, J.; Labarthe, E.; et al. (E)-1,1,4,4-Tetramethyl-2-tetrazene (TMTZ): A Prospective Alternative to Hydrazines in Rocket Propulsion. Chem. Eur. J. 2017, 23, 9897–9907. [Google Scholar] [CrossRef]
- Yungman, V.S. (Ed.) Thermal Constants of Substances; Wiley: New York, NY, USA, 1999; Volume 1–8. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Chachkov, D.V.; Mikhailov, O.V. Structure of (5656)macrotetracyclic chelates in the ternary systems M(II)-ethanedithioamide-acetone (M = Mn, Fe, Co, Ni, Cu, Zn) according to DFT calculations. Russ. J. Inorg. Chem. 2013, 58, 1073–1078. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Quantum-chemical calculation of molecular structures of (5656)macrotetracyclic 3d-metal complexes self-assembled inquaternary systems M(II) ion-ethanedithioamide-formaldehyde-ammonia by the density functional theory method. Russ. J. Inorg. Chem. 2014, 59, 218–223. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. Int. J. Mol. Sci. 2020, 21, 9085. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 0618–0622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Pople, J.A.; Seeger, R.; Krishnan, R. Variational Configuration Interaction Methods and Comparison with Perturbation Theory. Int. J. Quant. Chem. 1977, S11, 149–163. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Revs. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).