



Genetics of Multiple System Atrophy and Progressive Supranuclear Palsy: A Systemized Review of the Literature


Abstract
:1. Introduction
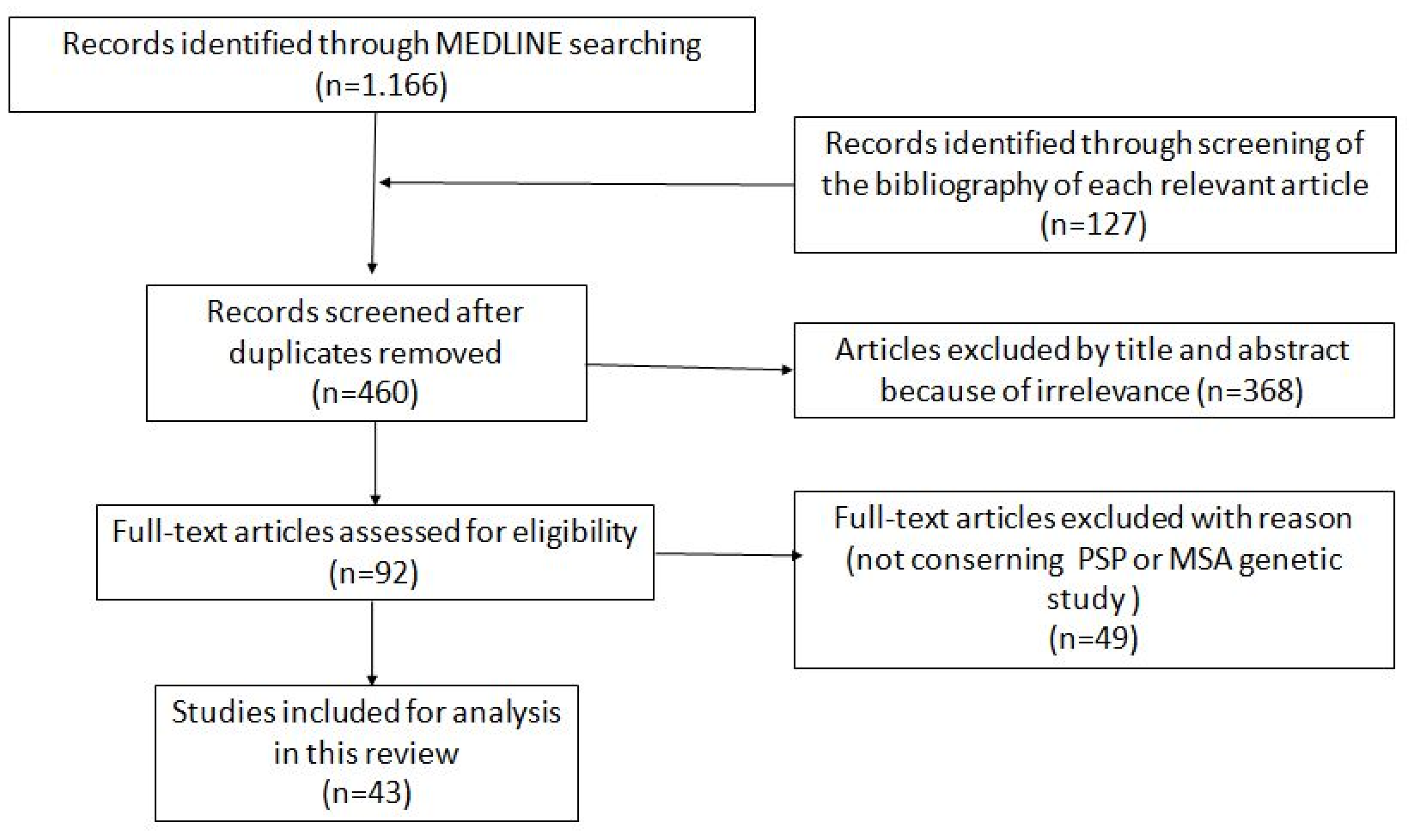
2. Materials and Methods
2.1. Study Design
2.2. Inclusion Criteria
2.3. Exclusion Criteria
3. Results
3.1. SNCA—α-Synuclein and MSA
3.2. COQ2—Coenzyme Q2 Polyprenyltransferaseand MSA
3.3. MAPT—Microtubule-Associated Protein Tauand MSA
3.4. GBA1—Beta-Glucocerebrosidaseand MSA
3.5. LRRK2—Leucine-Rich Repeat Kinase 2 and MSA
3.6. C9orf72—Chromosome 9 Open Reading Frame and MSA
3.7. MAPT Mutations and PSP
3.8. LRRK2 Mutations in PSP
3.9. Other Genes and PSP
3.10. Genetic Factors Identified by GWAS
4. Discussion
5. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Respondek, G.; Levin, J.; Höglinger, G.U. Progressive supranuclear palsy and multiple system atrophy: Clinicopathological concepts and therapeutic challenges. Curr. Opin. Neurol. 2018, 31, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Progressive Supranuclear Palsy-Parkinsonism Predominant (PSP-P)-A Clinical Challenge at the Boundaries of PSP and Parkinson’s Disease (PD). Front. Neurol. 2020, 11, 180. [Google Scholar] [CrossRef]
- Palma, J.-A.; Norcliffe-Kaufmann, L.; Kaufmann, H. Diagnosis of multiple system atrophy. Auton. Neurosci. 2018, 211, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.; Pernaute, R.S.; Fontan, A.; Ruiz, P.G.; Honnorat, J.; Lynch, T.; Chin, S.; Gonzalo, I.; Rabano, A.; Martınez, A.; et al. Clinical genetics of familial progressive supranuclear palsy. Brain 1999, 122, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Contreras, M.Y.S.; Strongosky, A.J.; Ogaki, K.; Whaley, N.R.; Tacik, P.M.; van Gerpen, J.A.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K.; et al. Three sib-pairs of autopsy-confirmed progressive supranuclear palsy. Park. Relat. Disord. 2015, 21, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Ros, R.; Gomez Garre, P.; Hirano, M.; Tai, Y.F.; Ampuero, I.; Vidal, L.; Rojo, A.; Fontan, A.; Vazquez, A.; Fanjul, S.; et al. Genetic linkage of autosomal dominant progressive supranuclear palsy to 1q31.1. Ann. Neurol. 2005, 57, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Momose, Y.; Tokiguchi, S.; Shimohata, M.; Terajima, K.; Onodera, O.; Kakita, A.; Yamada, M.; Takahashi, H.; Hirasawa, M.; et al. Multiplex families with multiple system atrophy. Arch. Neurol. 2007, 64, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Wüllner, U.; Schmitt, I.; Kammal, M.; Kretzschmar, H.A.; Neumann, M. Definite multiple system atrophy in a German family. J. Neurol. Neurosurg. Psychiatry 2009, 80, 449–450. [Google Scholar] [CrossRef] [Green Version]
- Hohler, A.D.; Singh, V.J. Probable hereditary multiple system atrophy-autonomic (MSA-A) in a family in the United States. J. Clin. Neurosci. 2012, 19, 479–480. [Google Scholar] [CrossRef]
- Soma, H.; Yabe, I.; Takei, A.; Fujiki, N.; Yanagihara, T.; Sasaki, H. Heredity in multiple system atrophy. J. Neurol. Sci. 2006, 240, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Yabe, I.; Yaguchi, H.; Kato, Y.; Miki, Y.; Takahashi, H.; Tanikawa, S.; Shirai, S.; Takahashi, I.; Kimura, M.; Hama, Y.; et al. Mutations in bassoon in individuals with familial and sporadic progressive supranuclear palsy-like syndrome. Sci. Rep. 2018, 8, 819. [Google Scholar] [CrossRef] [Green Version]
- Ygland, E.; van Westen, D.; Englund, E.; Rademakers, R.; Wszolek, Z.K.; Nilsson, K.; Nilsson, C.; Landqvist Waldo, M.; Alafuzoff, I.; Hansson, O.; et al. Slowly progressive dementia caused by MAPT R406W mutations: Longitudinal report on a new kindred and systematic review. Alzheimer’s Res. Ther. 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; Sima, A.A.F.; Junck, L.; Kluin, K.J.; Koeppe, R.A.; Ba, M.E.L.; Little, R.J.A. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann. Neurol. 1996, 39, 241–255. [Google Scholar] [CrossRef]
- Fujioka, S.; Algom, A.A.; Murray, M.E.; Strongosky, A.; Soto-Ortolaza, A.I.; Rademakers, R.; Ross, O.A.; Wszolek, Z.K.; Dickson, D.W. Similarities between familial and sporadic autopsy-proven progressive supranuclear palsy. Neurology 2013, 80, 2076–2078. [Google Scholar] [CrossRef] [Green Version]
- Donker Kaat, L.; Boon, A.J.W.; Azmani, A.; Kamphorst, W.; Breteler, M.M.B.; Anar, B.; Heutink, P.; van Swieten, J.C. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 2009, 73, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, J.A.; Shatunov, A.; Jones, A.; Bs, S.N.K.; Huang, A.Y.; Lawrence, L.; Lowe, J.K.; Lewis, C.; Payan, C.A.M.; et al. Genome-wide survey of copy number variants finds MAPT duplications in progressive supranuclear palsy. Mov. Disord. 2019, 34, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Sailer, A.; Scholz, S.W.; Nalls, M.A.; Schulte, C.; Federoff, M.; Price, T.R.; Lees, A.; Ross, O.A.; Dickson, D.W.; Mok, K.; et al. European Multiple System Atrophy Study Group and the UK Multiple System Atrophy Study Group A genome-wide association study in multiple system atrophy. Neurology 2016, 87, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Katzeff, J.S.; Phan, K.; Purushothuman, S.; Halliday, G.M.; Kim, W.S. Cross-examining candidate genes implicated in multiple system atrophy. Acta Neuropathol. Commun. 2019, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Zhou, Y.; Bin Jiao, B.; Shen, L. Genetics of Progressive Supranuclear Palsy: A Review. J. Park. Dis. 2021, 11, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.J.; Booth, A. A typology of reviews: An analysis of 14 review types and associated methodologies. Health Inf. Libr. J. 2009, 26, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, T.; Peacock, R. Effectiveness and efficiency of search methods in systematic reviews of complex evidence: Audit of primary sources. BMJ 2005, 331, 1064–1065. [Google Scholar] [CrossRef] [Green Version]
- Bougea, A. Synuclein in neurodegeneration. Adv. Clin. Chem. 2020, 103, 97–134. [Google Scholar]
- Stefanova, N.; Reindl, M.; Poewe, W.; Wenning, G.K. In vitro models of multiple system atrophy. Mov. Disord. 2005, 20 (Suppl. S12), S53–S56. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. Alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiely, A.P.; Ling, H.; Asi, Y.T.; Kara, E.; Proukakis, C.; Schapira, A.H.; Morris, H.R.; Roberts, H.C.; Lubbe, S.; Limousin, P.; et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol. Neurodegener. 2015, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2015, 35, e2181–e2185. [Google Scholar]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Dürr, A.; Wood, N.W.; Parkinson, M.H.; Camuzat, A.; Hulot, J.S.; Morrison, K.E.; Renton, A.; Sussmuth, S.D.; Landwehrmeyer, B.G.; et al. NNIPPS Genetic Study Group. Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS ONE 2009, 4, e7114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, S.W.; Houlden, H.; Schulte, C.; Sharma, M.; Li, A.; Berg, D.; Melchers, A.; Paudel, R.; Gibbs, J.R.; Simon-Sanchez, J.; et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol. 2009, 65, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wei, Q.Q.; Ou, R.; Cao, B.; Chen, X.; Zhao, B.; Guo, X.; Yang, Y.; Chen, K.; Wu, Y.; et al. Genetic Variants of SNCA Are Associated with Susceptibility to Parkinson’s Disease but Not Amyotrophic Lateral Sclerosis or Multiple System Atrophy in a Chinese Population. PLoS ONE 2015, 10, e0133776. [Google Scholar] [CrossRef] [Green Version]
- Mantle, D.; Hargreaves, I.P. Coenzyme Q10: Role in Less Common Age-Related Disorders. Antioxidants 2022, 11, 2293. [Google Scholar] [CrossRef]
- Porto, K.J.; Hirano, M.; Mitsui, J.; Chikada, A.; Matsukawa, T.; Ishiura, H.; Toda, T.; Kusunoki, S.; Tsuji, S. COQ2 V393A confers high risk susceptibility for multiple system atrophy in East Asian population. J. Neurol. Sci. 2021, 429, 117623. [Google Scholar] [CrossRef]
- Jeon, B.S.; Farrer, M.J.; Bortnick, S.F. Mutant COQ2 in multiplesystem atrophy. N. Engl. J. Med. 2014, 371, 80. [Google Scholar]
- Mikasa, M.; Kanai, K.; Li, Y.; Yoshino, H.; Mogushi, K.; Hayashida, A.; Ikeda, A.; Kawajiri, S.; Okuma, Y.; Kashihara, K.; et al. COQ2 variants in Parkinson’s disease and multiple system atrophy. J. Neural Transm. 2018, 125, 937–944. [Google Scholar] [CrossRef]
- Schottlaender, L.V.; Houlden, H.; Multiple-System Atrophy (MSA) Brain Bank Collaboration. Mutant COQ2 in multiple-system atrophy. N. Engl. J. Med. 2014, 371, 81. [Google Scholar]
- Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 2013, 369, 233–244. [Google Scholar] [CrossRef]
- Sun, Z.; Ohta, Y.; Yamashita, T.; Sato, K.; Takemoto, M.; Hishikawa, N.; Abe, K. New susceptible variant ofCOQ2gene in Japanese patients with sporadic multiple system atrophy. Neurol. Genet. 2016, 2, e54. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.-D.; Li, H.-F.; Wang, H.-X.; Ni, W.; Dong, Y.; Wu, Z.-Y. Mutation analysis of COQ2 in Chinese patients with cerebellar subtype of multiple system atrophy. CNS Neurosci. Ther. 2015, 21, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Kasai, T.; Tokuda, T.; Ohmichi, T.; Ishii, R.; Tatebe, H.; Nakagawa, M.; Mizuno, T. Serum Levels of Coenzyme Q10 in Patients with Multiple System Atrophy. PLoS ONE 2016, 11, e0147574. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Huang, P.; Cui, S.; Gao, C.; Lin, Y.; Fu, R.; Shen, J.; He, Y.; Tan, Y.; et al. Clinical correlates of decreased plasma coenzyme Q10 levels in patients with multiple system atrophy. Park. Relat. Disord. 2018, 57, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Compta, Y.; Giraldo, D.M.; Muñoz, E.; Antonelli, F.; Fernández, M.; Bravo, P.; Soto, M.; Cámara, A.; Torres, F.; Martí, M.J.; et al. Cerebrospinal fluid levels of coenzyme Q10 are reduced in multiple system atrophy. Park. Relat. Disord. 2017, 46, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Barca, E.; Kleiner, G.; Tang, G.; Ziosi, M.; Tadesse, S.; Masliah, E.; Louis, E.D.; Faust, P.; Kang, U.J.; Torres, J.; et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J. Neuropathol. Exp. Neurol. 2016, 75, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar]
- Cairns, N.J.; Atkinson, P.F.; Hanger, D.P.; Anderton, B.H.; Daniel, S.E.; Lantos, P.L. Tau protein in the glial cytoplasmic inclusions of multiple system atrophy can be distinguished from abnormal tau in Alzheimer’s disease. Neurosci. Lett. 1997, 230, 49–52. [Google Scholar] [CrossRef]
- Ezquerra, M.; Pastor, P.; Gaig, C.; Vidal-Taboada, J.M.; Cruchaga, C.; Muñoz, E.; Martí, M.J.; Valldeoriola, F.; Aguilar, M.; Calopa, M.; et al. Different MAPT haplotypes are associated with Parkinson’s disease and progressive supranuclear palsy. Neurobiol. Aging 2011, 32, 547.e11–547.e16. [Google Scholar] [CrossRef]
- Allen, M.; Kachadoorian, M.; Quicksall, Z.; Zou, F.; Chai, H.S.; Younkin, C.; Crook, J.E.; Pankratz, V.S.; Carrasquillo, M.M.; Krishnan, S.; et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res. Ther. 2014, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Vilarino-Guell, C.; Soto-Ortolaza, A.I.; Rajput, A.; Mash, D.C.; Papapetropoulos, S.; Pahwa, R.; Lyons, K.E.; Uitti, R.J.; Wszolek, Z.K.; Dickson, D.W.; et al. MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology 2011, 76, 670–672. [Google Scholar] [CrossRef] [Green Version]
- Labbé, C.; Heckman, M.G.; Lorenzo-Betancor, O.; Murray, M.E.; Ogaki, K.; Soto-Ortolaza, A.I.; Walton, R.L.; Fujioka, S.; Koga, S.; Uitti, R.J.; et al. MAPT haplotype diversity in multiple system atrophy. Park. Relat. Disord. 2016, 30, 40–45. [Google Scholar]
- Vieira, S.R.L.; Schapira, A.H.V. Glucocerebrosidase mutations and Parkinson disease. J. Neural Transm. 2022, 129, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Matsukawa, T.; Sasaki, H.; Yabe, I.; Matsushima, M.; Dürr, A.; Brice, A.; Takashima, H.; Kikuchi, A.; Aoki, M.; et al. Variants associated with Gaucher disease in multiple system atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Sklerov, M.; Kang, U.J.; Liong, C.; Clark, L.; Marder, K.; Pauciulo, M.; Nichols, W.C.; Chung, W.K.; Honig, L.S.; Cortes, E.; et al. Frequency of GBA Variants in Autopsy-proven Multiple System Atrophy. Mov. Disord. Clin. Pract. 2017, 4, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segarane, B.; Li, A.; Paudel, R.; Scholz, S.; Neumann, J.; Lees, A.; Revesz, T.; Hardy, J.; Mathias, C.J.; Wood, N.W.; et al. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology 2009, 72, 1185–1186. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.-Y.; Guo, J.-F.; Han, W.-W.; Zuo, X.; Wang, L.; Yao, L.-Y.; Pan, Q.; Xia, K.; Yan, X.-X.; Tang, B.-S. Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China. J. Clin. Neurosci. 2013, 20, 217–219. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Schottlaender, L.; Chelban, V.; Meissner, W.G.; Federoff, M.; Singleton, A.; Houlden, H. MSA Exome Consortium Lysosomal storage disorder gene variants in multiple system atrophy. Brain 2018, 141, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernick, A.I.; Walton, R.L.; Koga, S.; Soto-Beasley, A.I.; Heckman, M.G.; Gan-Or, Z.; Ren, Y.; Rademakers, R.; Uitti, R.J.; Wszolek, Z.K.; et al. GBA variation and susceptibility to multiple system atrophy. Park. Relat. Disord. 2020, 77, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Iannotta, L.; Greggio, E. LRRK2 signaling in neurodegeneration: Two decades of progress. Essays Biochem. 2021, 65, 859–872. [Google Scholar]
- Usmani, A.; Shavarebi, F.; Hiniker, A. The Cell Biology of LRRK2 in Parkinson’s Disease. Mol. Cell. Biol. 2021, 41, e00660-20. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Foroud, T.; May, S.; Senthil, G.; Sandroni, P.; Low, P.A.; Reich, S.; Colcher, A.; Stern, M.B.; Ondo, W.G.; et al. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov. Disord. 2007, 22, 546–549. [Google Scholar] [CrossRef]
- Yuan, X.; Chen, Y.; Cao, B.; Zhao, B.; Wei, Q.; Guo, X.; Yang, Y.; Yuan, L.; Shang, H. An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Park. Relat. Disord. 2015, 21, 147–149. [Google Scholar] [CrossRef]
- Heckman, M.G.; Schottlaender, L.; Soto-Ortolaza, A.I.; Diehl, N.N.; Rayaprolu, S.; Ogaki, K.; Fujioka, S.; Murray, M.E.; Cheshire, W.P.; Uitti, R.J.; et al. LRRK2 exonic variants and risk of multiple system atrophy. Neurology 2014, 83, 2256–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboldi, G.M.; Palma, J.; Cortes, E.; Iida, M.A.; Sikder, T.; Rn, B.H.; Raj, T.; Walker, R.H.; Crary, J.F.; Kaufmann, H.; et al. Early-onset pathologically proven multiple system atrophy with LRRK2 G2019S mutation. Mov. Disord. 2019, 34, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, S.W.; Majounie, E.; Revesz, T.; Holton, J.L.; Okun, M.S.; Houlden, H.; Singleton, A.B. Multiple system atrophy is not caused by C9orf72 hexanucleotide repeat expansions. Neurobiol. Aging 2015, 36, 1223.e1–1223.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, Y.; Wei, Q.; Ou, R.; Cao, B.; Zhao, B.; Shang, H.-F. C9ORF72 repeat expansions in Chinese patients with Parkinson’s disease and multiple system atrophy. J. Neural Transm. 2016, 123, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Schottlaender, L.V.; Polke, J.M.; Ling, H.; MacDoanld, N.D.; Tucci, A.; Nanji, T.; Pittman, A.; de Silva, R.; Holton, J.L.; Revesz, T.; et al. Analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonism. Neurobiol. Aging. 2015, 36, e1–e1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, J.S.; Quinzii, C.; Dunning-Broadbent, J.; Waters, C.; Mitsumoto, H.; Brannagan, T.H., 3rd; Cosentino, S.; Huey, E.D.; Nagy, P.; Kuo, S.H. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol. 2014, 71, 771–774. [Google Scholar] [CrossRef] [Green Version]
- Cannas, A.; Solla, P.; Borghero, G.; Floris, G.L.; Chio, A.; Mascia, M.M.; Modugno, N.; Muroni, A.; Orofino, G.; Di Stefano, F.; et al. C9ORF72 intermediate repeat expansion in patients affected by atypical parkinsonian syndromes or Parkinson’s disease complicated by psychosis or dementia in a Sardinian population. J. Neurol. 2015, 262, 2498–2503. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, G.; Gagliardi, M.; Procopio, R.; Morelli, M.; Quattrone, A.; Brighina, L.; Quattrone, A.; Annesi, G. Multiple system atrophy and C9orf72 hexanucleotide repeat expansions in a cohort of Italian patients. Neurobiol. Aging 2021, 112, 12–15. [Google Scholar] [CrossRef]
- King, A.; Lee, Y.K.; Jones, S.; Troakes, C. A pathologically confirmed case of combined amyotrophic lateral sclerosis with C9orf72 mutation and multiple system atrophy. Neuropathology 2022, 42, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Muma, N.A.; Zhukareva, V.; Cochran, E.J.; Shannon, K.M.; Hurtig, H.; Koller, W.C.; Bird, T.D.; Trojanowski, J.Q.; Lee, V.M.; et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol. 2002, 52, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Kawamata, T.; Komure, O.; Kuno, S.; D’Souza, I.; Poorkaj, P.; Kawai, J.; Tanimukai, S.; Yamamoto, Y.; Hasegawa, H.; et al. A mutation in the microtubule-associated protein tau in pallido-nigro-luysian degeneration. Neurology 1999, 53, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Delisle, M.B.; Murrell, J.R.; Richardson, R.; Trofatter, J.A.; Rascol, O.; Soulages, X.; Mohr, M.; Calvas, P.; Ghetti, B. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 1999, 98, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Soliveri, P.; Rossi, G.; Monza, D.; Tagliavini, F.; Piacentini, S.; Albanese, A.; Bugiani, O.; Girotti, F. A Case of Dementia Parkinsonism Resembling Progressive Supranuclear Palsy Due to Mutation in the Tau Protein Gene. Arch. Neurol. 2003, 60, 1454–1456. [Google Scholar] [CrossRef] [Green Version]
- Ogaki, K.; Li, Y.; Takanashi, M.; Ishikawa, K.-I.; Kobayashi, T.; Nonaka, T.; Hasegawa, M.; Kishi, M.; Yoshino, H.; Funayama, M.; et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Park. Relat. Disord. 2013, 19, 15–20. [Google Scholar] [CrossRef]
- Ogaki, K.; Motoi, Y.; Li, Y.; Tomiyama, H.; Shimizu, N.; Takanashi, M.; Nakanishi, A.; Yokoyama, K.; Hattori, N. Visual grasping in frontotemporal dementia and parkinsonism linked to chromosome 17 (microtubule-associated with protein tau): A comparison of N-Isopropyl-p- [(123)I]-iodoamphetamine brain perfusion single photon emission computed tomography analysis with progressive supranuclear palsy. Mov. Disord. 2011, 26, 561–563. [Google Scholar]
- Rohrer, J.D.; Paviour, D.; Vandrovcova, J.; Hodges, J.; de Silva, R.; Rossor, M.N. Novel L284R MAPT Mutation in a Family with an Autosomal Dominant Progressive Supranuclear Palsy Syndrome. Neurodegener. Dis. 2010, 8, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Pastor, P.; Pastor, E.; Carnero, C.; Vela, R.; Garcia, T.; Amer, G.; Tolosa, E.; Oliva, R. Familial atypical progres sive supranuclear palsy associated with homozigosity for the deln296 mutation in the tau gene. Ann. Neurol. 2001, 49, 263–267. [Google Scholar] [CrossRef]
- Rossi, G.; Gasparoli, E.; Pasquali, C.; Di Fede, G.; Testa, D.; Albanese, A.; Bracco, F.; Tagliavini, F. Progressive supranuclear palsy and Parkinson’s disease in a family with a new mutation in the tau gene. Ann. Neurol. 2004, 55, 448. [Google Scholar] [CrossRef]
- Nakayama, S.; Shimonaka, S.; Elahi, M.; Nishioka, K.; Oji, Y.; Matsumoto, S.-E.; Li, Y.; Yoshino, H.; Mogushi, K.; Hatano, T.; et al. Tau aggregation and seeding analyses of two novel MAPT variants found in patients with motor neuron disease and progressive parkinsonism. Neurobiol. Aging 2019, 84, 240.e13–240.e22. [Google Scholar] [CrossRef]
- Bird, T.D.; Nochlin, D.; Poorkaj, P.; Cherrier, M.; Kaye, J.; Payami, H.; Peskind, E.; Lampe, T.H.; Nemens, E.; Boyer, P.J.; et al. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 1999, 122, 741–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, R.; Thobois, S.; Streichenberger, N.; Kopp, N.; Sánchez, M.P.; Pérez, M.; Hoenicka, J.; Avila, J.; Honnorat, J.; De Yébenes, J.G. A New Mutation of the τ Gene, G303V, in Early-Onset Familial Progressive Supranuclear Palsy. Arch. Neurol. 2005, 62, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Stanford, P.M.; Halliday, G.M.; Brooks, W.S.; Kwok, J.B.; Storey, C.E.; Creasey, H.; Morris, J.G.; Fulham, M.J.; Schofield, P.R. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: Expansion of the disease phenotype caused by tau gene mutations. Brain 2000, 123, 880–893. [Google Scholar] [CrossRef] [Green Version]
- Spina, S.; Farlow, M.R.; Unverzagt, F.W.; Kareken, D.A.; Murrell, J.R.; Fraser, G.; Epperson, F.; Crowther, R.A.; Spillantini, M.G.; Goedert, M.; et al. The tauopathy associated with mutation+3 in intron 10 of Tau: Characterization of the MSTD family. Brain 2008, 131, 72–89. [Google Scholar] [CrossRef]
- Omoto, M.; Suzuki, S.; Ikeuchi, T.; Ishihara, T.; Kobayashi, T.; Tsuboi, Y.; Ogasawara, J.; Koga, M.; Kawai, M.; Iwaki, T.; et al. Autosomal dominant tauopathy with parkinsonism and central hypoventilation. Neurology 2012, 78, 762–764. [Google Scholar] [CrossRef]
- Morris, H.; Osaki, Y.; Holton, J.; Lees, A.; Wood, N.; Revesz, T.; Quinn, N. Tau exon 10 +16 mutation FTDP-17 presenting clinically as sporadic young onset PSP. Neurology 2003, 61, 102–104. [Google Scholar] [CrossRef]
- Rossi, G.; Bastone, A.; Piccoli, E.; Morbin, M.; Mazzoleni, G.; Fugnanesi, V.; Beeg, M.; Del Favero, E.; Cantù, L.; Motta, S.; et al. Different mutations at V363 MAPT codon are associated with atypical clinical phenotypes and show unusual structural and functional features. Neurobiol. Aging 2014, 35, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.Y.; Kouri, N.; Cook, C.N.; Serie, D.J.; Heckman, M.G.; Finch, N.A.; Caselli, R.J.; Uitti, R.J.; Wszolek, Z.K.; Graff-Radford, N.; et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol. Neurodegener. 2018, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.A.; Chen, Z.; Won, H.; Huang, A.Y.; Lowe, J.K.; Wojta, K.; Yokoyama, J.S.; Bensimon, G.; Leigh, P.N.; Payan, C.; et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener. 2018, 13, 41. [Google Scholar] [CrossRef]
- Höglinger, G.; Melhem, N.M.; Dickson, D.W.; Sleiman, P.M.A.; Wang, L.-S.; Klei, L.; Rademakers, R.; De Silva, R.; Litvan, I.; Riley, D.E.; et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011, 43, 699–705. [Google Scholar] [CrossRef]
- Jabbari, E.; Koga, S.; Valentino, R.R.; Reynolds, R.H.; Ferrari, R.; Tan, M.M.; Rowe, J.B.; Dalgard, C.L.; Scholz, S.W.; Dickson, D.W.; et al. Genetic determinants of survival in progressive supranuclear palsy: A genome-wide association study. Lancet Neurol. 2021, 20, 107–116. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Wszolek, Z.K.; Pfeiffer, R.F.; Tsuboi, Y.; Uitti, R.J.; McComb, R.D.; Stoessl, A.J.; Strongosky, A.J.; Zimprich, A.; Müller-Myhsok, B.; Farrer, M.J.; et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 2004, 62, 1619–1622. [Google Scholar] [CrossRef]
- Spanaki, C.; Latsoudis, H.; Plaitakis, A. LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology 2006, 67, 1518–1519. [Google Scholar] [CrossRef]
- Madžar, D.; Schulte, C.; Gasser, T. Screening forLRRK2R1441 mutations in a cohort of PSP patients from Germany. Eur. J. Neurol. 2009, 16, 1230–1232. [Google Scholar] [CrossRef]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.; Dachsel, J.C.; Lincoln, S.J.; Cobb, S.A.; Rajput, M.L.; Farrer, M. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef]
- Ruffmann, C.; Giaccone, G.; Canesi, M.; Bramerio, M.; Goldwurm, S.; Gambacorta, M.; Rossi, G.; Tagliavini, F.; Pezzoli, G. Atypical tauopathy in a patient with LRRK2-G2019S mutation and tremor-dominant Parkinsonism. Neuropathol. Appl. Neurobiol. 2011, 38, 382–386. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, B.; Ahmed, S.; et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 2018, 76, e211–e214. [Google Scholar] [CrossRef]
- Trinh, J.; Guella, I.; McKenzie, M.; Gustavsson, E.K.; Szu-Tu, C.; Petersen, M.S.; Rajput, A.; Rajput, A.H.; McKeown, M.; Jeon, B.S.; et al. Novel LRRK2 mutations in Parkinsonism. Park. Relat. Disord. 2015, 21, 1119–1121. [Google Scholar] [CrossRef]
- Sanchez-Contreras, M.; Heckman, M.G.; Tacik, P.; Diehl, N.; Brown, P.H.; Soto-Ortolaza, A.I.; Christopher, E.A.; Walton, R.L.; Ross, O.A.; Golbe, L.I.; et al. Study of LRRK2 variation in tauopathy: Progressive supranuclear palsy and corticobasal degeneration. Mov. Disord. 2016, 32, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments α-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Ross, O.A.; Teive, H.A.; Sławek, J.; Dickson, D.W.; Wszolek, Z.K. DCTN1-related neurodegeneration: Perry syndrome and beyond. Park. Relat. Disord. 2017, 41, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Caroppo, P.; Le Ber, I.; Clot, F.; Rivaud-Péchoux, S.; Camuzat, A.; De Septenville, A.; Boutoleau-Bretonnière, C.; Mourlon, V.; Sauvée, M.; Lebouvier, T.; et al. DCTN1Mutation Analysis in Families with Progressive Supranuclear Palsy–Like Phenotypes. JAMA Neurol. 2014, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, E.K.; Trinh, J.; Guella, I.; Szu-Tu, C.; Khinda, J.; Lin, C.-H.; Wu, R.-M.; Stoessl, J.; Appel-Cresswell, S.; McKeown, M.; et al. DCTN1 p.K56R in progressive supranuclear palsy. Park. Relat. Disord. 2016, 28, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Sasagasako, N.; Shen, C.; Shijo, M.; Hamasaki, H.; Suzuki, S.O.; Tsuboi, Y.; Fujii, N.; Iwaki, T. DCTN1 F52L mutation case of Perry syndrome with progressive supranuclear palsy-like tauopathy. Park. Relat. Disord. 2018, 51, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Herbst, S.; Lewis, P.A.; Morris, H.R. The emerging role of LRRK2 in tauopathies. Clin. Sci. 2022, 136, 1071–1079. [Google Scholar] [CrossRef]

{kind=link}
| GBA-MSA | LRRK2-MSA | MAPT-PSP | LRRK2-PSP |
|---|---|---|---|
| L444P-A456P-V460V or RecNciI | G2019S | R5L | R1441C |
| N370S, T369M and R496H | N279K | ||
| R262H | R1441H | ||
| L444P | L284R | ||
| N409S L483P | S285R | G2019S | |
| ΔN296 | |||
| T2310M | |||
| K298_H299insQ | A1413T | ||
| G2019S | |||
| P301L | |||
| G303V | |||
| S305S | |||
| IVS10+3G>A | |||
| IVS10+14C>T | |||
| IVS10+16C>T | |||
| V363A | |||
| R406W |
| Study | Sample | Mutation Type | Clinical Phenotypes | Family History | Clinical Diagnosis | Neuropathologic Diagnosis |
|---|---|---|---|---|---|---|
| Mitsui et al. [52] | 969 MSA (574 Japanese, 223 European, 172 N. American) | L444P-A456P-V460V or RecNciI | MSA-Parkinsonism and MSA-Cerebellar | Yes | MSA | NA |
| Sklerov et al. [53] | 17 MSA + AD | N370S, T369M, and R496H (4 pts/ 3 Ashkenazi Jewish) | Parkinsonism, orthostasis, urinary symptoms, constipation, erectile dysfunction, RBD | Yes (2 pts) No (2 pts) | MSA | MSA |
| Segarane et al. [54] | 108 MSA + 257 C (British origin) | R262H | NA | NA | MSA | MSA |
| Sun et al. [55] | 54 MSA, 109 ET, 657 C (all Chinese) | L444P | NA | No | MSA | NA |
| Wernick et al. [56] | 167 MSA + 834 C | N409S L483P | NA | NA | MSA | MSA |
| Study | Sample | Mutation Type | Clinical Phenotypes | Family History | Clinical Diagnosis | Neuropathologic Diagnosis |
|---|---|---|---|---|---|---|
| Poorkaj et al. [72] | 96 PSP + 4 C | R5L | falls, dysarthria, micrographia | No | PSP | PSP |
| Yasuda et al. [73] | N279K | Parkinsonism | Yes | PNLD | PSP-mimicker | |
| Delisle et al. [74] | N279K | apathy, memory disorder, Parkinsonism | Yes | FTDP-17 | NA | |
| indifference, attention disturbances | Yes | FTDP-17 | NA | |||
| Soliveri et al. [75] | N279K | personality and behavior changes | Yes | PSP | NA | |
| Ogaki et al. [77] | N279K | Parkinsonism, micrographia | Yes | PSP | PSP-mimicker | |
| oscillopsia, shuffling gait, bradykinesia | Yes | PSP | NA | |||
| Ogaki et al. [76] | N279N | Parkinsonism | Yes | PSP | NA | |
| Rohrer et al. [78] | L284R | falls, personality changes | Yes | PSP | NA | |
| Ogaki et al. [76] | S285R | speech and breathing disorder | No | PSP | NA | |
| Fujioka et al. [5] | S285R | dystonia and supranuclear gaze palsy | Yes | PSP | PSP-AD | |
| bradykinesia | Yes | PSP | PSP | |||
| Pastor et al. [79] | ΔN296 | speech and memory disorder | Yes | Atypical PSP | NA | |
| Rossi et al. [80] | ΔN296 | falls, antecollis, dysarthria | Yes | PSP-mimicker | NA | |
| Nakayama et al. [81] | K298_H299insQ | neck stiffness, postural instability | Yes | PSP | NA | |
| gait difficulty, cognitive dysfunction | NA | NA | NA | |||
| Bird et al. [82] | P301L | tremor, speech impairment | Yes | APD | PSP-mimicker | |
| Kaat et al. [15] | P301L | NA | Yes | PSP | NA | |
| Ros et al. [83] | G303V | Parkinsonism, falls, micrographia, dysarthria, ocular motor damage | Yes | PSP | PSP | |
| Stanford et al. [84] | S305S | dystonia, dysarthria, falls, bradykinesia | Yes | PSP | PSP | |
| Spina et al. [85] | IVS10+3G>A | dizziness, neck rigidity | Yes | Atypical PSP | PSP-mimicker | |
| Omoto et al. [86] | IVS10+14C>T | clumsiness, tremor, apathy | Yes | Perry syndrome | PSP-mimicker | |
| Morris et al. [87] | IVS10+16C>T | fatigue, micrographia, withdrawal | Yes | PSP | Tauopathy | |
| Rossi et al. [88] | V363A | diplopia, falls, bradykinesia | Yes | PSP | NA | |
| Ygland et al. [12] | R406W | dyscalculia, social isolation, apathy | Yes | AD | PSP-mimicker |
| Study | Sample | Mutation Type | Clinical Phenotypes | Family History | MAPT Haplotypes | Clinical Diagnosis | Neuropathologic Diagnosis |
|---|---|---|---|---|---|---|---|
| Zimprich et al. [93] Wszolek et al. [94] | Family pedigree (22 PD/190 members) | R1441C (1Fpt) | Parkinsonism, supranuclear gaze palsy | Yes | H1/H1 | PD | SN neuronal loss + PSP-like changes |
| Spanaki et al. [95] | 266 PD + 13 PSP | R1441H (1Fpt) | Parkinsonism, bulbar impairment dysfunction, major cognitive disorder | Yes | NA | PD | NA |
| Rajput et al. [97] | 85 LBD + 16 PSP + 16 ET + 11 MSA + 4 CBD + 1 PiD + 22 C | G2019S (1Mpt) | Parkinsonism | Yes | H1/H1 | PD | PSP + early-stage AD |
| Ruffmann et al. [98] | 1 PD | G2019S (1Mpt) | tremor | No | H1/H1 | PSP | PSP + early-stage AD |
| Trinh et al. [100] | 948 PD + 189 (PSP + MSA + DLB) + HC | T2310M | NA | No | NA | PSP | NA |
| Sanchez-Contreras et al. [101] | 1039 PSP + 145 CBD + 1790 C | A1413T (1Mpt) | Parkinsonism, memory disorder, postural instability, eyelid apraxia | No | H1/H1 | PSP | PSP + AGD |
| G2019S (1Fpt) | Bulbar dysfunction, tremor, retrocolitis | No | H1/H1 | PSP | PSP + early-stage AD | ||
| Blauwendraat et al. [99] | 624 AD + 216 LBD + 27 AD/ALS-FTD + 26 (AD + AP) + 40 complex cases + 50 PD + 49 LBD + 84 ALS-FTD + 13 MSA + 37 PSP + 7 CBD + 9 rare syndromes + 47 unclassified + 14 HS | G2019S (1Fpt) | NA | Yes | NA | PD | PSP + AD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bougea, A. Genetics of Multiple System Atrophy and Progressive Supranuclear Palsy: A Systemized Review of the Literature. Int. J. Mol. Sci. 2023, 24, 5281. https://doi.org/10.3390/ijms24065281
Bougea A. Genetics of Multiple System Atrophy and Progressive Supranuclear Palsy: A Systemized Review of the Literature. International Journal of Molecular Sciences. 2023; 24(6):5281. https://doi.org/10.3390/ijms24065281
Chicago/Turabian StyleBougea, Anastasia. 2023. "Genetics of Multiple System Atrophy and Progressive Supranuclear Palsy: A Systemized Review of the Literature" International Journal of Molecular Sciences 24, no. 6: 5281. https://doi.org/10.3390/ijms24065281
APA StyleBougea, A. (2023). Genetics of Multiple System Atrophy and Progressive Supranuclear Palsy: A Systemized Review of the Literature. International Journal of Molecular Sciences, 24(6), 5281. https://doi.org/10.3390/ijms24065281