
Transcriptomic Signatures of Single-Suture Craniosynostosis Phenotypes

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Study Participant Characteristics
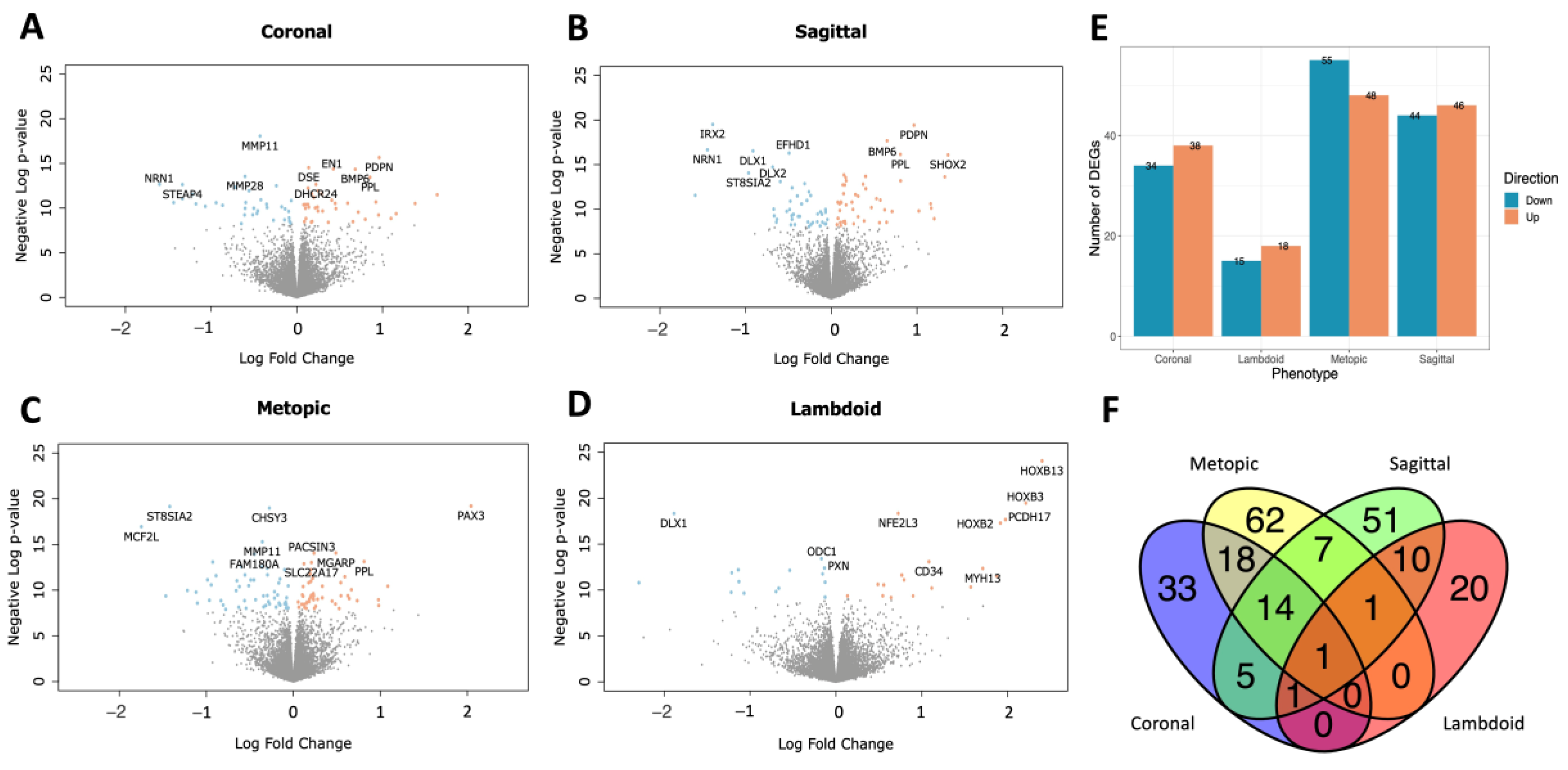
2.2. DEGs and Pathway Analysis by Phenotype
2.3. Sex-Stratified DEGs and Pathways by Phenotype
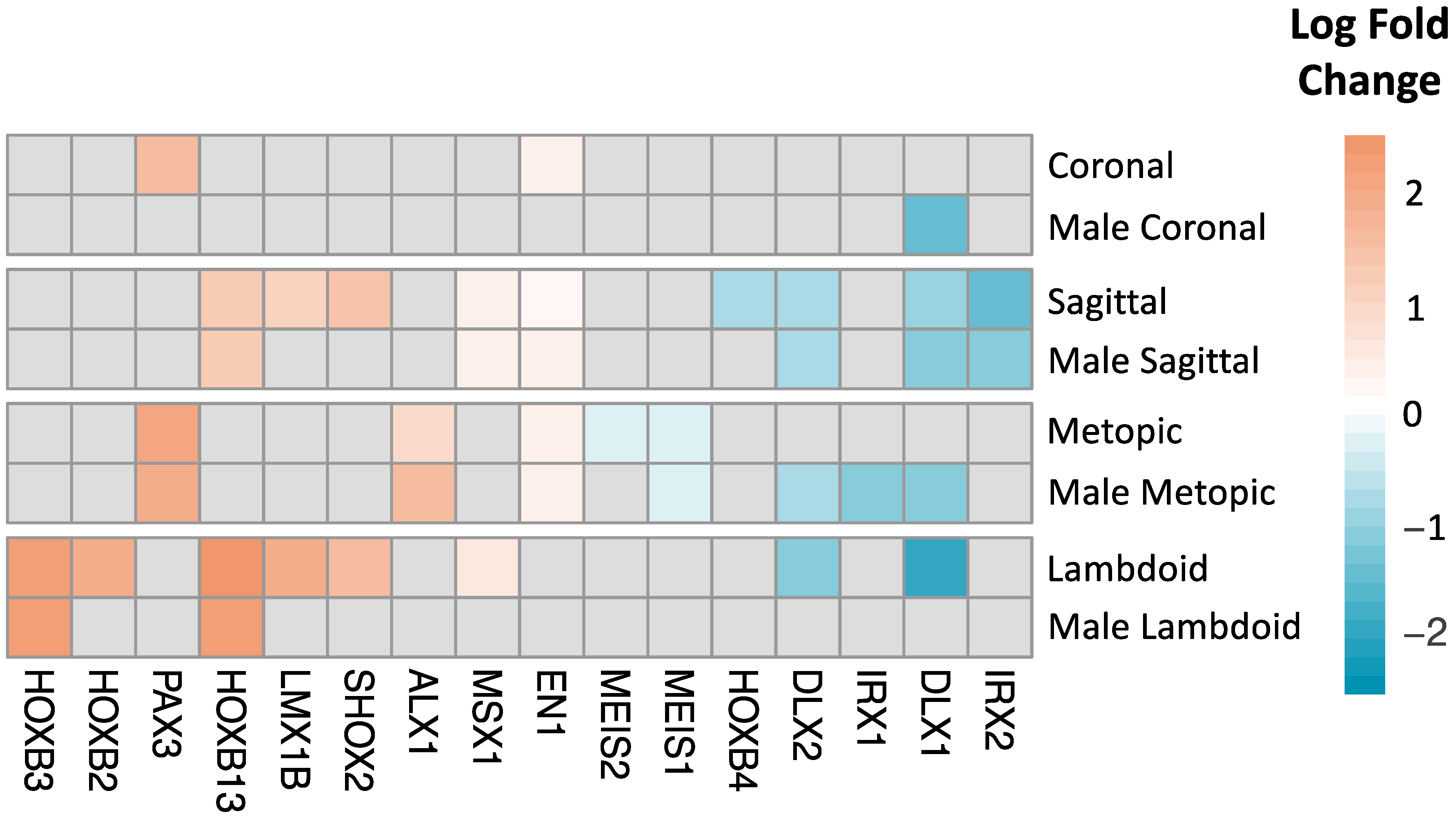
2.4. Transcription Factor Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Study Participants
4.3. Sample Processing
4.4. RNA Sequencing
4.5. Differential Gene Expression Analysis
4.6. Pathway Enrichment Analysis
4.7. Transcription Factor Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boulet, S.L.; Rasmussen, S.A.; Honein, M.A. A Population-Based Study of Craniosynostosis in Metropolitan Atlanta, 1989–2003. Am. J. Med. Genet. A 2008, 146A, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Wilkie, A.O.M. Craniosynostosis. Eur. J. Hum. Genet. 2011, 19, 369–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, A.O.M.; Byren, J.C.; Hurst, J.A.; Jayamohan, J.; Johnson, D.; Knight, S.J.L.; Lester, T.; Richards, P.G.; Twigg, S.R.F.; Wall, S.A. Prevalence and Complications of Single-Gene and Chromosomal Disorders in Craniosynostosis. Pediatrics 2010, 126, e391–e400. [Google Scholar] [CrossRef] [Green Version]
- Kabbani, H.; Raghuveer, T.S. Craniosynostosis. Am. Fam. Physician 2004, 69, 2863–2870. [Google Scholar] [PubMed]
- Ye, X.; Guilmatre, A.; Reva, B.; Peter, I.; Heuzé, Y.; Richtsmeier, J.T.; Fox, D.J.; Goedken, R.J.; Jabs, E.W.; Romitti, P.A. Mutation Screening of Candidate Genes in Patients with Nonsyndromic Sagittal Craniosynostosis. Plast. Reconstr. Surg. 2016, 137, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuzé, Y.; Holmes, G.; Peter, I.; Richtsmeier, J.T.; Jabs, E.W. Closing the Gap: Genetic and Genomic Continuum from Syndromic to Nonsyndromic Craniosynostoses. Curr. Genet. Med. Rep. 2014, 2, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.M.; MacLean, R.E. Craniosynostosis: Diagnosis, Evaluation, and Management; Oxford University Press: New York, NY, USA, 2000; p. 464. [Google Scholar]
- Boyadjiev, S.A. International Craniosynostosis Consortium Genetic Analysis of Non-Syndromic Craniosynostosis. Orthod. Craniofac. Res. 2007, 10, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Lajeunie, E.; Le Merrer, M.; Bonaïti-Pellie, C.; Marchac, D.; Renier, D. Genetic Study of Scaphocephaly. Am. J. Med. Genet. 1996, 62, 282–285. [Google Scholar] [CrossRef]
- Cohen, M.M. Etiopathogenesis of Craniosynostosis. Neurosurg. Clin. N. Am. 1991, 2, 507–513. [Google Scholar] [CrossRef]
- Graham, J.M.; Smith, D.W. Metopic Craniostenosis as a Consequence of Fetal Head Constraint: Two Interesting Experiments of Nature. Pediatrics 1980, 65, 1000–1002. [Google Scholar] [CrossRef]
- Jaskoll, T.; Melnick, M. The Effects of Long-Term Fetal Constraint in Vitro on the Cranial Base and Other Skeletal Components. Am. J. Med. Genet. 1982, 12, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Lakin, G.E.; Sinkin, J.C.; Chen, R.; Koltz, P.F.; Girotto, J.A. Genetic and Epigenetic Influences of Twins on the Pathogenesis of Craniosynostosis: A Meta-Analysis. Plast. Reconstr. Surg. 2012, 129, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lara, P.A.; Carmichael, S.L.; Graham, J.M.; Lammer, E.J.; Shaw, G.M.; Ma, C.; Rasmussen, S.A. National Birth Defects Prevention Study Fetal Constraint as a Potential Risk Factor for Craniosynostosis. Am. J. Med. Genet. A 2010, 152A, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Stanton, E.; Urata, M.; Chen, J.-F.; Chai, Y. The Clinical Manifestations, Molecular Mechanisms and Treatment of Craniosynostosis. Dis. Model. Mech. 2022, 15, dmm049390. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.W.H.; Booth, H.A.F.; Bruford, E.A. Classification and Nomenclature of All Human Homeobox Genes. BMC Biol. 2007, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Liu, X. PRC2, Chromatin Regulation, and Human Disease: Insights From Molecular Structure and Function. Front. Oncol. 2022, 12, 894585. [Google Scholar] [CrossRef]
- Marchant, T.W.; Johnson, E.J.; McTeir, L.; Johnson, C.I.; Gow, A.; Liuti, T.; Kuehn, D.; Svenson, K.; Bermingham, M.L.; Drögemüller, M.; et al. Canine Brachycephaly Is Associated with a Retrotransposon-Mediated Missplicing of SMOC2. Curr. Biol. 2017, 27, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Homayounfar, N.; Park, S.S.; Afsharinejad, Z.; Bammler, T.K.; MacDonald, J.W.; Farin, F.M.; Mecham, B.H.; Cunningham, M.L. Transcriptional Analysis of Human Cranial Compartments with Different Embryonic Origins. Arch. Oral Biol. 2015, 60, 1450–1460. [Google Scholar] [CrossRef] [Green Version]
- Milunsky, J.M.; Maher, T.A.; Zhao, G.; Roberts, A.E.; Stalker, H.J.; Zori, R.T.; Burch, M.N.; Clemens, M.; Mulliken, J.B.; Smith, R.; et al. TFAP2A Mutations Result in Branchio-Oculo-Facial Syndrome. Am. J. Hum. Genet. 2008, 82, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Weismann, C.G.; Satoda, M.; Pierpont, M.E.; Sweeney, E.; Thompson, E.M.; Gelb, B.D. Novel TFAP2B Mutations That Cause Char Syndrome Provide a Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2001, 69, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hagopian-Donaldson, S.; Serbedzija, G.; Elsemore, J.; Plehn-Dujowich, D.; McMahon, A.P.; Flavell, R.A.; Williams, T. Neural Tube, Skeletal and Body Wall Defects in Mice Lacking Transcription Factor AP-2. Nature 1996, 381, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-M.; Lin, Y.-L.; Chou, C.-W. GATA-3 Transduces Survival Signals in Osteoblasts through Upregulation of Bcl-x(L) Gene Expression. J. Bone Miner. Res. 2010, 25, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-H.; Lin, P.-I.; Ho, W.-P.; Chan, W.P.; Chen, T.-L.; Chen, R.-M. Participation of GATA-3 in Regulation of Bone Healing through Transcriptional Upregulation of Bcl-XL Expression. Exp. Mol. Med. 2017, 49, e398. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Beyer, R.P.; Smyth, M.D.; Clarke, C.M.; Timms, A.E.; Bammler, T.K.; Stamper, B.D.; Mecham, B.H.; Gustafson, J.A.; Cunningham, M.L. Osteoblast Differentiation Profiles Define Sex Specific Gene Expression Patterns in Craniosynostosis. Bone 2015, 76, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Tønne, E.; Due-Tønnessen, B.J.; Wiig, U.; Stadheim, B.F.; Meling, T.R.; Helseth, E.; Heimdal, K.R. Epidemiology of Craniosynostosis in Norway. J Neurosurg Pediatr 2020, 26, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Lajeunie, E.; Le Merrer, M.; Bonaïti-Pellie, C.; Marchac, D.; Renier, D. Genetic Study of Nonsyndromic Coronal Craniosynostosis. Am. J. Med. Genet. 1995, 55, 500–504. [Google Scholar] [CrossRef]
- Clarke, C.M.; Fok, V.T.; Gustafson, J.A.; Smyth, M.D.; Timms, A.E.; Frazar, C.D.; Smith, J.D.; Birgfeld, C.B.; Lee, A.; Ellenbogen, R.G.; et al. Single Suture Craniosynostosis: Identification of Rare Variants in Genes Associated with Syndromic Forms. Am. J. Med. Genet. A 2018, 176, 290–300. [Google Scholar] [CrossRef]
- May, R.D.; Frauchiger, D.A.; Albers, C.E.; Tekari, A.; Benneker, L.M.; Klenke, F.M.; Hofstetter, W.; Gantenbein, B. Application of Cytokines of the Bone Morphogenetic Protein (BMP) Family in Spinal Fusion—Effects on the Bone, Intervertebral Disc and Mesenchymal Stromal Cells. Curr. Stem Cell Res. Ther. 2019, 14, 618–643. [Google Scholar] [CrossRef]
- Chen, C.-P.; Chen, M.; Chen, C.-Y.; Chern, S.-R.; Wu, P.-S.; Chang, S.-P.; Kuo, Y.-L.; Chen, W.-L.; Pan, C.-W.; Wang, W. Prenatal Diagnosis and Molecular Cytogenetic Characterization of de Novo Pure Partial Trisomy 6p Associated with Microcephaly, Craniosynostosis and Abnormal Maternal Serum Biochemistry. Gene 2014, 536, 425–429. [Google Scholar] [CrossRef]
- Timberlake, A.T.; Kiziltug, E.; Jin, S.C.; Nelson-Williams, C.; Loring, E.; Yale Center for Genome Analysis; Allocco, A.; Marlier, A.; Banka, S.; Stuart, H.; et al. De Novo Mutations in the BMP Signaling Pathway in Lambdoid Craniosynostosis. Hum. Genet. 2023, 142, 21–32. [Google Scholar] [CrossRef]
- Ueharu, H.; Pan, H.; Hayano, S.; Zapien-Guerra, K.; Yang, J.; Mishina, Y. Augmentation of Bone Morphogenetic Protein Signaling in Cranial Neural Crest Cells in Mice Deforms Skull Base Due to Premature Fusion of Intersphenoidal Synchondrosis. Genesis 2023, e23509. [Google Scholar] [CrossRef]
- Lindsey, R.C.; Xing, W.; Pourteymoor, S.; Godwin, C.; Gow, A.; Mohan, S. Novel Role for Claudin-11 in the Regulation of Osteoblasts via Modulation of ADAM10-Mediated Notch Signaling. J. Bone Miner. Res. 2019, 34, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.M.; Cheon, Y.-H.; Kwak, S.C.; Jun, H.Y.; Yoon, K.-H.; Lee, M.S.; Kim, J.-Y. Claudin 11 Regulates Bone Homeostasis via Bidirectional EphB4-EphrinB2 Signaling. Exp. Mol. Med. 2018, 50, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, G.; Walter, I.; Helmreich, M.; Egerbacher, M. Localization of Matrix Metalloproteinases, (MMPs) Their Tissue Inhibitors, and Vascular Endothelial Growth Factor (VEGF) in Growth Plates of Children and Adolescents Indicates a Role for MMPs in Human Postnatal Growth and Skeletal Maturation. Calcif. Tissue Int. 2005, 76, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Alappat, S.; Zhang, Z.Y.; Chen, Y.P. Msx Homeobox Gene Family and Craniofacial Development. Cell Res. 2003, 13, 429–442. [Google Scholar] [CrossRef]
- Jabs, E.W.; Müller, U.; Li, X.; Ma, L.; Luo, W.; Haworth, I.S.; Klisak, I.; Sparkes, R.; Warman, M.L.; Mulliken, J.B. A Mutation in the Homeodomain of the Human MSX2 Gene in a Family Affected with Autosomal Dominant Craniosynostosis. Cell 1993, 75, 443–450. [Google Scholar] [CrossRef]
- Liu, Y.H.; Tang, Z.; Kundu, R.K.; Wu, L.; Luo, W.; Zhu, D.; Sangiorgi, F.; Snead, M.L.; Maxson, R.E. Msx2 Gene Dosage Influences the Number of Proliferative Osteogenic Cells in Growth Centers of the Developing Murine Skull: A Possible Mechanism for MSX2-Mediated Craniosynostosis in Humans. Dev. Biol. 1999, 205, 260–274. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jiang, W.; Xie, C.; Wu, X.; Ren, Q.; Wang, F.; Shen, X.; Hong, Y.; Wu, H.; Liao, Y.; et al. Msx1+ Stem Cells Recruited by Bioactive Tissue Engineering Graft for Bone Regeneration. Nat. Commun. 2022, 13, 5211. [Google Scholar] [CrossRef]
- Mavrogiannis, L.A.; Taylor, I.B.; Davies, S.J.; Ramos, F.J.; Olivares, J.L.; Wilkie, A.O.M. Enlarged Parietal Foramina Caused by Mutations in the Homeobox Genes ALX4 and MSX2: From Genotype to Phenotype. Eur. J. Hum. Genet. 2006, 14, 151–158. [Google Scholar] [CrossRef]
- Yagnik, G.; Ghuman, A.; Kim, S.; Stevens, C.G.; Kimonis, V.; Stoler, J.; Sanchez-Lara, P.A.; Bernstein, J.A.; Naydenov, C.; Drissi, H.; et al. ALX4 Gain-of-Function Mutations in Nonsyndromic Craniosynostosis. Hum. Mutat. 2012, 33, 1626–1629. [Google Scholar] [CrossRef] [Green Version]
- Kayserili, H.; Uz, E.; Niessen, C.; Vargel, I.; Alanay, Y.; Tuncbilek, G.; Yigit, G.; Uyguner, O.; Candan, S.; Okur, H.; et al. ALX4 Dysfunction Disrupts Craniofacial and Epidermal Development. Hum. Mol. Genet. 2009, 18, 4357–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, M.E.; Lacassie, Y.; Azamian, M.; Franciskovich, R.; Zapata, G.; Hernandez, P.P.; Liu, P.; Campbell, I.M.; Bostwick, B.L.; Lalani, S.R. Vertical Transmission of a Large Calvarial Ossification Defect Due to Heterozygous Variants of ALX4 and TWIST1. Am. J. Med. Genet. A 2021, 185, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, S.; Chen, E.; Yu, L.; Wang, J.; Wu, M. ALX1-Transcribed LncRNA AC132217.4 Promotes Osteogenesis and Bone Healing via IGF-AKT Signaling in Mesenchymal Stem Cells. Cell. Mol. Life Sci. 2022, 79, 328. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.N.; Kristensen, E.; Morck, D.W.; Boyd, S.; Hallgrímsson, B. Effects of Growth Hormone on the Ontogenetic Allometry of Craniofacial Bones. Evol. Dev. 2013, 15, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howie, R.N.; Durham, E.L.; Black, L.; Bennfors, G.; Parsons, T.E.; Elsalanty, M.E.; Yu, J.C.; Weinberg, S.M.; Cray, J.J. Effects of In Utero Thyroxine Exposure on Murine Cranial Suture Growth. PLoS ONE 2016, 11, e0167805. [Google Scholar] [CrossRef] [Green Version]
- Durham, E.; Howie, R.N.; Parsons, T.; Bennfors, G.; Black, L.; Weinberg, S.M.; Elsalanty, M.; Yu, J.C.; Cray, J.J. Thyroxine Exposure Effects on the Cranial Base. Calcif. Tissue Int. 2017, 101, 300–311. [Google Scholar] [CrossRef]
- German, B.; Ellis, L. Polycomb Directed Cell Fate Decisions in Development and Cancer. Epigenomes 2022, 6, 28. [Google Scholar] [CrossRef]
- Dudakovic, A.; Camilleri, E.T.; Xu, F.; Riester, S.M.; McGee-Lawrence, M.E.; Bradley, E.W.; Paradise, C.R.; Lewallen, E.A.; Thaler, R.; Deyle, D.R.; et al. Epigenetic Control of Skeletal Development by the Histone Methyltransferase Ezh2. J. Biol. Chem. 2015, 290, 27604–27617. [Google Scholar] [CrossRef] [Green Version]
- Pribadi, C.; Camp, E.; Cakouros, D.; Anderson, P.; Glackin, C.; Gronthos, S. Pharmacological Targeting of KDM6A and KDM6B, as a Novel Therapeutic Strategy for Treating Craniosynostosis in Saethre-Chotzen Syndrome. Stem Cell Res. Ther. 2020, 11, 529. [Google Scholar] [CrossRef]
- Zhan, J.; Wang, P.; Li, S.; Song, J.; He, H.; Wang, Y.; Liu, Z.; Wang, F.; Bai, H.; Fang, W.; et al. HOXB13 Networking with ABCG1/EZH2/Slug Mediates Metastasis and Confers Resistance to Cisplatin in Lung Adenocarcinoma Patients. Theranostics 2019, 9, 2084–2099. [Google Scholar] [CrossRef]
- Cray, J.J.; Durham, E.L.; Smalley, M.A.; Finegold, D.N.; Siegel, M.I.; Losee, J.E.; Mooney, M.P.; Cooper, G.M. The Effects of Testosterone on Craniosynostotic Calvarial Cells: A Test of the Gene/Environmental Model of Craniofacial Anomalies. Orthod. Craniofac. Res. 2011, 14, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.C.; Slemp, A.E.; Hwang, C.; Karmacharya, J.; Gordon, A.D.; Kirschner, R.E. Immunolocalization of Androgen Receptor in the Developing Craniofacial Skeleton. J. Craniofac. Surg. 2004, 15, 922–927; discussion 928–929. [Google Scholar] [CrossRef] [PubMed]
- Cray, J.; Burrows, A.M.; Vecchione, L.; Lensie, E.; Decesare, G.E.; Campbell, A.; Finegold, D.N.; Losee, J.E.; Siegel, M.I.; Cooper, G.M.; et al. Effects of Flutamide Therapy on Craniofacial Growth and Development in a Model of Craniosynostosis. J. Craniofac. Surg. 2010, 21, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.C.; Slemp, A.E.; Hwang, C.; Sena-Esteves, M.; Nah, H.-D.; Kirschner, R.E. Dihydrotestosterone Stimulates Proliferation and Differentiation of Fetal Calvarial Osteoblasts and Dural Cells and Induces Cranial Suture Fusion. Plast. Reconstr. Surg. 2007, 120, 1137–1147. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T. Svaseq: Removing Batch Effects and Other Unwanted Noise from Sequencing Data. Nucleic Acids Res. 2014, 42, e161. [Google Scholar] [CrossRef]
- Wu, D.; Lim, E.; Vaillant, F.; Asselin-Labat, M.-L.; Visvader, J.E.; Smyth, G.K. ROAST: Rotation Gene Set Tests for Complex Microarray Experiments. Bioinformatics 2010, 26, 2176–2182. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Coronal | Sagittal | Metopic | Lambdoid | All | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | |
| Sex | ||||||||||||
| Male | 59 | (18.55) | 25 | (7.86) | 148 | (46.54) | 72 | (22.64) | 14 | (4.40) | 318 | (100) |
| Female | 26 | (16.77) | 55 | (35.48) | 48 | (30.96) | 20 | (12.90) | 6 | (3.87) | 155 | (100) |
| Sample Origin | ||||||||||||
| Seattle, WA | 55 | (20.07) | 45 | (16.42) | 113 | (41.24) | 50 | (18.25) | 11 | (4.01) | 274 | (100) |
| St. Louis, MO | 30 | (25.42) | 13 | (11.02) | 49 | (41.53) | 21 | (17.80) | 5 | (4.24) | 118 | (100) |
| Chicago, IL | 0 | (0) | 21 | (38.18) | 18 | (32.73) | 12 | (21.82) | 4 | (7.27) | 55 | (100) |
| Atlanta, GA | 0 | (0) | 1 | (3.85) | 16 | (61.54) | 9 | (34.62) | 0 | (0) | 26 | (100) |
| Cohort | ||||||||||||
| 1 | 48 | (19.05) | 49 | (19.44) | 96 | (38.10) | 48 | (19.05) | 11 | (4.37) | 252 | (100) |
| 2 | 37 | (16.74) | 31 | (14.03) | 100 | (45.25) | 44 | (19.91) | 9 | (4.07) | 221 | (100) |
| Total | 85 | (17.97) | 80 | (16.91) | 196 | (41.44) | 92 | (19.45) | 20 | (4.23) | 473 | (100) |
| Control | Coronal | Sagittal | Metopic | Lambdoid | All | ||
|---|---|---|---|---|---|---|---|
| Proband Age (months) | Median | 9 | 10 | 5.5 | 9 | 9 | 8 |
| Standard Error | 3.17 | 0.75 | 0.40 | 0.46 | 0.68 | 0.66 | |
| Range | 0–120 | 1–59 | 1–36 | 1–25 | 2–15 | 0–120 | |
| Culture Time (days) | Median | 44 | 42 | 34.5 | 37 | 49 | 39 |
| Standard Error | 2.05 | 3.66 | 2.44 | 3.39 | 7.25 | 1.44 | |
| Range | 16–117 | 17–171 | 15–189 | 12–160 | 21–146 | 12–189 |
| Phenotype | Model | Pathway | Direction | FDR |
|---|---|---|---|---|
| Coronal | Primary (N = 473) | Fatty acid degradation | Up | 0.017 |
| Valine, leucine and isoleucine degradation | Up | 0.017 | ||
| Butanoate metabolism | Up | 0.019 | ||
| Metopic | Male (N = 318) | Growth hormone synthesis, secretion and action | Up | 0.028 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapehn, S.; Gustafson, J.A.; Timms, A.E.; Cunningham, M.L.; Paquette, A.G. Transcriptomic Signatures of Single-Suture Craniosynostosis Phenotypes. Int. J. Mol. Sci. 2023, 24, 5353. https://doi.org/10.3390/ijms24065353
Lapehn S, Gustafson JA, Timms AE, Cunningham ML, Paquette AG. Transcriptomic Signatures of Single-Suture Craniosynostosis Phenotypes. International Journal of Molecular Sciences. 2023; 24(6):5353. https://doi.org/10.3390/ijms24065353
Chicago/Turabian StyleLapehn, Samantha, Jonas A. Gustafson, Andrew E. Timms, Michael L. Cunningham, and Alison G. Paquette. 2023. "Transcriptomic Signatures of Single-Suture Craniosynostosis Phenotypes" International Journal of Molecular Sciences 24, no. 6: 5353. https://doi.org/10.3390/ijms24065353
APA StyleLapehn, S., Gustafson, J. A., Timms, A. E., Cunningham, M. L., & Paquette, A. G. (2023). Transcriptomic Signatures of Single-Suture Craniosynostosis Phenotypes. International Journal of Molecular Sciences, 24(6), 5353. https://doi.org/10.3390/ijms24065353