
The Effect of Aldosterone on Cardiorenal and Metabolic Systems

Abstract
:1. Introduction
2. Aldosterone Synthesis and Physiology
2.1. Basic Mechanism of Aldosterone Synthesis and Physiological Processes
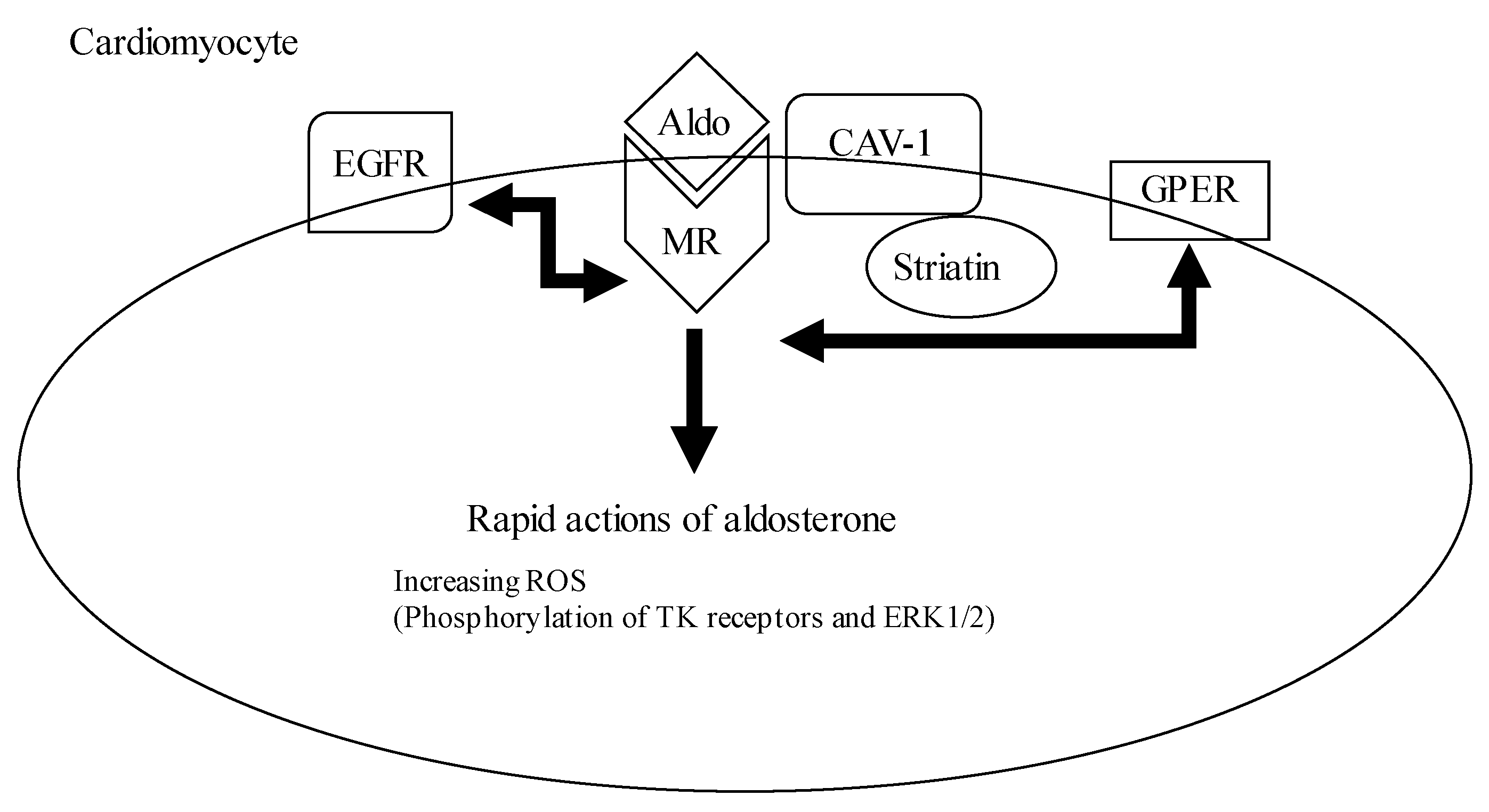
2.2. Pathophysiological Effect of Aldosterone
3. Metabolic and Other Pathophysiological Alterations Caused by Aldosterone
3.1. Aldosterone and Metabolic Alterations
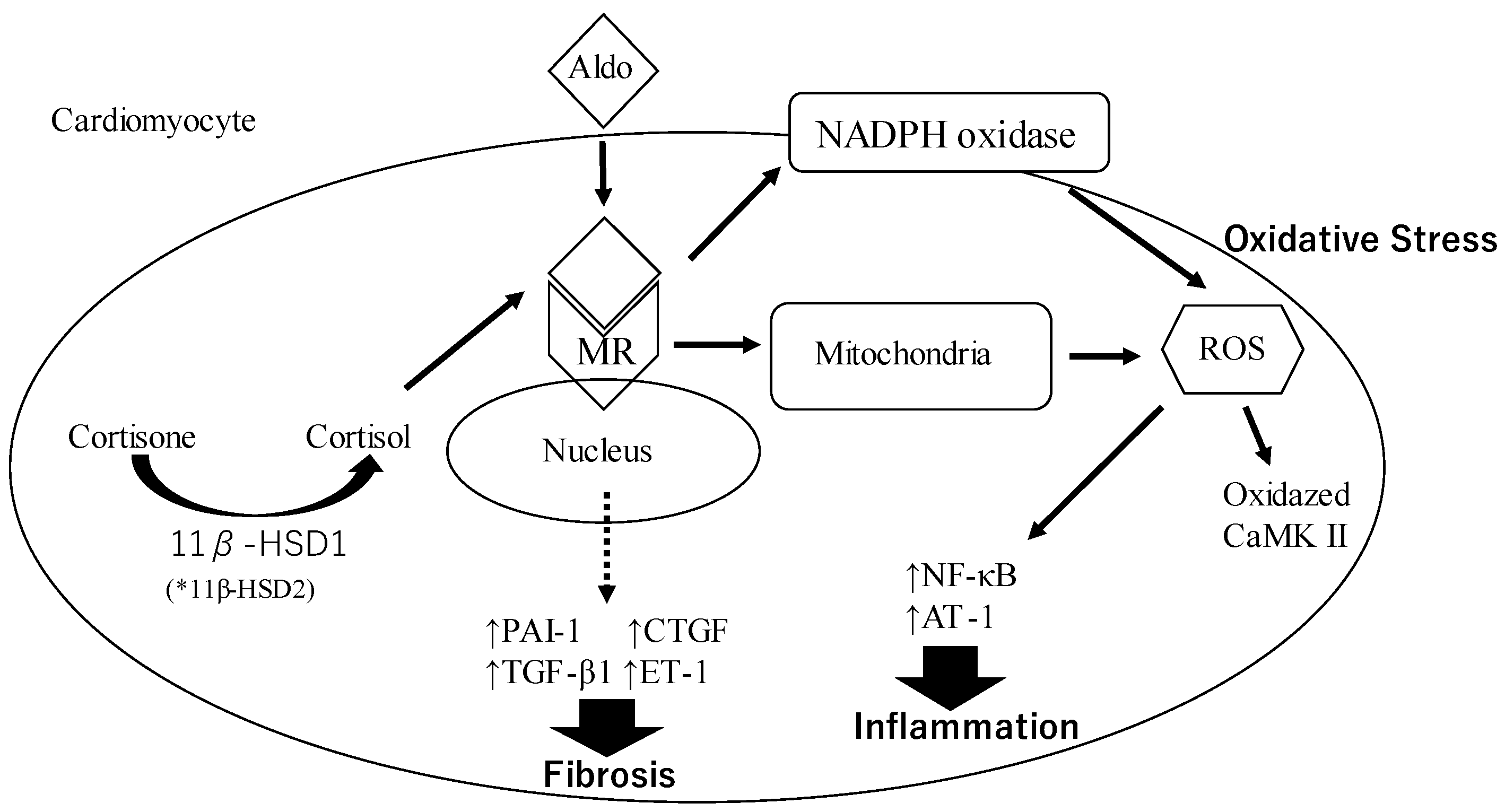
3.2. Oxidative Stress and Inflammation
3.2.1. Oxidative Stress
3.2.2. Inflammation
4. Vascular Stiffness and Thrombosis
4.1. Aldosterone and Vascular Stiffness
4.2. Aldosterone and Thrombosis
5. Aldosterone and Major Cardiovascular Disease
5.1. Coronary Artery Disease
5.2. Left Ventricular Remodeling and Heart Failure
5.3. Atrial Fibrillation
6. Aldosterone and Renal Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Argüeso, M.; Pascual-Corrales, E.; Bengoa Rojano, N.; García Cano, A.; Jiménez Mendiguchía, L.; Araujo-Castro, M. Higher risk of chronic kidney disease and progressive kidney function impairment in primary aldosteronism than in essential hypertension. Case-control study. Endocrine 2021, 73, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Bernini, G.; Caliumi, C.; Desideri, G.; Fabris, B.; Ferri, C.; Ganzaroli, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J. Am. Coll. Cardiol. 2006, 48, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Sone, M.; Inagaki, N.; Yamasaki, T.; Ogawa, O.; Takeda, Y.; Kurihara, I.; Umakoshi, H.; Ichijo, T.; Katabami, T.; et al. Obesity as a key factor underlying idiopathic hyperaldosteronism. J. Clin. Endocrinol. Metab. 2018, 103, 4456–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, S.; Fosam, A.; Chalk, L.; Deven, A.; Suzuki, M.; Correa, R.R.; Blau, J.E.; Demidowich, A.P.; Stratakis, C.A.; Muniyappa, R. Insulin sensitivity and pancreatic β-cell function in patients with primary aldosteronism. Endocrine 2021, 72, 96–103. [Google Scholar] [CrossRef]
- Reincke, M.; Fischer, E.; Gerum, S.; Merkle, K.; Schulz, S.; Pallauf, A.; Quinkler, M.; Hanslik, G.; Lang, K.; Hahner, S.; et al. Observational study mortality in treated primary aldosteronism: The German Conn’s registry. Hypertension 2012, 60, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Katsuragawa, S.; Tsurutani, Y.; Takiguchi, T.; Saito, J.; Nishikawa, T. Impact of primary aldosteronism on renal function in patients with type 2 diabetes. J. Diabetes Investig. 2021, 12, 217–225. [Google Scholar] [CrossRef]
- Chen, Z.W.; Tsai, C.H.; Pan, C.T.; Chou, C.H.; Liao, C.W.; Hung, C.S.; Wu, V.C.; Lin, Y.H.; TAIPAI Study Group. Endothelial dysfunction in primary aldosteronism. Int. J. Mol. Sci. 2019, 20, 5214. [Google Scholar] [CrossRef] [Green Version]
- Struthers, A.D. Aldosterone: Cardiovascular assault. Am. Heart J. 2002, 144, S2–S7. [Google Scholar] [CrossRef]
- Bauersachs, J.; López-Andrés, N. Mineralocorticoid receptor in cardiovascular diseases-Clinical trials and mechanistic insights. Br. J. Pharmacol. 2022, 179, 3119–3134. [Google Scholar] [CrossRef]
- Dudenbostel, T.; Calhoun, D.A. Use of aldosterone antagonists for treatment of uncontrolled resistant hypertension. Am. J. Hypertens. 2016, 30, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.S.; Giebisch, G. Renal potassium transport: Contributions of individual nephron segments and populations. Am. J. Physiol. 1978, 235, F515–F527. [Google Scholar] [CrossRef] [PubMed]
- Pácha, J.; Frindt, G.; Antonian, L.; Silver, R.B.; Palmer, L.G. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J. Gen. Physiol. 1993, 102, 25–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelis, A.; Jühlen, R.; Dieterich, P.; Peitzsch, M.; Lenders, J.W.M.; Hahner, S.; Schirbel, A.; Eisenhofer, G. A steady state system for in vitro evaluation of steroidogenic pathway dynamics: Application for CYP11B1, CYP11B2 and CYP17 inhibitors. J. Steroid Biochem. Mol. Biol. 2019, 188, 38–47. [Google Scholar] [CrossRef]
- Grundy, H.M.; Simpson, S.A.; Tait, J.F. Isolation of a highly active mineralocorticoid from beef adrenal extract. Nature 1952, 169, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Bravo, E.L. Regulation of aldosterone secretion: Current concepts and newer aspects. Adv. Nephrol. Necker Hosp. 1977, 7, 105–120. [Google Scholar]
- Farkash, Y.; Timberg, R.; Orly, J. Preparation of antiserum to rat cytochrome P-450 cholesterol side chain cleavage, and its use for ultrastructural localization of the immunoreactive enzyme by protein A-gold technique. Endocrinology 1986, 118, 1353–1365. [Google Scholar] [CrossRef]
- Hume, R.; Kelly, R.W.; Taylor, P.L.; Boyd, G.S. The catalytic cycle of cytochrome P-450scc and intermediates in the conversion of cholesterol to pregnenolone. Eur. J. Biochem. 1984, 140, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, K.; Fujita, H. Light and electron microscopic immunohistochemistry of the localization of adrenal steroidogenic enzymes. Microsc. Res. Tech. 1997, 36, 445–453. [Google Scholar] [CrossRef]
- Ichimura, T.; Yamamura, H.; Sasamoto, K.; Tominaga, Y.; Taoka, M.; Kakiuchi, K.; Shinkawa, T.; Takahashi, N.; Shimada, S.; Isobe, T. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 2005, 280, 13187–13194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrey, F. Transcriptional control of sodium transport in tight epithelial by adrenal steroids. J. Membr. Biol. 1995, 144, 93–110. [Google Scholar] [CrossRef]
- Wagner, C.A.; Mohebbi, N.; Uhlig, U.; Giebisch, G.H.; Breton, S.; Brown, D.; Geibel, J.P. Angiotensin II stimulates H⁺-ATPase activity in intercalated cells from isolated mouse connecting tubules and cortical collecting ducts. Cell. Physiol. Biochem. 2011, 28, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumz, M.L.; Lynch, I.J.; Greenlee, M.M.; Cain, B.D.; Wingo, C.S. The renal H+-K+-ATPases: Physiology, regulation, and structure. Am. J. Physiol. Renal Physiol. 2010, 298, F12–F21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, N.M.; Seckl, J.R. 11beta-hydroxysteroid dehydrogenase type 1 and obesity. Front. Horm. Res. 2008, 36, 146–164. [Google Scholar] [PubMed]
- Buonafine, M.; Bonnard, B.; Jaisser, F. Mineralocorticoid receptor and cardiovascular disease. Am. J. Hypertens. 2018, 31, 1165–1174. [Google Scholar] [CrossRef]
- Wehling, M. Rapid actions of aldosterone revisited: Receptors in the limelight. J. Steroid Biochem. Mol. Biol. 2018, 176, 94–98. [Google Scholar] [CrossRef]
- Haas, E.; Bhattacharya, I.; Brailoiu, E.; Damjanović, M.; Brailoiu, G.C.; Gao, X.; Mueller-Guerre, L.; Marjon, N.A.; Gut, A.; Minotti, R.; et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ. Res. 2009, 104, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1870–1882. [Google Scholar] [CrossRef]
- Cheng, S.B.; Dong, J.; Pang, Y.; LaRocca, J.; Hixon, M.; Thomas, P.; Filardo, E.J. Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol. Cell. Endocrinol. 2014, 382, 950–959. [Google Scholar] [CrossRef]
- Li, Y.C.; Ding, X.S.; Li, H.M.; Zhang, Y.; Bao, J. Role of G protein-coupled estrogen receptor 1 in modulating transforming growth factor-β stimulated mesangial cell extracellular matrix synthesis and migration. Mol. Cell. Endocrinol. 2014, 391, 50–59. [Google Scholar] [CrossRef]
- Chang, Y.; Han, Z.; Zhang, Y.; Zhou, Y.; Feng, Z.; Chen, L.; Li, X.R.; Li, L.; Si, J.Q. G protein-coupled estrogen receptor activation improves contractile and diastolic functions in rat renal interlobular artery to protect against renal ischemia reperfusion injury. Biomed. Pharmacother. 2019, 112, 108666. [Google Scholar] [CrossRef]
- Ruhs, S.; Nolze, A.; Hübschmann, R.; Grossmann, C. 30 years of the mineralocorticoid receptor: Nongenomic effects via the mineralocorticoid receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef] [Green Version]
- Reincke, M.; Bancos, I.; Mulatero, P.; Scholl, U.I.; Stowasser, M.; Williams, T.A. Diagnosis and treatment of primary aldosteronism. Lancet Diabetes Endocrinol. 2021, 9, 876–892. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, K.; Ando, K.; Fujita, T.; Hasebe, N.; Higaki, J.; Horiuchi, M.; Imai, Y.; Imaizumi, T.; Ishimitsu, T.; Ito, M.; et al. The japanese society of hypertension guidelines for the management of hypertension (JSH 2014). Hypertens. Res. 2014, 37, 253–390. [Google Scholar]
- Funder, J.W. Aldosterone and mineralocorticoid receptors-physiology and pathophysiology. Int J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [Green Version]
- Catena, C.; Colussi, G.; Sechi, L.A. Aldosterone, organ damage and dietary salt. Clin. Exp. Pharmacol. Physiol. 2013, 40, 922–928. [Google Scholar] [CrossRef]
- Goodfriend, T.L.; Kelley, D.E.; Goodpaster, B.H.; Winters, S.J. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes. Res. 1999, 7, 355–362. [Google Scholar] [CrossRef]
- Kidambi, S.; Kotchen, J.M.; Grim, C.E.; Raff, H.; Mao, J.; Singh, R.J.; Kotchen, T.A. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension 2007, 49, 704–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colussi, G.; Catena, C.; Lapenna, R.; Nadalini, E.; Chiuch, A.; Sechi, L.A. Insulin resistance and hyperinsulinemia are related to plasma aldosterone levels in hypertensive patients. Diabetes Care 2007, 30, 2349–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherajee, S.J.; Fujita, Y.; Rafiq, K.; Nakano, D.; Mori, H.; Masaki, T.; Hara, T.; Kohno, M.; Nishiyama, A.; Hitomi, H. Aldosterone induces vascular insulin resistance by increasing insulin-like growth factor-1 receptor and hybrid receptor. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.B.; McGraw, A.P.; Jaffe, I.Z.; Sowers, J.R. Mineralocorticoid receptor-mediated vascular insulin resistance: An early contributor to diabetes-related vascular disease? Diabetes 2013, 62, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Balasubramanian, K. Excess aldosterone-induced changes in insulin signaling molecules and glucose oxidation in gastrocnemius muscle of adult male rat. Mol. Cell. Biochem. 2013, 372, 113–126. [Google Scholar] [CrossRef]
- Luther, J.M. Effects of aldosterone on insulin sensitivity and secretion. Steroids 2014, 91, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Ohshima, S.; Fujisawa, E.; Koya, D.; Tsuneki, H.; Sasaoka, T. Aldosterone inhibits insulin-induced glucose uptake by degradation of insulin receptor substrate (IRS) 1 and IRS2 via a reactive oxygen species-mediated pathway in 3T3-L1 adipocytes. Endocrinology 2009, 150, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Hirata, A.; Maeda, N.; Hiuge, A.; Hibuse, T.; Fujita, K.; Okada, T.; Kihara, S.; Funahashi, T.; Shimomura, I. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc. Res. 2009, 84, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Mosso, L.M.; Carvajal, C.A.; Maiz, A.; Ortiz, E.H.; Castillo, C.R.; Artigas, R.A.; Fardella, C.E. A possible association between primary aldosteronism and a lower β-cell function. J. Hypertens. 2007, 25, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.; Bochud, M.; Burnier, M. The association of aldosterone with obesity-related hypertension and the metabolic syndrome. Semin. Nephrol. 2007, 27, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Belin de Chantemèle, E.J.; Mintz, J.D.; Rainey, W.E.; Stepp, D.W. Impact of leptin-mediated sympatho-activation on cardiovascular function in obese mice. Hypertension 2011, 58, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemèle, E.J. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.C.; Otvos, L., Jr.; Belin de Chantemèle, E.J. Leptin induces hypertension and endothelial dysfunction via aldosterone-dependent mechanisms in obese female mice. Hypertension 2016, 67, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.H.; Kim, K.Y.; Kim, J.H.; Baek, A.; Cho, H.; Lee, Y.H.; Kim, J.W.; Kim, D.; Han, S.H.; Lim, J.-S.; et al. A novel adipokine CTRP1 stimulates aldosterone production. FASEB J. 2008, 22, 1502–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowska, J.D. C1q/TNF-related Protein 1, a Multifunctional adipokine: An overview of current data. Am. J. Med. Sci. 2020, 360, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Muendlein, A.; Leiherer, A.; Saely, C.; Ebner, J.; Geiger, K.; Brandtner, E.M.; Vonbank, A.; Fraunberger, P.; Drexel, H. The novel adipokine CTRP1 is significantly associated with the incidence of major adverse cardiovascular events. Atherosclerosis 2019, 286, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, D.; Goicoechea, M.; Sánchez-Niño, M.D.; Ortiz, A.; Verde, E.; Verdalles, U.; Pérez de José, A.; Delgado, A.; Hurtado, E.; Sánchez-Cámara, L.; et al. Obesity and chronic kidney disease progression-the role of a new adipocytokine: C1q/tumour necrosis factor-related protein-1. Clin. Kidney J. 2019, 12, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Schiffrin, E.L. Vascular actions of aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, inflammation, immune system, and hypertension. Am. J. Hypertens. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- Parasiliti-Caprino, M.; Lopez, C.; Prencipe, N.; Lucatello, B.; Settanni, F.; Giraudo, G.; Rossato, D.; Mengozzi, G.; Ghigo, E.; Benso, A.; et al. Prevalence of primary aldosteronism and association with cardiovascular complications in patients with resistant and refractory hypertension. J. Hypertens. 2020, 38, 1841–1848. [Google Scholar] [CrossRef]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.B.; Turcu, A.F.; Auchus, R.J. Primary aldosteronism: Practical approach to diagnosis and management. Circulation 2018, 138, 823–835. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Keidar, S.; Kaplan, M.; Pavlotzky, E.; Coleman, R.; Hayek, T.; Hamoud, S.; Aviram, M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: A possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation 2004, 109, 2213–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwashima, F.; Yoshimoto, T.; Minami, I.; Sakurada, M.; Hirono, Y.; Hirata, Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology 2008, 149, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- He, B.J.; Joiner, M.L.; Singh, M.V.; Luczak, E.D.; Swaminathan, P.D.; Koval, O.M.; Kutschke, W.; Allamargot, C.; Yang, J.; Guan, X.; et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011, 17, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Rocha, R.; Rudolph, A.E.; Frierdich, G.E.; Nachowiak, D.A.; Kekec, B.K.; Blomme, E.A.; McMahon, E.G.; Delyani, J.A. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1802–H1810. [Google Scholar] [CrossRef]
- Blasi, E.R.; Rocha, R.; Rudolph, A.E.; Blomme, E.A.; Polly, M.L.; McMahon, E.G. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003, 63, 1791–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, Y.; Ueda, S.; Hamada, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin. Exp. Nephrol. 2012, 16, 81–88. [Google Scholar] [CrossRef]
- Brown, N.J.; Kim, K.S.; Chen, Y.Q.; Blevins, L.S.; Nadeau, J.H.; Meranze, S.G.; Vaughan, D.E. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J. Clin. Endocrinol. Metab. 2000, 85, 336–344. [Google Scholar] [CrossRef]
- Chun, T.Y.; Pratt, J.H. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol. Cell. Endocrinol. 2005, 239, 55–61. [Google Scholar] [CrossRef]
- Somanna, N.K.; Yariswamy, M.; Garagliano, J.M.; Siebenlist, U.; Mummidi, S.; Valente, A.J.; Chandrasekar, B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 2015, 27, 1928–1938. [Google Scholar] [CrossRef]
- Bunda, S.; Wang, Y.; Mitts, T.F.; Liu, P.; Arab, S.; Arabkhari, M.; Hinek, A. Aldosterone stimulates elastogenesis in cardiac fibroblasts via mineralocorticoid receptor-independent action involving the consecutive activation of Gα13, c-Src, the insulin-like growth factor-I receptor, and phosphatidylinositol 3-kinase/Akt. J. Biol. Chem. 2009, 284, 16633–16647. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial sodium channel in aldosterone-induced endothelium stiffness and aortic dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef]
- Gromotowicz, A.; Szemraj, J.; Stankiewicz, A.; Zakrzeska, A.; Mantur, M.; Jaroszewicz, E.; Rogowski, F.; Chabielska, E. Study of the mechanisms of aldosterone prothrombotic effect in rats. J. Renin. Angiotensin Aldosterone Syst. 2011, 12, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Gromotowicz-Poplawska, A.; Stankiewicz, A.; Kramkowski, K.; Gradzka, A.; Wojewodzka-Zelezniakowicz, M.; Dzieciol, J.; Szemraj, J.; Chabielska, E. The acute prothrombotic effect of aldosterone in rats is partially mediated via angiotensin II receptor type 1. Thromb. Res. 2016, 138, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Gromotowicz-Poplawska, A.; Flaumenhaft, R.; Gholami, S.K.; Merrill-Skoloff, G.; Chabielska, E.; Williams, G.H.; Romero, J.R. Enhanced thrombotic responses are associated with striatin deficiency and aldosterone. J. Am. Heart Assoc. 2021, 10, e022975. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.E.; Rariy, C.M.; Sun, B.; Williams, J.; Lasky-Su, J.; Baudrand, R.; Yao, T.; Moize, B.; Hafiz, W.M.; Romero, J.R.; et al. Variants in striatin gene are associated with salt-sensitive blood pressure in mice and humans. Hypertension 2015, 65, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Garza, A.E.; Pojoga, L.H.; Moize, B.; Hafiz, W.M.; Opsasnick, L.A.; Siddiqui, W.T.; Horenstein, M.; Adler, G.K.; Williams, G.H.; Khalil, R.A. Critical role of striatin in blood pressure and vascular responses to dietary sodium intake. Hypertension 2015, 66, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Gromotowicz-Poplawska, A.; Marcinczyk, N.; Misztal, T.; Golaszewska, A.; Aleksiejczuk, M.; Rusak, T.; Chabielska, E. Rapid effects of aldosterone on platelets, coagulation, and fibrinolysis lead to experimental thrombosis augmentation. Vascul. Pharmacol. 2019, 122–123, 106598. [Google Scholar] [CrossRef]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta 2020, 507, 167–173. [Google Scholar] [CrossRef]
- Lippi, G.; South, A.M.; Henry, B.M. Electrolyte imbalances in patients with severe coronavirus disease 2019 (COVID-19). Ann. Clin. Biochem. 2020, 57, 262–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawathiparnich, P.; Kumar, S.; Vaughan, D.E.; Brown, N.J. Spironolactone abolishes the relationship between aldosterone and plasminogen activator inhibitor-1 in humans. J. Clin. Endocrinol. Metab. 2002, 87, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jia, R.; Bao, Y. Aldosterone up-regulates production of plasminogen activator inhibitor-1 by renal mesangial cells. J. Biochem. Mol. Biol. 2007, 40, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Sone, M.; Inagaki, N.; Yamasaki, T.; Ogawa, O.; Takeda, Y.; Kurihara, I.; Itoh, H.; Umakoshi, H.; Tsuiki, M.; et al. Prevalence of cardiovascular disease and its risk factors in primary aldosteronism: A multicenter study in Japan. Hypertension 2018, 71, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Mulatero, P.; Monticone, S.; Bertello, C.; Viola, A.; Tizzani, D.; Iannaccone, A.; Crudo, V.; Burrello, J.; Milan, A.; Rabbia, F.; et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2013, 98, 4826–4833. [Google Scholar] [CrossRef] [Green Version]
- Savard, S.; Amar, L.; Plouin, P.F.; Steichen, O. Cardiovascular complications associated with primary aldosteronism: A controlled cross-sectional study. Hypertension 2013, 62, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.T.; Liao, C.W.; Tsai, C.H.; Chang, Y.Y.; Chen, Z.W.; Pan, C.T.; Lin, L.-C.; Wu, V.; Kuo, S.-F.; Wu, X.-M.; et al. U-shaped relationship between left ventricular mass index and estimated glomerular filtration rate in patients with primary aldosteronism. J. Investig. Med. 2020, 68, 371–377. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Liao, C.W.; Tsai, C.H.; Chen, C.W.; Pan, C.T.; Chen, Z.W.; Chen, Y.-L.; Lin, L.-C.; Chang, Y.-R.; Wu, V.-C.; et al. Left ventricular dysfunction in patients with primary aldosteronism: A propensity score-matching follow-up study with tissue doppler imaging. J. Am. Heart Assoc. 2019, 8, e013263. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.T.; Liao, C.W.; Tsai, C.H.; Chen, Z.W.; Chen, L.; Hung, C.S.; Liu, Y.-C.; Lin, P.-C.; Chang, C.-C.; Chang, Y.-Y.; et al. Influence of different treatment strategies on new-onset atrial fibrillation among patients with primary aldosteronism: A nationwide longitudinal cohort-based study. J. Am. Heart Assoc. 2020, 9, e013699. [Google Scholar] [CrossRef]
- Burrello, J.; Monticone, S.; Losano, I.; Cavaglià, G.; Buffolo, F.; Tetti, M.; Covella, M.; Rabbia, F.; Veglio, F.; Pasini, B.; et al. Prevalence of hypokalemia and primary aldosteronism in 5100 patients referred to a tertiary hypertension unit. Hypertension 2020, 75, 1025–1033. [Google Scholar] [CrossRef]
- Ohno, Y.; Sone, M.; Inagaki, N.; Kawashima, A.; Takeda, Y.; Yoneda, T.; Kurihara, I.; Itoh, H.; Tsuiki, M.; Ichijo, T.; et al. Nadir aldosterone levels after confirmatory tests are correlated with left ventricular hypertrophy in primary aldosteronism. Hypertension 2020, 75, 1475–1482. [Google Scholar] [CrossRef]
- Seccia, T.M.; Letizia, C.; Muiesan, M.L.; Lerco, S.; Cesari, M.; Bisogni, V.; Petramala, L.; Maiolino, G.; Volpin, R.; Rossi, G.P. Atrial fibrillation as presenting sign of primary aldosteronism: Results of the prospective appraisal on the prevalence of primary aldosteronism in hypertensive (PAPPHY) study. J. Hypertens. 2020, 38, 332–339. [Google Scholar] [CrossRef]
- Hu, J.; Shen, H.; Huo, P.; Yang, J.; Fuller, P.J.; Wang, K.; Yang, Y.; Ma, L.; Cheng, Q.; Gong, L.; et al. Heightened cardiovascular risk in hypertension associated with renin-independent aldosteronism versus renin-dependent aldosteronism: A collaborative study. J. Am. Heart Assoc. 2021, 10, e023082. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Ren, Y.; Wang, W.; Cheng, W.; Zhou, F.; He, S.; Liu, X.; Li, L.; Tang, L.; Deng, Q.; et al. Left ventricular remodeling in patients with primary aldosteronism: A prospective cardiac magnetic resonance imaging study. Korean J. Radiol. 2021, 22, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Aune, A.; Kokorina, M.; Grytaas, M.A.; Midtbø, H.; Løvås, K.; Gerdts, E. Preclinical cardiac disease in women and men with primary aldosteronism. Blood Press 2021, 30, 230–236. [Google Scholar] [CrossRef]
- Gan, L.; Li, N.; Heizhati, M.; Lin, M.; Zhu, Q.; Yao, X.; Wu, T.; Wang, M.; Luo, Q.; Zhang, D.; et al. Higher plasma aldosterone is associated with increased risk of cardiovascular events in hypertensive patients with suspected OSA: UROSAH data. Front. Endocrinol. 2022, 13, 1017177. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Vaidya, A.; Subudhi, S.; Waikar, S.S. Aldosterone in chronic kidney disease and renal outcomes. Eur. Heart J. 2022, 43, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Pan, C.T.; Chang, Y.Y.; Chen, Z.W.; Wu, V.C.; Hung, C.S.; Lin, Y.-H. Left ventricular remodeling and dysfunction in primary aldosteronism. J. Hum. Hypertens. 2021, 35, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Naruse, M.; Naruse, K.; Hase, M.; Yoshimoto, T.; Tanaka, M.; Seki, T.; Demura, R.; Demura, H. Left ventricular hypertrophy is more prominent in patients with primary aldosteronism than in patients with other types of secondary hypertension. Hypertens. Res. 1997, 20, 85–90. [Google Scholar] [CrossRef]
- Matsumura, K.; Fujii, K.; Oniki, H.; Oka, M.; Iida, M. Role of aldosterone in left ventricular hypertrophy in hypertension. Am. J. Hypertens. 2006, 19, 13–18. [Google Scholar] [CrossRef] [Green Version]
- López, N.; Díez, J.; Fortuño, M.A. Differential hypertrophic effects of cardiotrophin-1 on adult cardiomyocytes from normotensive and spontaneously hypertensive rats. J. Mol. Cell. Cardiol. 2006, 41, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Andrés, N.; Martin-Fernandez, B.; Rossignol, P.; Zannad, F.; Lahera, V.; Fortuno, M.A.; Cachofeiro, V.; Díez, J. A role for cardiotrophin-1 in myocardial remodeling induced by aldosterone. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2372–H2382. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Do, H.; Pham, T.; Vu, L.T.; Zuin, M.; Rigatelli, G. Left ventricular dysfunction causing ischemia in patients with patent coronary arteries. Perfusion 2018, 33, 115–122. [Google Scholar] [CrossRef]
- Reil, J.C.; Hohl, M.; Selejan, S.; Lipp, P.; Drautz, F.; Kazakow, A.; Münz, B.M.; Müller, P.; Steendijk, P.; Reil, G.-H.; et al. Aldosterone promotes atrial fibrillation. Eur. Heart J. 2012, 33, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.T.; Chiang, F.T.; Tseng, C.D.; Hwang, J.J.; Kuo, K.T.; Wu, C.K.; Yu, C.-C.; Wang, Y.-C.; Lai, L.P.; Lin, J.-L. Increased expression of mineralocorticoid receptor in human atrial fibrillation and a cellular model of atrial fibrillation. J. Am. Coll. Cardiol. 2010, 55, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.F.; Yang, S.F.; Chu, H.J.; Ueng, K.C. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int. J. Cardiol. 2013, 169, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, R.; Borowiec, A.; Smolis-Bak, E.; Kowalik, I.; Sosnowski, C.; Kraska, A.; Kazimierska, B.; Wozniak, J.; Zareba, W.; Szwed, H. Effect of combined spironolactone-β-blocker ± enalapril treatment on occurrence of symptomatic atrial fibrillation episodes in patients with a history of paroxysmal atrial fibrillation (SPIR-AF study). Am. J. Cardiol. 2010, 106, 1609–1614. [Google Scholar] [CrossRef]
- Swedberg, K.; Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Shi, H.; Vincent, J.; Pitt, B.; EMPHASIS-HF Study Investigators. Eplerenone and atrial fibrillation in mild systolic heart failure: Results from the EMPHASIS-HF (Eplerenone in mild patients hospitalization and survival study in heart failure) study. J. Am. Coll. Cardiol. 2012, 59, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Yao, L.; Nagai, Y.; Miyata, K.; Yoshizumi, M.; Kagami, S.; Kondo, S.; Kiyomoto, H.; Shokoji, T.; Kimura, S.; et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 2004, 43, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Reincke, M.; Rump, L.C.; Quinkler, M.; Hahner, S.; Diederich, S.; Lorenz, R.; Seufert, J.; Schirpenbach, C.; Beuschlein, F.; Bidlingmaier, M.; et al. Risk factors associated with a low glomerular filtration rate in primary aldosteronism. J. Clin. Endocrinol. Metab. 2009, 94, 869–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halimi, J.M.; Mimran, A. Albuminuria in untreated patients with primary aldosteronism or essential hypertension. J. Hypertens. 1995, 13, 1801–1802. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Chan, J.C.; Cooper, M.E.; Gansevoort, R.T.; Haller, H.; Remuzzi, G.; Rossing, P.; Schmieder, R.E.; Nowack, C.; et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: A randomized clinical trial. JAMA 2015, 314, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, S.D.; Bravo, E.L. Aldosterone breakthrough during angiotensin receptor blocker use: More questions than answers? Clin. J. Am. Soc. Nephrol. 2013, 8, 1637–1639. [Google Scholar] [CrossRef] [Green Version]
- Spencer, S.; Wheeler-Jones, C.; Elliott, J. Aldosterone and the mineralocorticoid receptor in renal injury: A potential therapeutic target in feline chronic kidney disease. J. Vet. Pharmacol. Ther. 2020, 43, 243–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. Pathophysiological roles of aldosterone and mineralocorticoid receptor in the kidney. J. Pharmacol. Sci. 2011, 115, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauersachs, J.; Jaisser, F.; Toto, R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015, 65, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Jaisser, F.; Farman, N. Emerging roles of the mineralocorticoid receptor in pathology: Toward new paradigms in clinical pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogawa, Y.; Nagasu, H.; Itano, S.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Kadoya, H.; Satoh, M.; Sasaki, T.; Kashihara, N. The eNOS-NO pathway attenuates kidney dysfunction via suppression of inflammasome activation in aldosterone-induced renal injury model mice. PLoS ONE 2018, 13, e0203823. [Google Scholar] [CrossRef] [Green Version]
- Martín-Fernández, B.; Rubio-Navarro, A.; Cortegano, I.; Ballesteros, S.; Alía, M.; Cannata-Ortiz, P.; Olivares-Álvaro, E.; Egido, J.; de Andrés, B.; Gaspar, M.L.; et al. Aldosterone induces renal fibrosis and inflammatory M1-macrophage subtype via mineralocorticoid receptor in rats. PLoS ONE 2016, 11, e0145946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, Y.; Miyata, K.; Sun, G.P.; Rahman, M.; Kimura, S.; Miyatake, A.; Kiyomoto, H.; Kohno, M.; Abe, Y.; Yoshizumi, M.; et al. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension 2005, 46, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Diah, S.; Zhang, G.X.; Nagai, Y.; Zhang, W.; Gang, L.; Kimura, S.; Hamid, M.R.; Tamiya, T.; Nishiyama, A.; Hitomi, H. Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells. Exp. Cell Res. 2008, 314, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Joharapurkar, A.; Jain, M. Role of mineralocorticoid receptor antagonists in kidney diseases. Drug Dev. Res. 2021, 82, 341–363. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Rossignol, P.; Pizard, A.; Machu, J.L.; Collier, T.; Girerd, N.; Huby, A.C.; Gonzalez, A.; Diez, J.; López, B.; et al. Potential spironolactone effects on collagen metabolism biomarkers in patients with uncontrolled blood pressure. Heart 2019, 105, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Kadoya, H.; Satoh, M.; Sasaki, T.; Taniguchi, S.; Takahashi, M.; Kashihara, N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015, 29, 3899–3910. [Google Scholar] [CrossRef]
- Kobayashi, H.; Abe, M.; Nakamura, Y.; Takahashi, K.; Fujita, M.; Takeda, Y.; Yoneda, T.; Kurihara, I.; Itoh, H.; Tsuiki, M.; et al. Association between acute fall in estimated glomerular filtration rate after treatment for primary aldosteronism and long-term decline in renal function. Hypertension 2019, 74, 630–638. [Google Scholar] [CrossRef]
- Baran, W.; Krzemińska, J.; Szlagor, M.; Wronka, M.; Młynarska, E.; Franczyk, B.; Rysz, J. Mineralocorticoid receptor antagonists-use in chronic kidney disease. Int. J. Mol. Sci. 2021, 22, 9995. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F.L. Pathogenesis and treatment of primary aldosteronism. Nat. Rev. Endocrinol. 2020, 16, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, W.B.; Relman, A.S. Effects of electrolyte disorders on renal structure and function. N. Engl. J. Med. 1967, 276, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, H.B.; Calp-Inal, S.; Zhou, X.J.; Choudhury, D. Hypokalemic nephropathy. Kidney Int. Rep. 2018, 3, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Reungjui, S.; Roncal, C.A.; Sato, W.; Glushakova, O.Y.; Croker, B.P.; Suga, S.; Ouyang, X.; Tungsanga, K.; Nakagawa, T.; Johnson, R.J.; et al. Hypokalemic nephropathy is associated with impaired angiogenesis. J. Am. Soc. Nephrol. 2008, 19, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Author (Year) | Country | Study Design (Number) | Findings |
|---|---|---|---|
| Cardiovascular disease | |||
| Youichi Ohno (2018) [84] | Japan | Retrospective cross-sectional study (n = 2582) | Patients with PA have a higher prevalence of CVD than those who are age- and sex-matched with EH. |
| Min-Tsun Liao (2019) [87] | Taiwan | Prospective observational and cross-sectional study (n = 336) | In PA patients, the relationship between eGFR and LVMI is not linear. |
| Yi-Yao Chang (2019) [88] | Taiwan | Prospective observational study (n = 249) | Adjusting by propensity score matching between the patients with PA and EH, patients with PA had worse LV diastolic function than patients with EH. |
| Chien-Ting Pan (2020) [89] | Taiwan | Retrospective matched case-control study (n = 11,010) | The patients with PA who underwent adrenalectomy had a lower incidence of NOAF compared with those with EH. |
| Jacopo Burrello (2020) [90] | Italy | Retrospective observational study (n = 5100) | PA and hypokalemia are associated with an increased risk of cardiovascular events. |
| Youichi Ohno (2020) [91] | Japan | Retrospective observational study (n = 1186) | Nadir PAC in the patients with PA after confirmatory tests is associated with LVMI, not the basal aldosterone level itself. |
| Teresa M Seccia (2020) [92] | Italy | Prospective observational study (n = 411) | Unexplained atrial fibrillation in the hypertensive patients shows a high prevalence of PA. |
| Jinbo Hu (2021) [93] | China | Cross-sectional study (n = 5521) | Patients with renin-independent aldosteronism are more closely associated with CVD risk than those with renin-dependent aldosteronism. |
| Tao Wu (2021) [94] | China | Prospective observational study (n = 70) | Compared with patients with EH, patients with PA have a higher degree of LV hypertrophy. |
| Arleen Aune (2021) [95] | Norway | Cross-sectional study (n = 198) | Patients with PA have a higher prevalence of LV hypertrophy both in women and men, compared to EH. |
| Lin Gun (2022) [96] | China | Retrospective cohort study (n = 3173) | Higher plasma aldosterone concentration is associated with increased risk of CVD and all-cause mortality in the hypertensive patients, even independent of OSA and PA. |
| Chronic kidney disease | |||
| María Fernández-Argüeso (2021) [2] | Spain | Case-control study (n = 100) | Compared with patients with EH, patients with PA have a higher prevalence of CKD at the time of diagnosis. |
| Ashish Verma (2022) [97] | United States of America | Prospective observational study (n = 3680) | Regardless of concomitant diabetes, high serum aldosterone levels in the serum of patients with CKD are independently associated with an increased risk for CKD progression. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otsuka, H.; Abe, M.; Kobayashi, H. The Effect of Aldosterone on Cardiorenal and Metabolic Systems. Int. J. Mol. Sci. 2023, 24, 5370. https://doi.org/10.3390/ijms24065370
Otsuka H, Abe M, Kobayashi H. The Effect of Aldosterone on Cardiorenal and Metabolic Systems. International Journal of Molecular Sciences. 2023; 24(6):5370. https://doi.org/10.3390/ijms24065370
Chicago/Turabian StyleOtsuka, Hiromasa, Masanori Abe, and Hiroki Kobayashi. 2023. "The Effect of Aldosterone on Cardiorenal and Metabolic Systems" International Journal of Molecular Sciences 24, no. 6: 5370. https://doi.org/10.3390/ijms24065370
APA StyleOtsuka, H., Abe, M., & Kobayashi, H. (2023). The Effect of Aldosterone on Cardiorenal and Metabolic Systems. International Journal of Molecular Sciences, 24(6), 5370. https://doi.org/10.3390/ijms24065370