1. Introduction
Cajal–Retzius cells (CRs) are transient excitatory pioneer neurons that migrate tangentially at the surface of the cortex during development and also populate the borders of the hippocampal fissure [
1]. They come in multiple types depending on their origins. At least four sources at the border of the pallium have been characterized: the cortical hem, the pallial septum, the ventral pallium, and the thalamic eminences [
2,
3]. Besides their well-known function in cortical layering through the secretion of Reelin, they also control multiple steps of cortical development from neuronal proliferation to dendritogenesis and in functional area formation [
1,
4]. In rodents, neocortical CRs are almost fully eliminated in the two first postnatal weeks according to a neuronal cell-death program, which remains to be explored. Our previous studies have shown that BAX-mediated apoptosis is responsible for the elimination of a fraction of CRs, especially those derived from the septum [
5]. In addition, this cell-death mechanism is dependent on neuronal activity, since blocking their excitatory inputs from inhibitory neurons [
6] or hyperpolarizing the CRs [
7] leads to their partial survival.
Cell death and survival are two Janus-faced components of cell life, and inhibiting cell survival leads to the same outcome. Amongst the most well-known pathways involved in cell survival is PI3K/AKT/mTOR, whose activation inhibits apoptosis, potentially leading to human cancer when overactivated in proliferating cells [
8]. Class I PI3K catalytic subunits transduce signals from either receptor tyrosine kinase (RTKs) or G protein-coupled receptors (GPCRs) to AKT, which in turn phosphorylates numerous regulators to activate the mTORC1 complex. This pathway is negatively controlled at different levels by various proteins such as PTEN (phosphatase and tensin homolog), which reduces PIP3 levels, or by the TSC1/2 complex (tuberous sclerosis complex), which acts as GTPase activating proteins to inhibit the GTPase RHEB, the main activator of mTORC1 [
9]. In addition to its phosphatase activity on PIP3 at the plasma membrane, PTEN also functions in different compartments such as the nucleus, the endoplasmic reticulum, and the mitochondria, where it regulates transcription and calcium release from the ER and structure, respectively [
10].
During brain development, the activation of the pathway secondary to mutations in its components is responsible for various cortical malformations, spanning from hemimegalencephaly to focal cortical dysplasia associated with intractable pediatric epilepsy [
11]. The severity of the phenotype correlates with the occurrence of the somatic mutation: the earlier the occurrence, the worse the phenotype [
12].
In the mouse brain, the conditional activation of the pathway leads to different outcomes depending on whether it is eliminated in proliferating cells or neurons [
13]. The deletion of
Pten in neural progenitors enhances cell proliferation [
14], whereas it causes hypertrophy in early post-mitotic neurons without altering specification and radial migration in the principal neurons of the neocortex [
15]. The pathway is also essential for synapse and dendritic development [
16] and participates in synaptic plasticity [
17]. PTEN removal in hippocampal granule cells leads to focal seizures or generalized seizures depending on the degree of mosaicism [
18].
During postnatal cortical development, active pyramidal neurons inhibit the PTEN expression in interneurons and protect them from cell death [
19]. Here, we investigated the role of the PI3K/AKT/mTOR pathway in the cell death of CRs derived from the ΔNp73 lineage, which includes 80% of all CRs in the neocortex. We show that this pathway is physiologically inhibited at the postnatal stage in CRs before they start to die through apoptosis. We then sustained its activation using a genetic approach to prevent CRs from death. Both gain and loss-of-function approaches of PI3K or PTEN, respectively, led to CR survival, whereas the activation of the mTOR pathway by TSC1 removal had little effect on the cell numbers. As expected for the activation of this pathway, the morphology and the intrinsic properties of the survival CRs were altered, but these cells were still active and expressed more Reelin in an overall preserved cortex. PTEN loss-of-function in ΔNp73-derived CRs reduced Reelin N-terminal cleavage in the cortex of young animals, leading to an increase in the full-length fraction with no change in the mTOR activity in the pyramidal neurons underneath. Finally, the persistence of CRs in layer I of the cortex did not alter the general behavior of the
Pten model, but increased its susceptibility to kainate-induced seizures.
3. Discussion
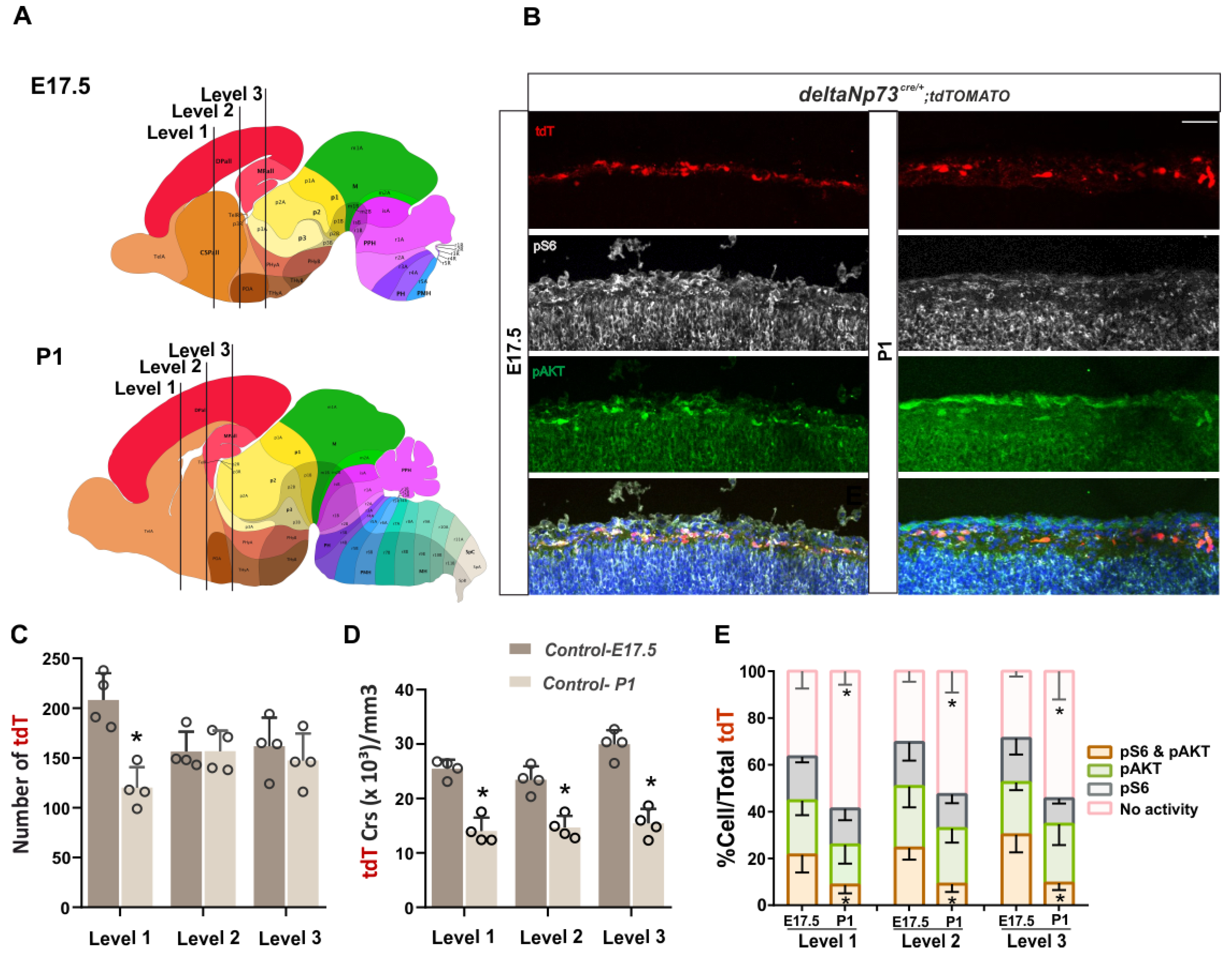
The PI3K/AKT/mTOR pathway is one of the major cell survival signaling cascades involved in the regulation of mitochondria-mediated apoptosis and has been shown, amongst others, to control interneuron cell death [
19]. Here, we focused our study on a particular type of excitatory neuron, CRs, which populate the surface of the cortex during embryogenesis and die postnatally after orchestrating cortical development. We previously showed that Bax-mediated apoptosis and neuronal activity are involved in CR death [
5,
7]. By investigating the PI3K/AKT/mTOR activity around postnatal stages, just before massive cell death occurs in ΔNp73-derived CRs, we found that it is decreased, suggesting its participation in the control of CR cell death as well (
Figure 1E). Besides this temporal regulation of its activity, we also highlighted a differential spatial activation of AKT and S6K along the medio–lateral axis at three rostro–caudal levels. Overall, AKT and mTOR activities are higher in the medial/dorsal and lateral cortex, respectively, at E17.5 and P1, suggesting that they are differentially regulated in the marginal zone above the future cortical areas (
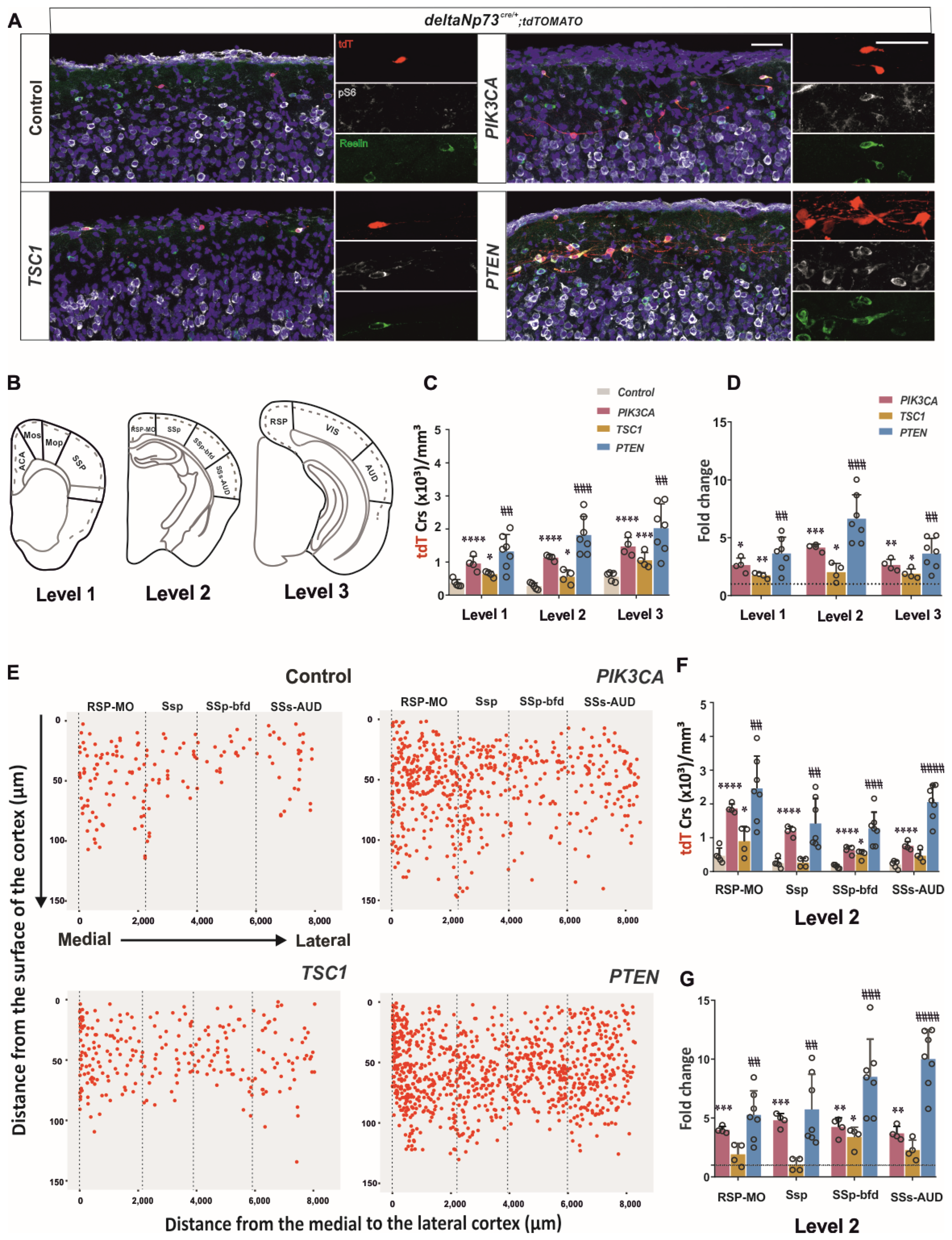
Figure S1). We then sustained the activation of the pathway in these cells in vivo using a genetic approach, and found that a fraction of them survived at P24 (
Figure 2) in the cortex up to 110 days, whereas they should be almost eliminated by day 15. By conditionally targeting
Pten and
Tsc1 in ΔNp73-derived CRs, we found that AKT activation contributes most to the survival CR phenotype compared to mTOR (
Figure 2A). In addition, the increase in the CRs in the
Pten model was higher in the lateral cortex than in the medio–dorsal (
Figure 2G), which correlates with the lower AKT pathway activation described at E17.5 and P1 in this region (
Figure S1). Altogether, the PI3K/AKT pathway is more inhibited in the lateral cortex compared to medio–lateral, which may confer to the lateral CRs’ greater susceptibility to cell death. The various timing of CR elimination along the medio–lateral axis may influence the development of the future brain areas as well. Since the secondary somatosensory auditory cortex presents with a 10-fold increase in CR at P24 in the
Pten model (
Figure 2G), it would be interesting in the future to challenge the animal behavioral response to various tests involving auditory stimulation (i.e., pre-pulse inhibition or fear conditioning test).
In this study, we also had the opportunity to test and compare two models of activation through either the expression of an activated PI3KCA mutant protein (p110*) or the deletion of PTEN. Both approaches should increase the PIP3 levels to activate AKT through PDK1 and one would have expected a phenocopy between the two mutants and even a stronger phenotype for the P110*, which has been shown to strongly activate AKT [
31]. Although the number of persistent CRs is similar between the two mutants (
Figure 2A,C), the
Pten mutant clearly exhibits a large increase in dendrite and/or axonal extensions, which is rarely seen in the p110* model. One hypothesis may be the compensation of PIP3 overproduction in P110* mutant CRs by endogenous PTEN, but one would expect to also have such a compensation effect for cell survival and fewer CRs in P110* compared to the
Pten model. Alternatively, PTEN may also have PIP3-independent functions [
10]. Axodendritic outgrowth requires some reorganization of actin/microtubule cytoskeleton and, besides the known regulation of F-actin dynamics by phosphoinositides through Rho GTPases, PTEN directly regulates the stability of the microtubules through their detyrosination [
32]. Apart from these morphological differences in CR projections, both mutant CRs present with an increase in the soma size together with an increase in pS6 staining. These two phenotypes are also observed in the
Tsc1 mutant CRs (
Figure 2A) and are linked to mTORC1 activation and the increase in protein synthesis [
9].
Surprisingly, whereas the loss of
Tsc1 in the cortex has been shown to increase the basal dendrite arborization of pyramidal neurons, a phenotype reversible upon rapamycin treatment during a critical period [
33], only bipolar CRs were observed in
Tsc1 cKO (
Figure 2A). This suggests that the activation of the mTORC1 pathway alone is not sufficient to alter the neuronal polarity and/or the axodendritic outgrowth in CRs, but it is probably necessary, since these morphological changes require an increase in protein synthesis [
16].
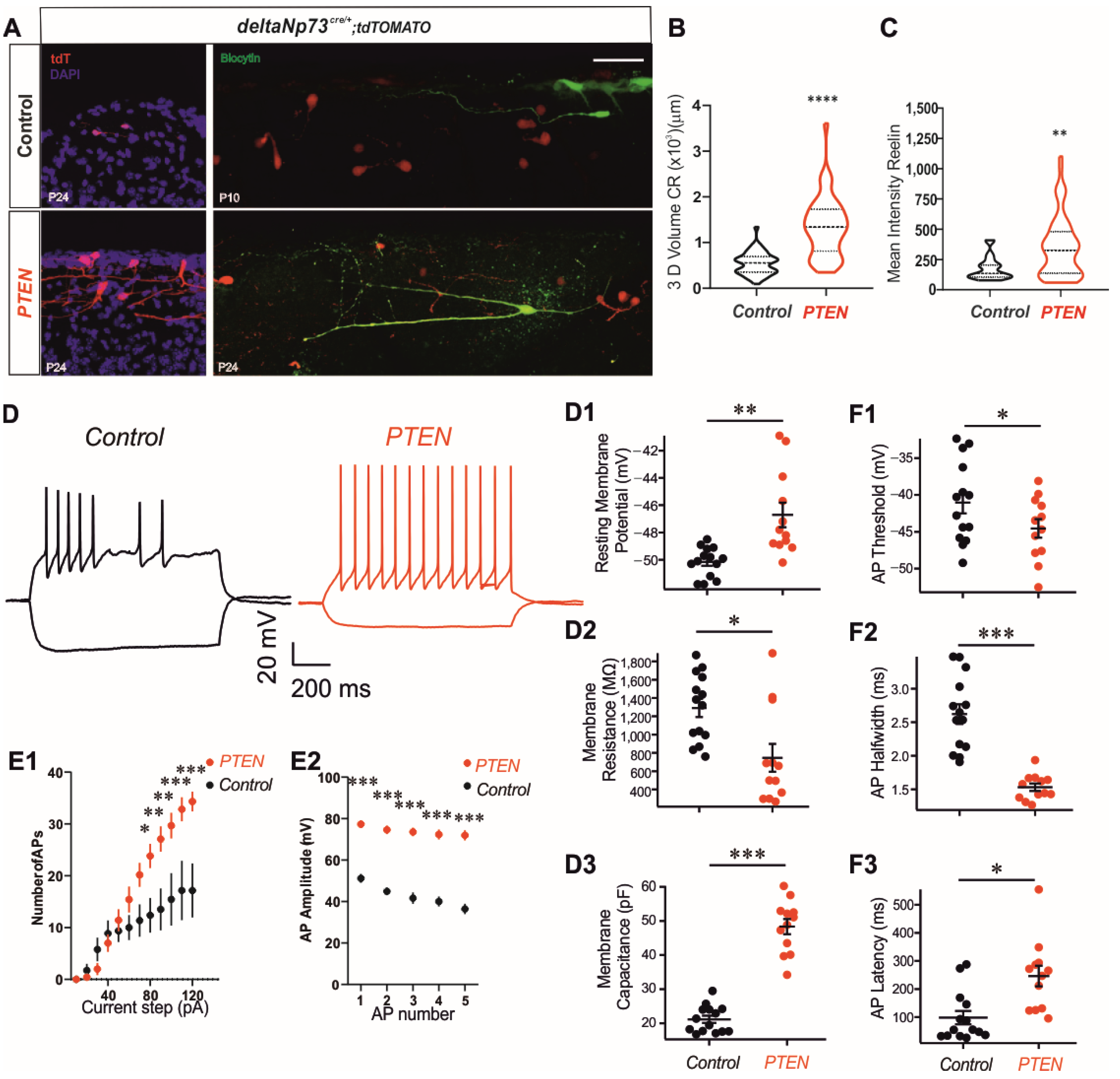
Altogether, these results strongly suggest that PTEN regulates the growth of CR extensions by both AKT-dependent and -independent pathways. This drastic morphological increase in dendritic arborization is consistent with a large increase in membrane capacitance (
Figure 3). We also observed several other changes consistent with a more excitable phenotype. This includes a decreased action potential threshold and input resistance, and increased number of action potentials at current steps higher than 60 pA. Previous reports have demonstrated altered hippocampal expression profiles of voltage-activated ion channels in PTEN knockout animals [
34]. We postulate that similar changes in membrane excitability occurred in the CRs in our preparation.
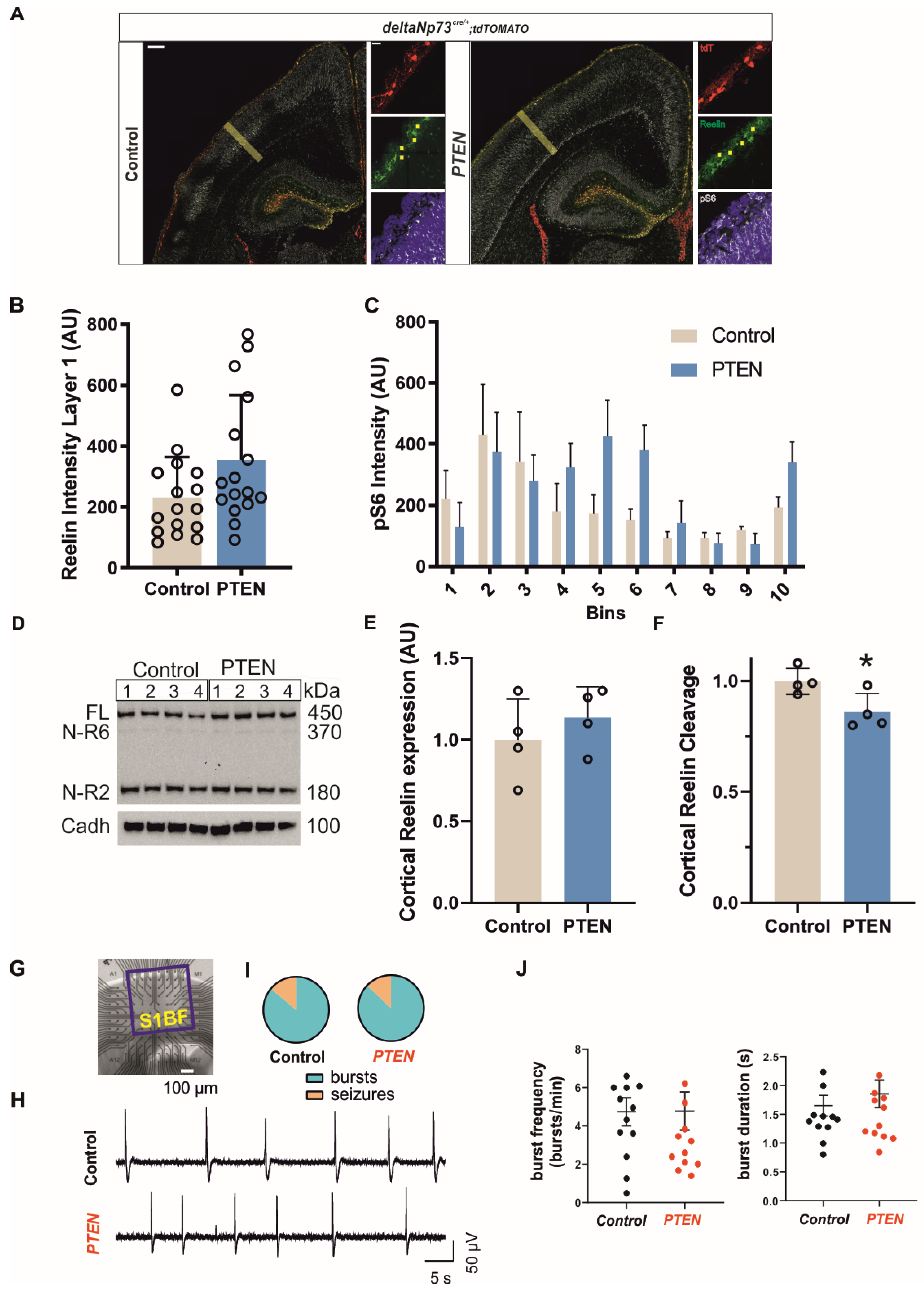
Our results, together with the well-known effects of AKT activation on cell survival [
35], strongly suggest that apoptosis is inhibited in our
Pten model, which leads to an increase in CR densities in postnatal cortex. In theory,
Pten and
Bax inactivation should have similar secondary phenotypes linked to CR persistence. They have similar increases in cell densities, about five-fold. The
Bax model presents with an increase in dendrites and dendritic spines in upper pyramidal neurons, which leads to an alteration of the excitatory to inhibitory balance and hyperexcitability together with an increase in susceptibility to kainate-induced seizures [
28]. Although we did not investigate the morphological changes of these pyramidal cells in the
Pten model, neuronal network activity is unaffected in the S1 barrel cortex. However, both
Bax and
Pten animals do present an increased susceptibility to seizures. Interestingly, both models showed an opposite gender differential effect, with the
Pten model affecting females. While the role of testosterone in models of temporal lobe epilepsy has already been investigated [
36], sex differences in the protein expression of the AKT/mTOR pathway have been shown in the hippocampus and cortex [
37], and these differential expressions may explain the increased duration in the seizures of
Pten females after kainate injection.
4. Materials and Methods
4.1. Animals
ΔNp73CreIRESGFP(ΔNp73Cre) [
20] and
Ai9 line (Strain# 007905)
ROSA26loxP-stop-loxP-Tomato(R26mT) [
38] transgenic mice were kept in a C57BL/6J background.
R26StopFlp110* mice (strain# 012343) were a gift from Dr. Guillaume Canaud (University Paris Cité) [
24].
PtenloxP mice [
39] and
Tsc1loxP mice [
40] were provided by Dr. Mario Pende (University Paris Cité). Animals were genotyped by PCR using specific primers (
Supplementary Table S9). Different cohorts of control and mutant animals were produced according to the experiments and are summarized in
Table 1.
All animals were handled in strict accordance with good animal practice as defined by national animal welfare bodies.
4.2. Tissue Preparation and Immunohistochemistry
The staging of animals was conducted considering midday of the vaginal plug formation as embryonic day 0.5 (E0.5) for the embryonic stage and the birth date as postnatal day 0 (P0) at the postnatal stage. Brain collection and fixation of both embryos and P1 animals were performed as previously described [
2]. For juvenile mouse (P24 days after birth) brain sections, the animals were anesthetized by the intraperitoneal administration of a mixture of sedative (xylazine 10 mg/kg) and anesthetic (ketamine 180 mg/kg) substances and were intracardially perfused by 4% paraformaldehyde (PFA) in PBS 1× (
v/
w), pH 7.4 and post-fixed 2 h in 4% PFA at 4 °C. The brains were rinsed in PBS for 1 h and placed in 30% sucrose in PBS 1× (
v/
w) overnight for cryoprotection, and then embedded in OCT compound (Sakura). Embedded tissues were sectioned on a cryostat with a 20 µm thickness for embryonic and P1 stage and 50 µm for P24 brains. Three sections from rostral, intermediate, and caudal brain levels were chosen from the controls and mutants. Immunostaining on embryonic and P1 sections was performed as previously described [
2]. The P24 brain sections were washed first in PBS 1× and then in 0.2% triton + PBS1X before immunostaining. Afterwards, the sections were permeabilized in PGT (PBS1× 0.25% triton 0.2% gelatin) for 1 h and then incubated with primary antibodies diluted in PGT at 4 °C overnight. The second day after washing, the sections were first incubated in PGT for 10 min and then were incubated in a secondary antibody diluted in PGT at room temperature for 1 h. Double immunostaining of two rabbit antibodies was carried out using a Zenon kit (Zenon Alexa Fluor 488 Rabbit IgG Labeling Kit, Z25302, Invitrogen by Thermo Fisher Scientific (Waltham, MA, USA)). The primary antibodies used for immunohistochemistry were: goat anti-Reelin antibody (R and D Systems (Minneapolis, MIN, USA) AF3820, 1:500), rabbit anti-phospho-AKT serine 473 antibody (Cell Signaling Technology (Danvers, MA, USA) 4060, 1:200), rabbit anti-phospho-S6 antibody (Ser 240/244 Cell Signaling Technology(Danvers, MA, USA) 5364, 1:1000), rat anti-ctip2 antibody (Abcam (Cambridge, UK) ab18465, 1:250), rabbit anti-cux1,2 antibody (Santa Cruz (Dallas, TX, USA) sc-13024, 1:250). The secondary antibodies used against the primary antibodies were: donkey anti-rabbit Alexa-488 (711-545-152, Jackson ImmunoResearch Laboratories(Cambridge, UK),1:500), donkey anti-goat Alexa-488 (A-11055, Molecular Probes(Eugene, OR, USA), 1:500), donkey anti-goat Cy5 (705-175-147, Jackson ImmunoResearch Laboratories (Cambridge, UK), 1:250), and donkey anti-rat Alexa-Cy5 (712-175-153, Jackson ImmunoResearch Laboratories(Cambridge, UK), 1:250). DAPI (D1306, ThermoFisher Scientific (Waltham, MA, USA), 1:1000) was used for nuclear staining. Sections were mounted using Vectashield (H-1000, Vector Labs(Burlingame, CA, USA)).
4.3. Image Acquisition and Analyses
Immunofluorescence images were acquired using a confocal Leica SP8 microscope (Leica Microsystems, Mannheim, Germany) at the E17.5 and P1 stages and a slide scanner Nanozoomer 2.0 (Hamamatsu) with a 20× objective for the quantification of CRs at P24. Confocal images were acquired using a 40× oil immersion objective, pinhole-sized “airy 2” and four z-stacks of 5 µm were defined in order to acquire sections of 20 µm thickness.
Then, the X and Y positions of each of the tdT-positive cells were recorded from a 150 µm depth straightened cortex using Fiji software [
41]. For each section, the density of CRs (tdTomato + CRs/mm
3) was calculated considering the thickness of the section, the neocortical length at P1 or the area lengths at P24 and the depth from the surface of the cortex.
Illustrated images with bigger magnification (
Figure 2 and
Figure S2) were acquired with a 40× and 3 digital zoom. 9 to 10 z-stacks with an optical section of 2 µm were acquired.
Illustrated images including biocytin-filled CRs (
Figure 3) were acquired using a 63× oil immersion objective. Then, 20 to 60 z-stacks of 2 µm optical section were defined depending on the thickness of the sections and the size of the neurons. tdTomato + neurons were counted and analyzed for their position using the ImageJ software. Three sections from each biological replicate at the embryonic, P1, and P24 stages were analyzed corresponding to three different brain levels.
4.4. Analysis of Reelin and pS6 Fluorescent Intensities in Layer I and in Cortex
Using Fiji software [
41], the fluorescent intensity signal of extracellular Reelin in layer I of the primary somatosensory cortex at the P1 stage was measured in four different regions of interest (ROIs) devoid of any cell bodies. From the same cortical area, pS6 fluorescent intensities were measured in a ROI rectangle divided into 10 bins and drawn perpendicular to the surface of the cortex from LI to L6 in the primary somatosensory cortex. In both quantifications, the mean intensities of the ROIs were corrected by subtracting from their value the mean intensity of an ROI outside the cortex where signals were minimal (from cerebral nuclei).
4.5. Western Blot Analysis
Cortices from the Pten and control mice (n = 4) were dissected from the brain at P7–P8 and snap-frozen in liquid nitrogen before extraction in RIPA buffer (ref: #R0278; Sigma-Aldrich (Saint-Louis, MI, USA) with protease (ref: #04693116001; Roche (Bâle, Switzerland) and phosphatase (ref: #5870; Cell Signaling (Danvers, MA, USA)) inhibitors. Following this, 25 µg of the total protein extracts were run on a 3–8% Tris–acetate gradient gel (NuPAGETM, Ref: #EA03785BOX; Invitrogen (Waltham, MA, USA)) and transferred to nitrocellulose membrane (ref: #GE10600002, AmershamTM ProtanTM, Sigma-Aldrich (Saint-Louis, MI, USA)) before overnight incubation with primary antibodies (mouse anti-Reelin G10 (1:500; MAB5364; Millipore (Burlington, MA, USA), mouse anti-pan Cadherin (1:1000; C1821; Sigma-Aldrich (Saint-Louis, MI, USA)) in a Tris buffer (0.1M; pH:8) sodium (1.5M) Tween 20 (0.05%) (TBST) solution containing 5% fat-free milk. After 45 min of incubation with horseradish peroxidase conjugated mouse secondary antibodies (1/20.000, ref: # 115-035-008; Jackson ImmunoResearch (Cambridge, UK)) and three washes in TBST, the membranes were incubated in chemiluminescent substrate (Super Signal Pico Plus ref: #34580; Thermo Fisher Scientist (Waltham, MA, USA)) before exposition and signal acquisition under a digital camera (ChemidocTM Touch Imaging System; Bio-Rad (Hercules, CA, USA). Signal intensities were analyzed using Image Lab Software (V6.1; Bio-Rad, (Hercules, CA, USA)). The Reelin expression (sum of full-length, NR-6, and NR-2 intensities) was normalized to pan-Cadherin and its cleavage products to total Reelin (sum of NR-6 plus NR-2 normalized to sum of full-length, NR-6, and NR-2).
4.6. Neuronal Soma Size Measurement
To extract the cell volume, we considered the entire z-stack to extract ROIs using the machine learning-based WEKA tool implemented on Fiji/ImageJ2 [
42]. Beforehand, we trained a WEKA classifier on fluorescent series of Td-tomato-loaded cells set using the following features: Gaussian blur, Derivatives, Structure, Minimum, Maximum, and Median. Classification was performed using the FastRandom Forest algorithm. Upon extraction of 3D ROIs, we recorded the volume using the 3D ROI manager tool developed by Ollion and colleagues [
43].
4.7. Patch-Cell Recording
Slice preparation: Coronal slices were prepared from Pten transgenic animals between P20 and P24. At these ages, the animals were anaesthetized with ketamine (100 mg/kg), xylazine (7 mg/kg), and isoflurane, and perfused transcardially with a sucrose-based cutting solution containing the following (in mM): sucrose 110, KCl 2.5, NaH2PO4 1.25, NaHCO3 30, HEPES 20, glucose 25, thiourea 2, Na-ascorbate 5, Na-Pyruvate 3, CaCl2 0.5, and MgCl2 10. Coronal slices were prepared from the control animals at P10, as CR cells were non-viable for electrophysiological recording after this age. At P10, the animals did not undergo cardiac perfusion. For both Pten and controls, the brains were rapidly removed, placed upright, and cut into 300 μm thick coronal slices (Leica Biosystems Vibratomes, VT1200S, (Wetzlar, Germany)) in the cutting solution at 4 °C. The slices were transferred to an immersed-type chamber and maintained in artificial cerebral spinal fluid (ACSF) containing the following (in mM): NaCl 125, KCl 2.5, NaH2PO4 1.25, NaHCO3 26, glucose 10, Na-pyruvate 2, CaCl2 2, and MgCl2 1. The slices were incubated at 32 °C for approximately 10 min and then maintained at room temperature for at least 45 min. Prior to recording, the slices were transferred to a recording chamber perfused with ACSF at 3 mL/min at 30 °C.
Electrophysiological recordings: CR cells were located using the fluorescence tdTomato tag with a 535 nm LED and long-pass mCherry filter set in an Olympus BX51 microscope (Olympus Corporation, Tokyo, Japan). A recording pipette with a resistance between 2 and 5 MΩ with positive pressure was inserted into layer I of the Barrel cortex. Whole-cell recordings were performed with intracellular solutions containing the following (in mM): K-methyl sulfonate 135, KCl 5, EGTA-KOH 0.1, HEPES 10, NaCl 2, MgATP 5, Na2GTP 0.4, Na2-phosphocreatine 10, and biocytin (4 mg/mL). Series resistance was <20 MΩ and was monitored throughout the recordings. Data were discarded if the series resistance changed more than 10% during the experiment. The bridge balance was measured every 20 sec and compensated with internal circuitry and monitored throughout the experiments. The liquid junction potential was not corrected. Data were obtained using a Multiclamp 700B amplifier and digitized using a Digidata 1440 ADDA board. Data were sampled at 10 kHz. Axon™ pCLAMP® 9 electrophysiology data acquisition and analysis software (Molecular Devices, (San Jose, CA, USA)) was used for data acquisition. Action potential firing properties were measured with a series of one-second-long depolarizing current steps.
Immunochemistry and cell identification: Post hoc confirmation of all cells was performed by labeling intracellular biocytin. Following overnight incubation in 4% PFA in PBS, the slices were permeabilized with 0.2% Triton in PBS and blocked for 48 h with 3% goat serum (50062Z, by Thermo Fisher Scientific (Waltham, MA, USA)) in PBS with 0.2% Triton. Streptavidin and Alexa Fluor™ 546 conjugate (S11225, by Thermo Fisher Scientific (Waltham, MA, USA), 1:300) incubation was carried out in block solution for 4 h at room temperature. The slices were mounted in ProLong™ Diamond Antifade Mountant (P36970, Invitrogen by Thermo Fisher Scientific, (Waltham, MA, USA)), resulting in a partial clarification of tissue following incubation at room temperature for 24 h. Images were acquired.
Data analysis: Electrophysiological recordings were analyzed using custom-written macros with IGORpro (WaveMetrics Inc. (Lake Oswego, OR, USA) and Axograph software (Axograph, RRID:SCR_014284). Action potentials (APs) were detected automatically by threshold crossing with Axograph (Axograph RRID:SCR_014284) followed by visual inspection. RM measurements were calculated from linear fits of the voltage responses to current step injections of 10 pA. Action potential firing properties were measured on a positive current step series starting at 20 pA with increasing 10 pA current steps. The threshold was measured at the first AP at rheobase. The AP full-width at half-maximal amplitude (AP width) and after-hyperpolarizing potential (AHP) amplitude were measured for the first AP at threshold. The latency of the first AP and number of APs was measured with increasing current steps over rheobase. The instantaneous firing frequency and amplitude of the first five APs were measured at current injection steps 1.5 times over rheobase. Sag potentials were measured with a protocol injecting a current as necessary to maintain a VM of −70 mV with a one-second-long negative current step resulting in a membrane hyperpolarization of −100 mV. The sag was calculated as the difference between the peak and steady-state voltage during the 1 s hyperpolarization.
4.8. Multi-Electrode Array (MEA) Recordings
All experiments were performed on adult (P75) female mice. The mice were sacrificed and the brain removed. Coronal slices (400 μm) of S1 cortex were cut at low speed (0.04 mm/s) and at a vibration frequency of 70 Hz in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) supplemented with sucrose (in mM: 87 NaCl, 2.5 KCl, 2.5 CaCl2, 7 MgCl2, 1 NaH2PO4, 25 NaHCO3, and 10 glucose, saturated with 95% O2 and 5% CO2). The slices were maintained at 32 °C in a storage chamber containing standard ACSF (in mM: 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2 and 5% CO2) for 20 min, and then stored for at least 1 h before recording in a pro-epileptic (0 mM Mg2+, 6 mM K+; 0Mg6K) ACSF. S1 cortical slices were transferred to planar MEA Petri dishes (200-30 ITO electrodes organized in a 12 × 12 matrix, with internal reference, 30 μm diameter and 200 μm inter-electrode distance; Multichannel Systems, Kusterdingen, Germany), and kept in place using a small platinum anchor. The slices were continuously perfused at a rate of 2 mL/min with 0Mg6K ACSF during the recordings. Images of the S1 cortical slices on MEAs were acquired with a video microscope table (MEA-VMT1; Multichannel Systems, Germany) through MEA Monitor software (Multichannel Systems, Kusterdingen, Germany) to identify the location of the electrodes on the cortex and to select the electrodes of interest. Data were sampled at 10 kHz and network activity was recorded at 32 °C by MEA2100-120 system (bandwidth 1–3000 Hz, gain 5×, Multichannel Systems, Kusterdingen, Germany) through the MC Rack 4.5.1 software (Multichannel Systems, Kusterdingen, Germany).
Multi-electrode (MEA) data analysis: Bursting raw data were analyzed with MC Rack (Multichannel Systems, Kusterdingen, Germany). The detection of bursts was performed using the “Spike Sorter” algorithm, which sets a threshold based on multiples of standard deviation of the noise (five-fold) calculated over the first 500 ms of recording free of electrical activity. A five-fold standard deviation threshold was used to automatically detect each event, which could be modified in real-time by the operator using a visual check if needed. Bursts were arbitrarily defined as discharges shorter than 5 s in duration. Typically, bursts were characterized by fast voltage oscillations followed by slow oscillations or negative shifts. To analyze seizure activity, the data were exported to Neuroexplorer (Nex Technologies, Colorado Springs, CO, USA). Paroxysmal events were identified as discharges lasting more than 5 s; successive paroxysmal discharges were considered separate events based on their waveform and on the presence of a minimum (>10 s) interval of silent or bursting activity between them [
44].
4.9. Behavior
Three independent cohorts were generated to produce a total of 15 to 20 mice per genotype (control and
Pten model) and per sex (males and females). Due to ΔNp73 expression in the epidermal and hair follicle stem cells [
45], the
Pten models start to develop skin accumulation under the paws and curly fur after 12 weeks that preclude later investigations for ethical considerations. Each cohort was submitted successively to an adapted SHIRPA test to clinically evaluate each animal at 8 weeks old, followed by the rearing activity (at 9 weeks old), the open field (at 10 weeks old), and the Y-Maze tests (at 11 weeks old) according to the International Mouse Phenotyping Resource of Standardised Screens (IMPReSS) recommendations (accessed on the 10 January 2020,
https://www.mousephenotype.org/impress/index). Finally, 12-weeks-old mice were processed for kainate injection to test the susceptibility to seizures before euthanasia.
4.9.1. Horizontal Activity
Spontaneous horizontal locomotor activity was assessed using the open field test (OFT). To do so, unacclimated mice were moved to the monitor area and immediately placed individually in the center of the motor activity boxes (40 cm L, 40 cm W, 40 cm H) placed on an infrared floor under ~50 lux of light measured in the center of the arena, for a 120 min period. The total distance traveled by the animal (horizontal activity) and the time spent in the center (25 cm × 25 cm) of the arena were recorded by an infrared camera system connected to the Videotrack v2.6 Automated Behavioural Analysis software (ViewPoint, Lyon, France). The results are indicated as distance traveled in centimeters.
4.9.2. Vertical Activity
Spontaneous locomotor activity was also evaluated with an infrared beam system fitted to rectangular cages (35 × 25cm) (ActiMot, TSE Systems GmbH, Berlin, Germany, version V5.1.9). Movement of the mice resulted in the interruption of the infrared beams. Each interruption was automatically counted, and the coordinates of the animal were determined in three dimensions (X–Y–Z axis). Each unacclimated animal was placed in the center of the cage and the number of beam breaks recorded during a 1-h period. We examined and analyzed the vertical (Z beam breaks) and horizontal (X + Y beam breaks) activities directly from the data collected by the system software.
4.9.3. Y-Maze
Short-term memory was assessed by spontaneous alternation behavior in the Y-maze task. Experiments were carried out using standard Y-maze apparatus made of three identical black plastic arms (60 cm arms; walls 10 cm) set at 120° angles from each other. Different visual cues (geometrical forms) were placed on the wall of two arms of the maze and were kept constant during the experiments. Mice were placed at the end of one arm of the Y-maze and the sequence of arm entries were recorded over 10 min. The introduction arm was randomly assigned for each animal. An arm visit was recorded when a mouse moved all four paws into the arm. An alternation was defined as consecutive entries into all three arms (e.g., A, B, C or A, C, B) and was automatically recorded using Videotrack v2.6 Automated Behavioural Analysis (ViewPoint, Lyon, France). The number of maximum alternations was the total number of arm entries minus two and the percentage of alternations was calculated as the ratio of actual to maximum alternations multiplied by 100: (actual alternations/maximum alternations) × 100. Persevering behavior was defined as subjects making significantly fewer alternations than would be expected by chance (50%).
4.9.4. Kainate Injection and Susceptibility to Seizures
Animals at 12 weeks old were injected with a single intraperitoneal dose of kainate (#0222/10, 10 mg, Bio-Techne (Minneapolis, MIN, USA) at 15 mg/kg and recorded for 2 h by Videotrack v2.6 Automated Behavioural Analysis (ViewPoint, Lyon, France) software. The latency, duration, and the number of seizures and their severity were recorded by two users blind to the animals.
4.10. Statistical Analyses
The results are reported as mean ± SD (histological and behavioral analyses) or SEM (slice and cell recording) in the figures and supplementary tables. The statistical significance of the linear variables was assessed using paired or unpaired Student’s t test, multiple t test with inequal variance, repeated-measures ANOVA followed by post hoc tests where appropriate, and a non-parametric Mann–Whitney U test when the distribution was not normal. Normality was tested with the Jarque–Bera test or Shapiro–Wilk test. Prism8 software (GraphPad, Boston, MA, USA) was used to test for outliers, normality, statistical differences, and for graphical representations of histograms and plots. Each test is detailed in the supplementary tables and in the figure legends.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}