
Genetic Background of Metabolically Healthy and Unhealthy Obesity Phenotypes in Hungarian Adult Sample Population



Abstract
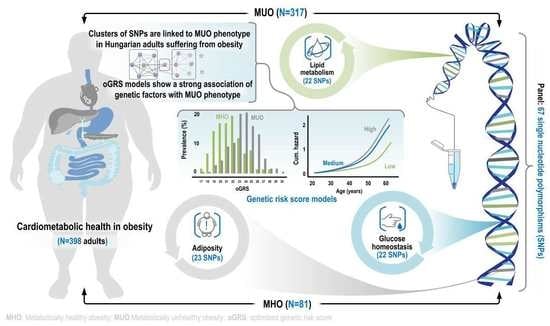
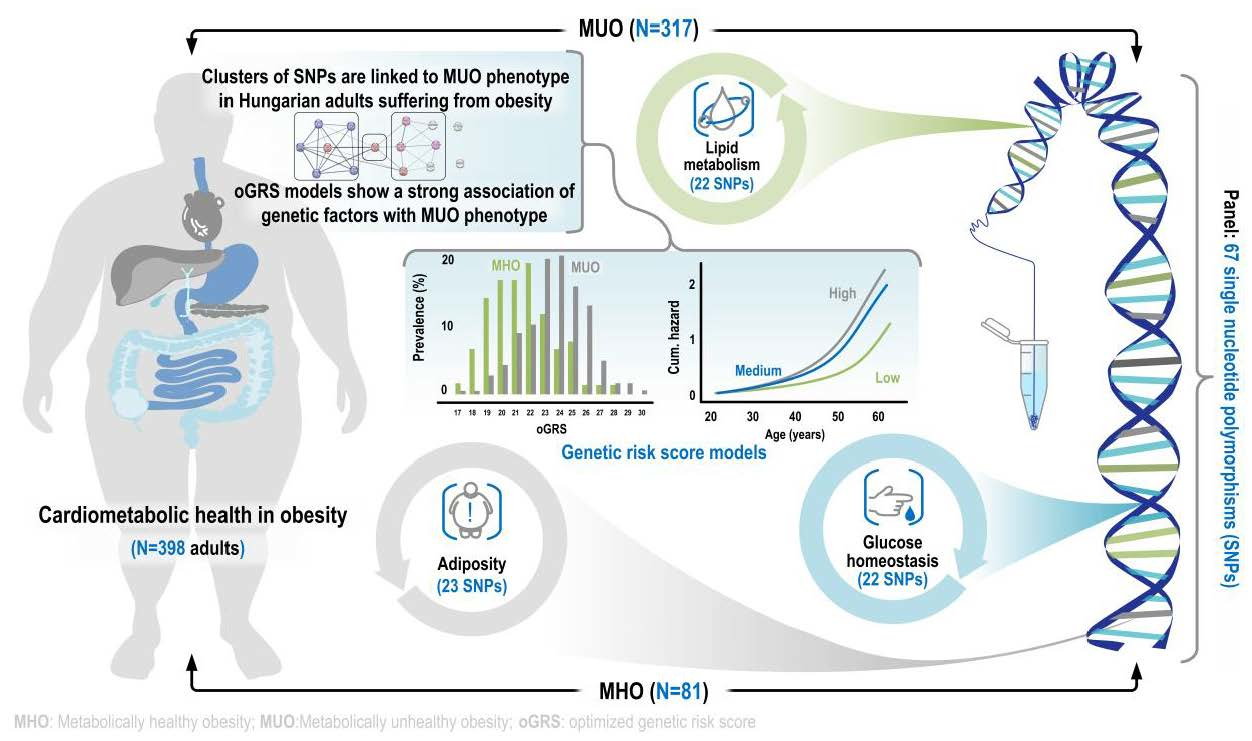
1. Introduction
2. Results
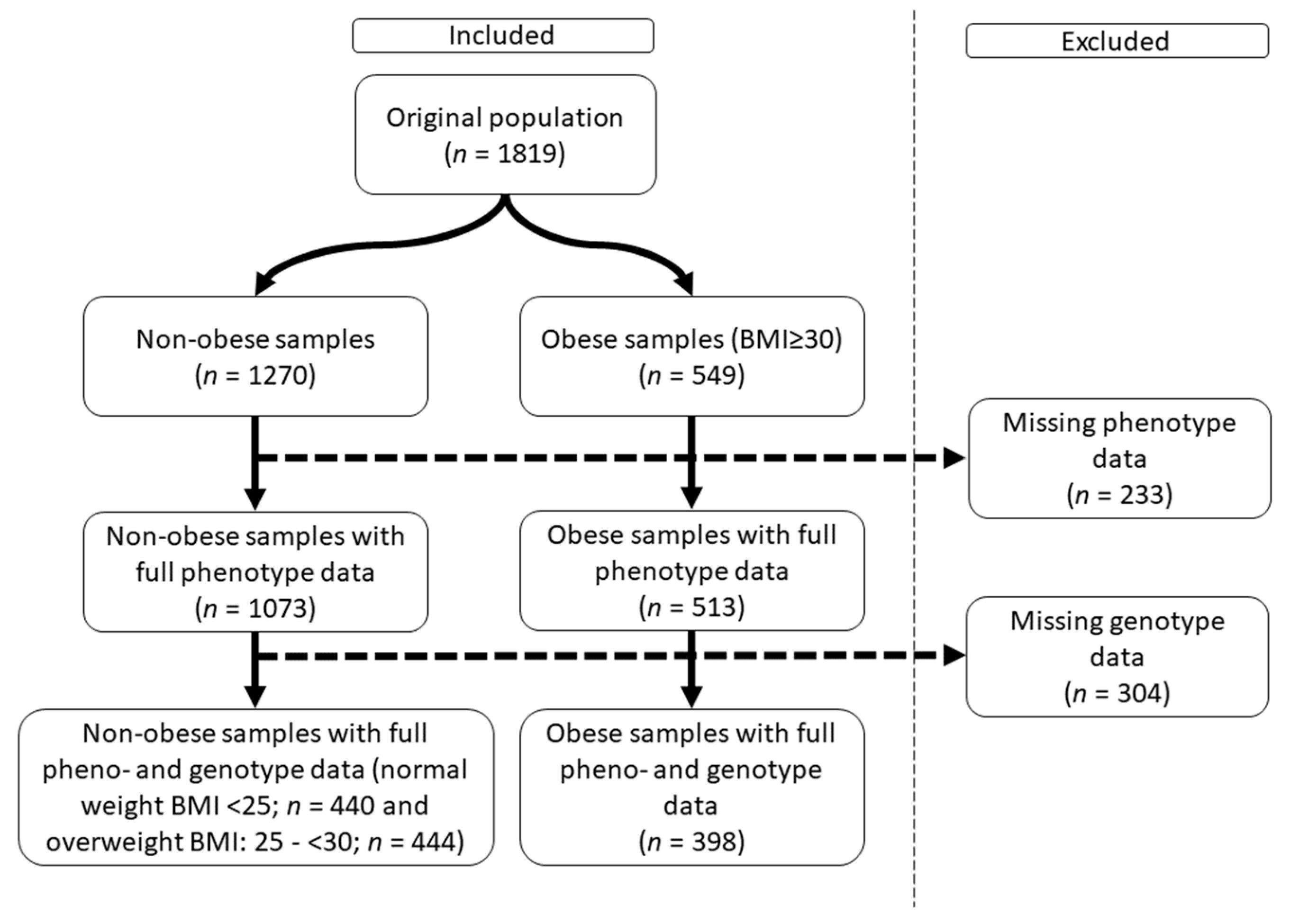
2.1. Characteristics of the Obese Study Samples
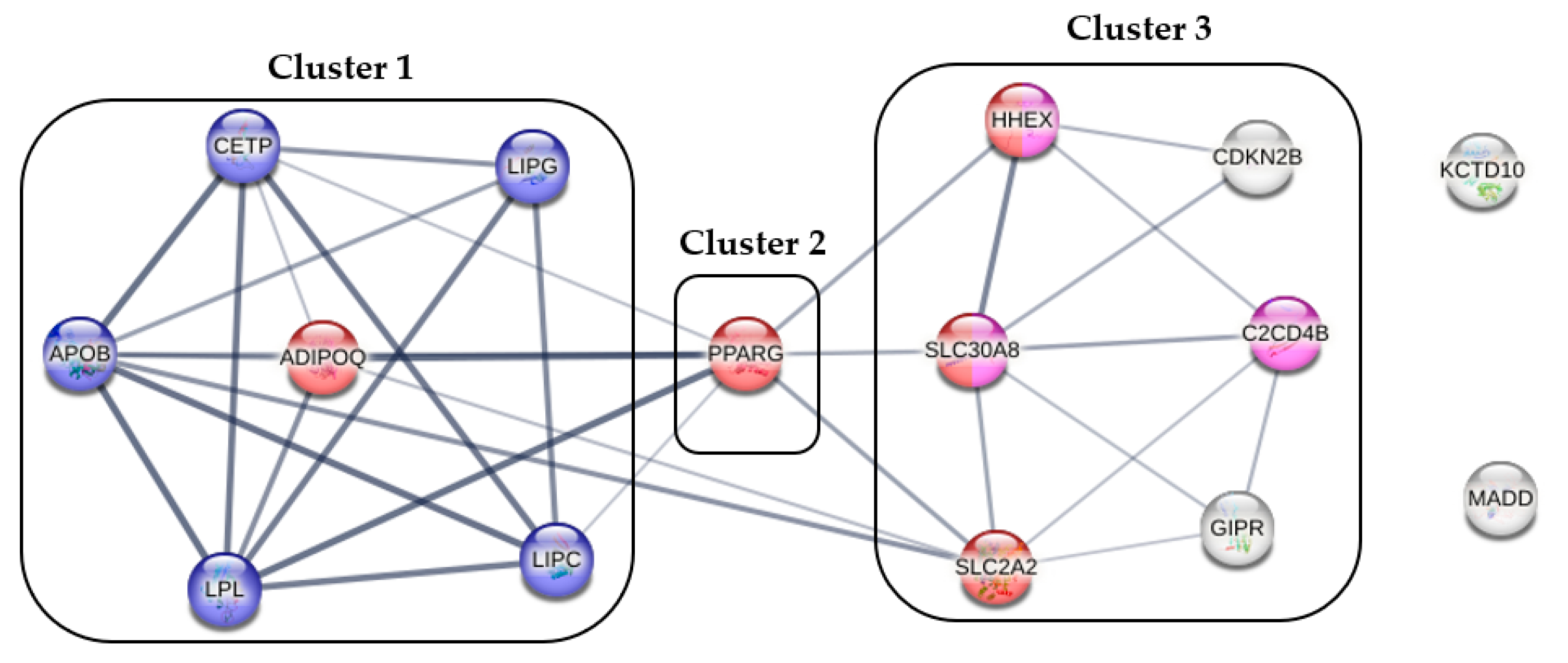
2.2. The Best-Fitting Genetic Models by SNPs
2.3. Optimization of Genetic Risk Score and the Association of Optimized Genetic Risk Score with MUO and Related Parameters
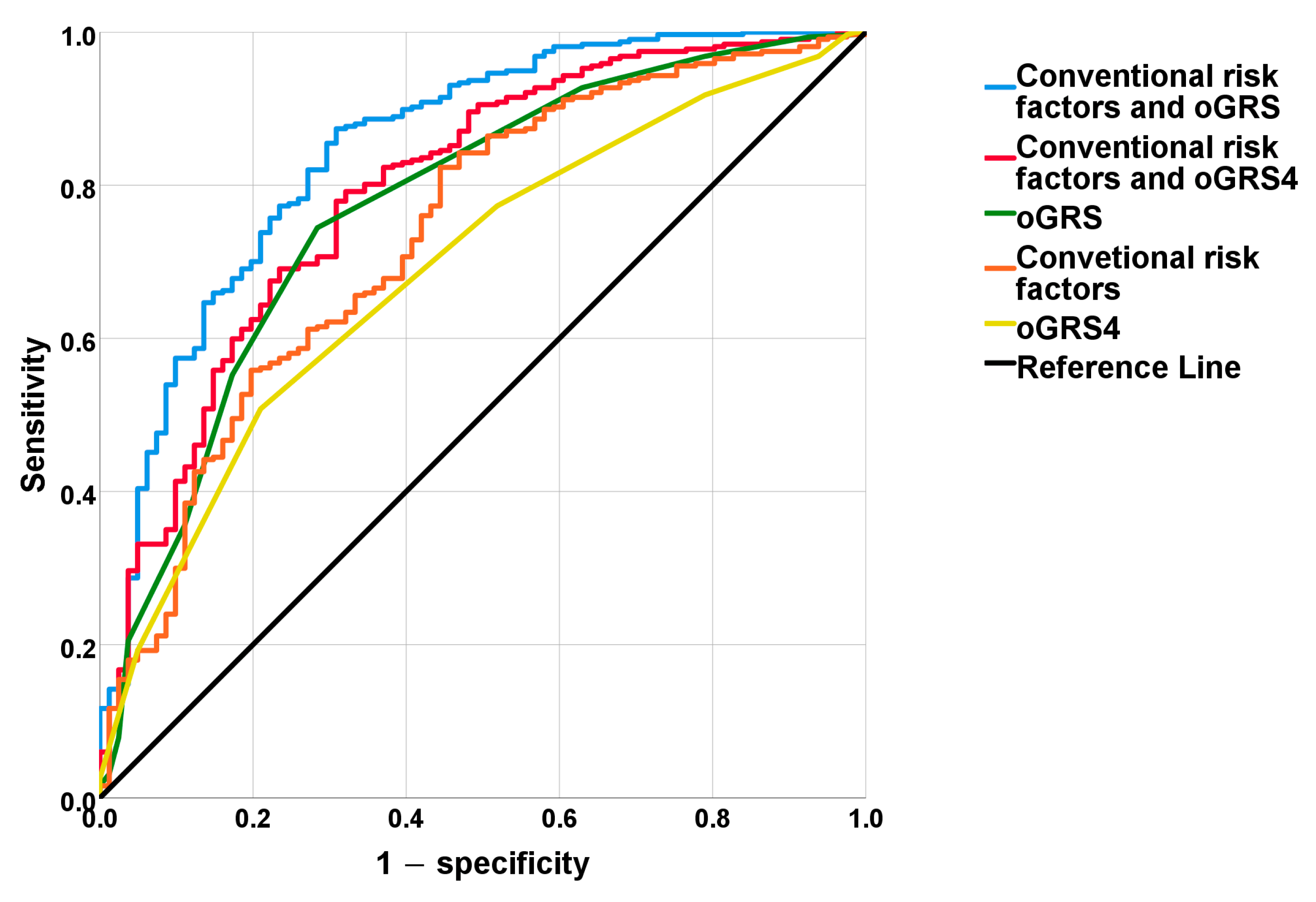
2.4. The Discriminatory Power of MUO-Associated Genetic and Non-Genetic Risk Factors Based on ROC Curve Analyses
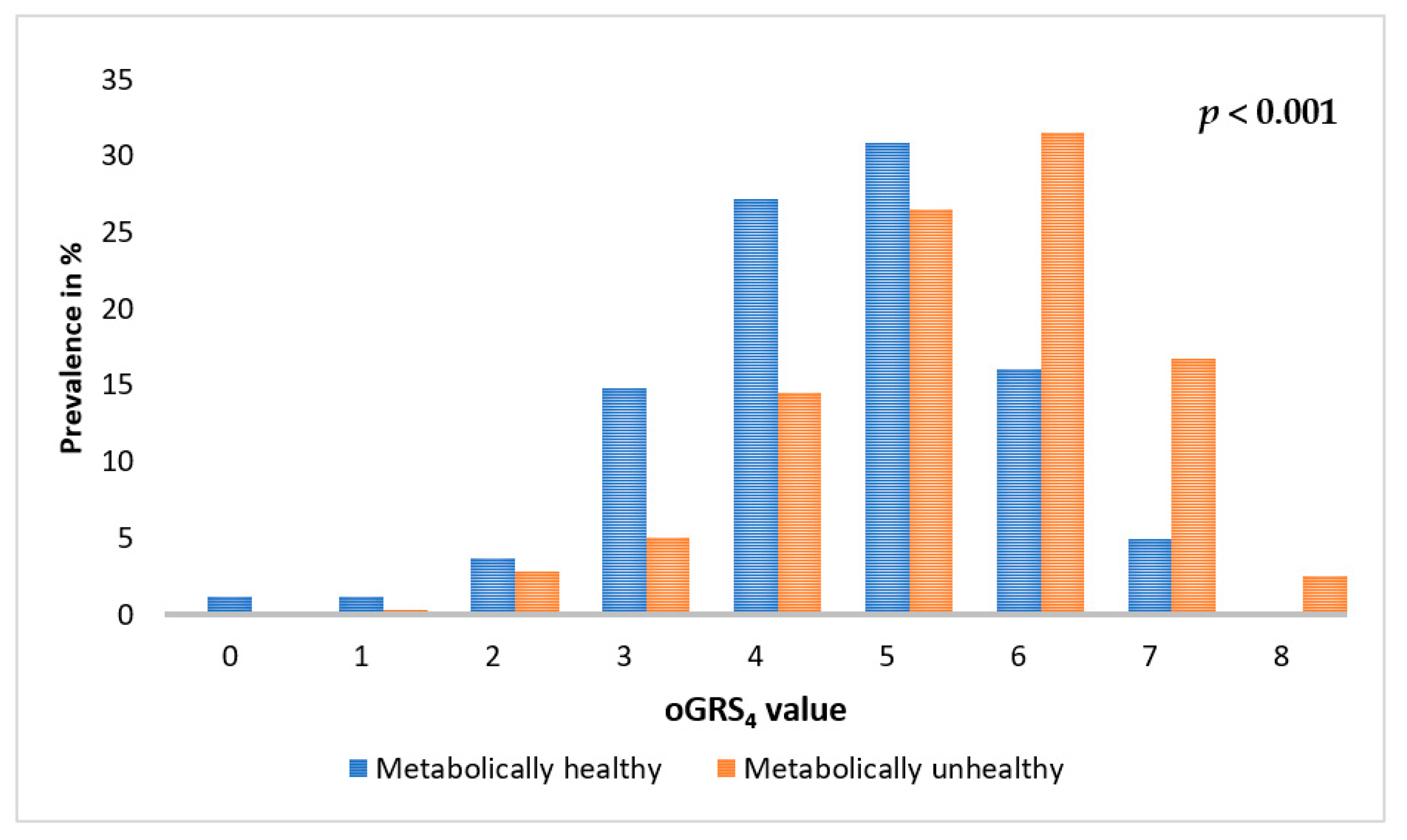
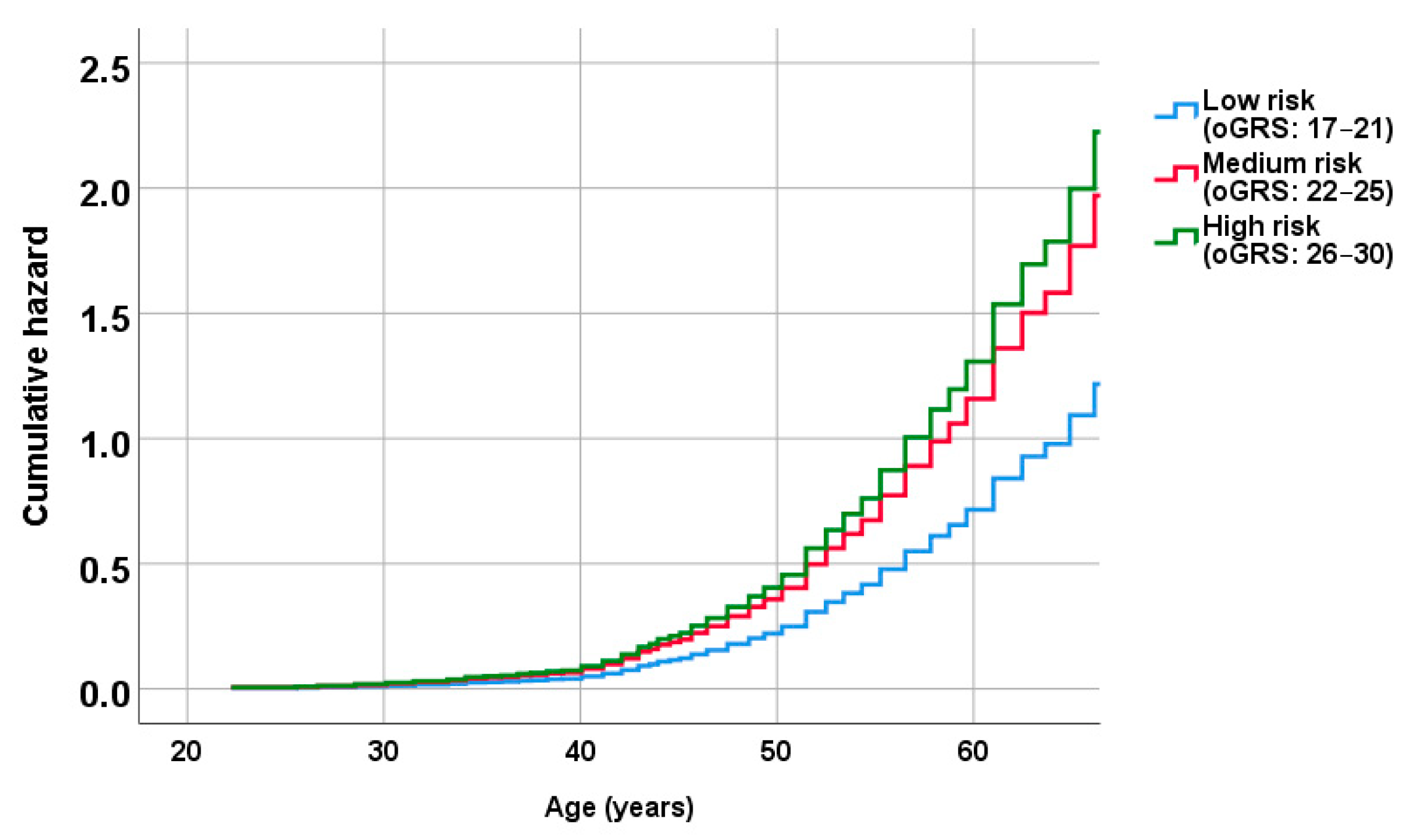
2.5. Association of oGRS with the Prevalence of Metabolically Unhealthy Obesity and with the Age of Individuals Affected
2.6. Results of Trend and Multivariate Logistic Regression Analyses on the Association of oGRSs with the Metabolic Status
3. Discussion
4. Materials and Methods
4.1. Sample Population and Relevant Parameters Defined
4.2. Defining Metabolically Healthy and Unhealthy Obesity
4.3. DNA Isolation, SNP Selection and Genotyping
4.4. Identification and Coding of the Genetic Model Best Associated with HOMA-IR by SNPs
- a)
- Codominant genetic model: homozygote genotype with risk allele was labelled as 2, whereas heterozygote gene labelled as 1 and 0 was coded for no risk allele.
- b)
- Dominant genetic model: 2 was coded for the presence of one or two risk alleles and 0 was coded for the absence of a risk allele.
- c)
- Recessive genetic model: 2 was counted for the presence of two risk alleles, while 0 was counted for the homozygote gene with the absence of a risk allele and for the heterozygote gene.
4.5. Calculation and Optimization of the Genetic Risk Score
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swinburn, B.A.; Kraak, V.I.; Allender, S.; Atkins, V.J.; Baker, P.I.; Bogard, J.R.; Brinsden, H.; Calvillo, A.; De Schutter, O.; Devarajan, R.; et al. The Global Syndemic of Obesity, Undernutrition, and Climate Change: The Lancet Commission report. Lancet 2019, 393, 791–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Southerland, J.; Wang, K.; Bailey, B.A.; Alamian, A.; Stevens, M.A.; Wang, Y. Ethnic Differences in Risk Factors for Obesity among Adults in California, the United States. J. Obes. 2017, 2017, 2427483. [Google Scholar] [CrossRef] [PubMed]
- Shai, I.; Jiang, R.; Manson, J.E.; Stampfer, M.J.; Willett, W.C.; Colditz, G.A.; Hu, F.B. Ethnicity, obesity, and risk of type 2 diabetes in women: A 20-year follow-up study. Diabetes Care 2006, 29, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.P.; David Cheng, T.Y.; Tsai, S.P.; Chan, H.T.; Hsu, H.L.; Hsu, C.C.; Eriksen, M.P. Are Asians at greater mortality risks for being overweight than Caucasians? Redefining obesity for Asians. Public Health Nutr. 2009, 12, 497–506. [Google Scholar] [CrossRef]
- Pischon, T.; Boeing, H.; Hoffmann, K.; Bergmann, M.; Schulze, M.B.; Overvad, K.; van der Schouw, Y.T.; Spencer, E.; Moons, K.G.; Tjonneland, A.; et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 2008, 359, 2105–2120. [Google Scholar] [CrossRef]
- Malik, V.S.; Willett, W.C.; Hu, F.B. Global obesity: Trends, risk factors and policy implications. Nat. Rev. Endocrinol. 2013, 9, 13–27. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Hu, F.B.; Schulze, M.B. Metabolically healthy obesity: Epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013, 1, 152–162. [Google Scholar] [CrossRef]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; World Obesity, F. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef]
- Sims, E.A. Are there persons who are obese, but metabolically healthy? Metabolism 2001, 50, 1499–1504. [Google Scholar] [CrossRef]
- Tsatsoulis, A.; Paschou, S.A. Metabolically Healthy Obesity: Criteria, Epidemiology, Controversies, and Consequences. Curr. Obes. Rep. 2020, 9, 109–120. [Google Scholar] [CrossRef]
- Bluher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, bnaa004. [Google Scholar] [CrossRef]
- Eckel, N.; Meidtner, K.; Kalle-Uhlmann, T.; Stefan, N.; Schulze, M.B. Metabolically healthy obesity and cardiovascular events: A systematic review and meta-analysis. Eur. J. Prev. Cardiol 2016, 23, 956–966. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Schulze, M.B. Metabolically healthy obesity: The low-hanging fruit in obesity treatment? Lancet Diabetes Endocrinol. 2018, 6, 249–258. [Google Scholar] [CrossRef]
- Zembic, A.; Eckel, N.; Stefan, N.; Baudry, J.; Schulze, M.B. An Empirically Derived Definition of Metabolically Healthy Obesity Based on Risk of Cardiovascular and Total Mortality. JAMA Netw. Open 2021, 4, e218505. [Google Scholar] [CrossRef]
- Rey-Lopez, J.P.; de Rezende, L.F.; Pastor-Valero, M.; Tess, B.H. The prevalence of metabolically healthy obesity: A systematic review and critical evaluation of the definitions used. Obes. Rev. 2014, 15, 781–790. [Google Scholar] [CrossRef]
- Albuquerque, D.; Nobrega, C.; Manco, L.; Padez, C. The contribution of genetics and environment to obesity. Br. Med. Bull. 2017, 123, 159–173. [Google Scholar] [CrossRef]
- Herrera, B.M.; Lindgren, C.M. The genetics of obesity. Curr. Diab Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef]
- Goodarzi, M.O. Genetics of obesity: What genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Navarro, E.; Funtikova, A.N.; Fito, M.; Schroder, H. Can metabolically healthy obesity be explained by diet, genetics, and inflammation? Mol. Nutr. Food Res. 2015, 59, 75–93. [Google Scholar] [CrossRef]
- Berezina, A.; Belyaeva, O.; Berkovich, O.; Baranova, E.; Karonova, T.; Bazhenova, E.; Brovin, D.; Grineva, E.; Shlyakhto, E. Prevalence, Risk Factors, and Genetic Traits in Metabolically Healthy and Unhealthy Obese Individuals. Biomed. Res. Int. 2015, 2015, 548734. [Google Scholar] [CrossRef] [PubMed]
- Sedaghati-Khayat, B.; Barzin, M.; Akbarzadeh, M.; Guity, K.; Fallah, M.S.; Pourhassan, H.; Azizi, F.; Daneshpour, M.S. Lack of association between FTO gene variations and metabolic healthy obese (MHO) phenotype: Tehran Cardio-metabolic Genetic Study (TCGS). Eat Weight Disord. 2020, 25, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.B. The effect of obesity on health outcomes. Mol. Cell Endocrinol. 2010, 316, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Wang, M.W. Obesity, a health burden of a global nature. Acta Pharmacol. Sin. 2012, 33, 145–147. [Google Scholar] [CrossRef]
- Okunogbe, A.; Nugent, R.; Spencer, G.; Ralston, J.; Wilding, J. Economic impacts of overweight and obesity: Current and future estimates for eight countries. BMJ Glob. Health 2021, 6, e006351. [Google Scholar] [CrossRef]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef]
- Meigs, J.B.; Wilson, P.W.; Fox, C.S.; Vasan, R.S.; Nathan, D.M.; Sullivan, L.M.; D’Agostino, R.B. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J. Clin. Endocrinol. Metab. 2006, 91, 2906–2912. [Google Scholar] [CrossRef]
- Huang, L.O.; Loos, R.J.F.; Kilpelainen, T.O. Evidence of genetic predisposition for metabolically healthy obesity and metabolically obese normal weight. Physiol. Genom. 2018, 50, 169–178. [Google Scholar] [CrossRef]
- Park, J.M.; Park, D.H.; Song, Y.; Kim, J.O.; Choi, J.E.; Kwon, Y.J.; Kim, S.J.; Lee, J.W.; Hong, K.W. Understanding the genetic architecture of the metabolically unhealthy normal weight and metabolically healthy obese phenotypes in a Korean population. Sci. Rep. 2021, 11, 2279. [Google Scholar] [CrossRef]
- Walton, R.G.; Zhu, B.B.; Unal, R.; Spencer, M.; Sunkara, M.; Morris, A.J.; Charnigo, R.; Katz, W.S.; Daugherty, A.; Howatt, D.A.; et al. Increasing Adipocyte Lipoprotein Lipase Improves Glucose Metabolism in High Fat Diet-induced Obesity. J. Biol. Chem. 2015, 290, 11547–11556. [Google Scholar] [CrossRef]
- VidalPuig, A.; JimenezLinan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef]
- AlSaleh, A.; Sanders, T.A.B.; O’Dell, S.D. Postgraduate Symposium Effect of interaction between PPARG, PPARA and ADIPOQ gene variants and dietary fatty acids on plasma lipid profile and adiponectin concentration in a large intervention study. Proc. Nutr. Soc. 2012, 71, 141–153. [Google Scholar] [CrossRef]
- Evseeva, M.N.; Dyikanov, D.T.; Karagyaur, M.N.; Prikazchikova, T.A.; Sheptulina, A.F.; Balashova, M.S.; Zatsepin, T.S.; Rubtsov, Y.P.; Kulebyakin, K.Y. Hematopoietically-expressed homeobox protein HHEX regulates adipogenesis in preadipocytes. Biochimie 2021, 185, 68–77. [Google Scholar] [CrossRef]
- Narasimhan, A.; Chinnaiyan, M.; Karundevi, B. Ferulic acid regulates hepatic GLUT2 gene expression in high fat and fructose-induced type-2 diabetic adult male rat. Eur. J. Pharmacol. 2015, 761, 391–397. [Google Scholar] [CrossRef]
- Mehramiz, M.; Ghasemi, F.; Esmaily, H.; Tayefi, M.; Hassanian, S.M.; Sadeghzade, M.; Sadabadi, F.; Moohebati, M.; Azarpazhooh, M.R.; Parizadeh, S.M.R.; et al. Interaction between a variant of CDKN2A/B-gene with lifestyle factors in determining dyslipidemia and estimated cardiovascular risk: A step toward personalized nutrition. Clin. Nutr. 2018, 37, 254–261. [Google Scholar] [CrossRef]
- Qi, Q.B.; Bray, G.A.; Hu, F.B.; Sacks, F.M.; Qi, L. Weight-loss diets modify glucose-dependent insulinotropic polypeptide receptor rs2287019 genotype effects on changes in body weight, fasting glucose, and insulin resistance: The Preventing Overweight Using Novel Dietary Strategies trial. Am. J. Clin. Nutr. 2012, 95, 506–513. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef]
- Piko, P.; Werissa, N.A.; Fiatal, S.; Sandor, J.; Adany, R. Impact of Genetic Factors on the Age of Onset for Type 2 Diabetes Mellitus in Addition to the Conventional Risk Factors. J. Pers. Med. 2020, 11, 6. [Google Scholar] [CrossRef]
- Park, M.H.; Kim, N.; Lee, J.Y.; Park, H.Y. Genetic loci associated with lipid concentrations and cardiovascular risk factors in the Korean population. J. Med. Genet. 2011, 48, 10–15. [Google Scholar] [CrossRef]
- Nikolajevic Starcevic, J.; Santl Letonja, M.; Praznikar, Z.J.; Makuc, J.; Vujkovac, A.C.; Petrovic, D. Polymorphisms XbaI (rs693) and EcoRI (rs1042031) of the ApoB gene are associated with carotid plaques but not with carotid intima-media thickness in patients with diabetes mellitus type 2. Vasa 2014, 43, 171–180. [Google Scholar] [CrossRef]
- Niu, C.; Luo, Z.; Yu, L.; Yang, Y.; Chen, Y.; Luo, X.; Lai, F.; Song, Y. Associations of the APOB rs693 and rs17240441 polymorphisms with plasma APOB and lipid levels: A meta-analysis. Lipids Health Dis. 2017, 16, 166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiao, W.; Zhao, X.; Tao, M. Quantitative assessment of the influence of hematopoietically expressed homeobox variant (rs1111875) on type 2 diabetes risk. Mol. Genet. Metab. 2011, 102, 194–199. [Google Scholar] [CrossRef]
- Abudureyimu, S.; Abulaiti, P.; Li, H.; Xing, Z.; Liu, S.; Li, W.; Gao, Y. Roles of endothelial lipase gene related single nucleotide polymorphisms in patients with coronary artery disease. Gene 2021, 788, 145669. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet-Ostaptchouk, J.V.; Nuotio, M.L.; Slagter, S.N.; Doiron, D.; Fischer, K.; Foco, L.; Gaye, A.; Gogele, M.; Heier, M.; Hiekkalinna, T.; et al. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: A collaborative analysis of ten large cohort studies. BMC Endocr. Disord. 2014, 14, 9. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Saeedi Moghaddam, S.; Masinaei, M.; Rezaei, N.; Mohammadi Fateh, S.; Dilmaghani-Marand, A.; Abdolhamidi, E.; Razi, F.; Khashayar, P.; Mahdavihezaveh, A.; et al. Association between being metabolically healthy/unhealthy and metabolic syndrome in Iranian adults. PLoS ONE 2022, 17, e0262246. [Google Scholar] [CrossRef]
- Gutierrez-Repiso, C.; Linares-Pineda, T.M.; Gonzalez-Jimenez, A.; Aguilar-Lineros, F.; Valdes, S.; Soriguer, F.; Rojo-Martinez, G.; Tinahones, F.J.; Morcillo, S. Epigenetic Biomarkers of Transition from Metabolically Healthy Obesity to Metabolically Unhealthy Obesity Phenotype: A Prospective Study. Int. J. Mol. Sci. 2021, 22, 10417. [Google Scholar] [CrossRef]
- Kramer, C.K.; Zinman, B.; Retnakaran, R. Are metabolically healthy overweight and obesity benign conditions?: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 758–769. [Google Scholar] [CrossRef]
- Mongraw-Chaffin, M.; Foster, M.C.; Anderson, C.A.M.; Burke, G.L.; Haq, N.; Kalyani, R.R.; Ouyang, P.; Sibley, C.T.; Tracy, R.; Woodward, M.; et al. Metabolically Healthy Obesity, Transition to Metabolic Syndrome, and Cardiovascular Risk. J. Am. Coll. Cardiol. 2018, 71, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Deedwania, P.; Lavie, C.J. Dangers and Long-Term Outcomes in Metabolically Healthy Obesity: The Impact of the Missing Fitness Component. J. Am. Coll. Cardiol. 2018, 71, 1866–1868. [Google Scholar] [CrossRef]
- Schroder, H.; Ramos, R.; Baena-Diez, J.M.; Mendez, M.A.; Canal, D.J.; Fito, M.; Sala, J.; Elosua, R. Determinants of the transition from a cardiometabolic normal to abnormal overweight/obese phenotype in a Spanish population. Eur. J. Nutr. 2014, 53, 1345–1353. [Google Scholar] [CrossRef]
- Soriguer, F.; Gutierrez-Repiso, C.; Rubio-Martin, E.; Garcia-Fuentes, E.; Almaraz, M.C.; Colomo, N.; Esteva de Antonio, I.; de Adana, M.S.; Chaves, F.J.; Morcillo, S.; et al. Metabolically healthy but obese, a matter of time? Findings from the prospective Pizarra study. J. Clin. Endocrinol. Metab. 2013, 98, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Appleton, S.L.; Seaborn, C.J.; Visvanathan, R.; Hill, C.L.; Gill, T.K.; Taylor, A.W.; Adams, R.J.; North West Adelaide Health Study, T. Diabetes and cardiovascular disease outcomes in the metabolically healthy obese phenotype: A cohort study. Diabetes Care 2013, 36, 2388–2394. [Google Scholar] [CrossRef]
- WHO. WHO European Regional Obesity Report 2022; WHO Regional Office for Europe: Copenhagen, Denmark, 2022. [Google Scholar]
- Guo, F.; Garvey, W.T. Cardiometabolic Disease Staging Predicts Effectiveness of Weight-Loss Therapy to Prevent Type 2 Diabetes: Pooled Results From Phase III Clinical Trials Assessing Phentermine/Topiramate Extended Release. Diabetes Care 2017, 40, 856–862. [Google Scholar] [CrossRef]
- Walley, A.J.; Asher, J.E.; Froguel, P. The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet. 2009, 10, 431–442. [Google Scholar] [CrossRef]
- Choquet, H.; Meyre, D. Genetics of Obesity: What have we Learned? Curr. Genom. 2011, 12, 169–179. [Google Scholar] [CrossRef]
- Herrera, B.M.; Keildson, S.; Lindgren, C.M. Genetics and epigenetics of obesity. Maturitas 2011, 69, 41–49. [Google Scholar] [CrossRef]
- Thompson, A.L. Developmental origins of obesity: Early feeding environments, infant growth, and the intestinal microbiome. Am. J. Hum. Biol. 2012, 24, 350–360. [Google Scholar] [CrossRef]
- Hall, K.D.; Farooqi, I.S.; Friedman, J.M.; Klein, S.; Loos, R.J.F.; Mangelsdorf, D.J.; O’Rahilly, S.; Ravussin, E.; Redman, L.M.; Ryan, D.H.; et al. The energy balance model of obesity: Beyond calories in, calories out. Am. J. Clin. Nutr. 2022, 115, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Aronne, L.J.; Astrup, A.; de Cabo, R.; Cantley, L.C.; Friedman, M.I.; Heymsfield, S.B.; Johnson, J.D.; King, J.C.; Krauss, R.M.; et al. The carbohydrate-insulin model: A physiological perspective on the obesity pandemic. Am. J. Clin. Nutr. 2021, 114, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Sorensen, T.I.A. An integrated model of obesity pathogenesis that revisits causal direction. Nat. Rev. Endocrinol. 2022, 18, 261–262. [Google Scholar] [CrossRef]
- Szeles, G.; Voko, Z.; Jenei, T.; Kardos, L.; Pocsai, Z.; Bajtay, A.; Papp, E.; Pasti, G.; Kosa, Z.; Molnar, I.; et al. A preliminary evaluation of a health monitoring programme in Hungary. Eur. J. Public Health 2005, 15, 26–32. [Google Scholar] [CrossRef]
- Szigethy, E.; Szeles, G.; Horvath, A.; Hidvegi, T.; Jermendy, G.; Paragh, G.; Blasko, G.; Adany, R.; Voko, Z. Epidemiology of the metabolic syndrome in Hungary. Public Health 2012, 126, 143–149. [Google Scholar] [CrossRef]
- Nagy, K.; Fiatal, S.; Sandor, J.; Adany, R. Distinct Penetrance of Obesity-Associated Susceptibility Alleles in the Hungarian General and Roma Populations. Obes. Facts 2017, 10, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Piko, P.; Fiatal, S.; Kosa, Z.; Sandor, J.; Adany, R. Genetic factors exist behind the high prevalence of reduced high-density lipoprotein cholesterol levels in the Roma population. Atherosclerosis 2017, 263, 119–126. [Google Scholar] [CrossRef]
- Werissa, N.A.; Piko, P.; Fiatal, S.; Kosa, Z.; Sandor, J.; Adany, R. SNP-Based Genetic Risk Score Modeling Suggests No Increased Genetic Susceptibility of the Roma Population to Type 2 Diabetes Mellitus. Genes 2019, 10, 942. [Google Scholar] [CrossRef] [PubMed]
- Salanti, G.; Southam, L.; Altshuler, D.; Ardlie, K.; Barroso, I.; Boehnke, M.; Cornelis, M.C.; Frayling, T.M.; Grallert, H.; Grarup, N.; et al. Underlying genetic models of inheritance in established type 2 diabetes associations. Am. J. Epidemiol. 2009, 170, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Templeton, G.F.; Pope, M.B.; Burney, L.L. The Usefulness of the Two-Step Normality Transformation in Retesting Existing Theories: Evidence on the Productivity Paradox. Data Base Adv. Inf. Syst. 2021, 52, 53–64. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Jonckheere, A.R. A Distribution-free k-sample test against ordered alternatives. Biometrika 1954, 41, 133–145. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MHO (n = 81) | MUO (n = 317) | p-Value | ||
|---|---|---|---|---|
| Average (95% CI) | ||||
| Age (years) | 45.5 (42.7–48.2) | 54.4 (53.2–55.5) | <0.001 | |
| BMI (kg/m2) | 33.1 (32.4–33.8) | 34.5 (34.1–35.0) | 0.001 | |
| Waist circumference (cm) | 106.9 (104.5–109.2) | 112.9 (111.5–114.2) | <0.001 | |
| Systolic blood pressure (mmHg) | 127.3 (123.9–130.7) | 141.0 (1392–142.8) | <0.001 | |
| Diastolic blood pressure (mmHg) | 80.7 (78.9–82.5) | 85.0 (84.0–86.1) | <0.001 | |
| Fasting TAG level (mM) | 1.20 (1.09–1.30) | 2.21 (2.02–2.39) | <0.001 | |
| Fasting HDL-C level (mM) | 1.45 (1.45–1.52) | 1.20 (1.17–1.24) | <0.001 | |
| Fasting glucose level (mM) | 4.33 (4.16–4.51) | 5.56 (5.33–5.79) | <0.001 | |
| Fasting insulin level (mM) | 186.57 (165.36–207.79) | 278.59 (256.62–300.57) | <0.001 | |
| HOMA-IR | 5.29 (4.59–5.98) | 10.98 (9.57–12.40) | <0.001 | |
| CRP concentration (nM) | 28.86 (23.66–34.06) | 45.52 (40.75–50.30) | <0.001 | |
| Prevalence in % (95% CI) | p-Value | |||
| Women | 46.9 (36.3–57.7) | 47.6 (42.2–53.1) | 0.908 | |
| Anti-hypertensive treatment | 0.0 | 75.4 (70.4–79.9) | <0.001 | |
| Anti-diabetic treatment | 0.0 | 16.1 (12.4–20.4) | <0.001 | |
| Lipid-lowering treatment | 6.2 (2.4–13.0) | 31.5 (26.6–36.8) | <0.001 | |
| Education | Less than primary or primary | 28.4 (19.5–38.8) | 28.7 (23.9–33.9) | 0.846 |
| High or vocational school | 63.0 (52.1–72.9) | 60.6 (55.1–65.8) | ||
| College or university | 8.6 (3.9–16.2) | 10.7 (7.7–14.5) | ||
| Normal Weight BMI < 25 | Overweight BMI: 25– < 30 | Obese BMI ≥ 30 | p for Trend | |
|---|---|---|---|---|
| Average oGRS Value (95% CI) | ||||
| Metabolically healthy | 23.18 (22.94–23.42) | 23.27 (22.96–23.58) | 21.44 (20.95–21.94) | <0.001 ** |
| Metabolically unhealthy | 22.65 (21.88–23.41) | 23.05 (22.72–23.39) | 23.72 (23.48–23.95) | <0.001 ** |
| Average oGRS4 Value (95% CI) | p for Trend | |||
| Metabolically healthy | 5.07 (4.94–5.20) | 5.10 (4.93–5.28) | 4.47 (4.17–4.77) | 0.033 * |
| Metabolically unhealthy | 4.92 (4.52–5.32) | 5.04 (4.85–5.23) | 5.38 (5.24–5.53) | <0.001 ** |
| OR (95% CI) | p-value | |
|---|---|---|
| oGRS | 1.10 (1.03–1.17) | 0.00257 ** |
| oGRS × BMI | 1.07 (1.05–1.08) | <0.001 ** |
| oGRS4 | 1.15 (1.03–1.28) | 0.012 * |
| oGRS4 × BMI | 1.01 (1.00–1.01) | 0.001 ** |
| Wildman et al. [26] | Meigs et al. [27] | |
|---|---|---|
| BP ≥ 130/85 mmHg | X | X |
| Use of blood pressure medication | X | X |
| Fasting TAG level ≥ 1.7 mM (≥150 mg/dL) | X | X |
| HDL cholesterol 1 mM (≥40 mg/dL) in men or 1.3 mM (50 mg/dL) in women | X | X |
| Lipid-lowering medication | X | |
| Fasting glucose level 5.6 mM (≥100 mg/dL) | X | X |
| Use of medications for diabetes | X | |
| Waist circumference > 102 cm in men or > 88 cm in women | X | |
| HOMA-IR ≥ 90% among those with BMI ≥ 30 kg/m2 | X | |
| C-reactive protein ≥ 90% among those with BMI ≥ 30 kg/m2 | X | |
| Criteria for MUO | >1 | >2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piko, P.; Llanaj, E.; Nagy, K.; Adany, R. Genetic Background of Metabolically Healthy and Unhealthy Obesity Phenotypes in Hungarian Adult Sample Population. Int. J. Mol. Sci. 2023, 24, 5209. https://doi.org/10.3390/ijms24065209
Piko P, Llanaj E, Nagy K, Adany R. Genetic Background of Metabolically Healthy and Unhealthy Obesity Phenotypes in Hungarian Adult Sample Population. International Journal of Molecular Sciences. 2023; 24(6):5209. https://doi.org/10.3390/ijms24065209
Chicago/Turabian StylePiko, Peter, Erand Llanaj, Karoly Nagy, and Roza Adany. 2023. "Genetic Background of Metabolically Healthy and Unhealthy Obesity Phenotypes in Hungarian Adult Sample Population" International Journal of Molecular Sciences 24, no. 6: 5209. https://doi.org/10.3390/ijms24065209
APA StylePiko, P., Llanaj, E., Nagy, K., & Adany, R. (2023). Genetic Background of Metabolically Healthy and Unhealthy Obesity Phenotypes in Hungarian Adult Sample Population. International Journal of Molecular Sciences, 24(6), 5209. https://doi.org/10.3390/ijms24065209