
F1·Fo ATP Synthase/ATPase: Contemporary View on Unidirectional Catalysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
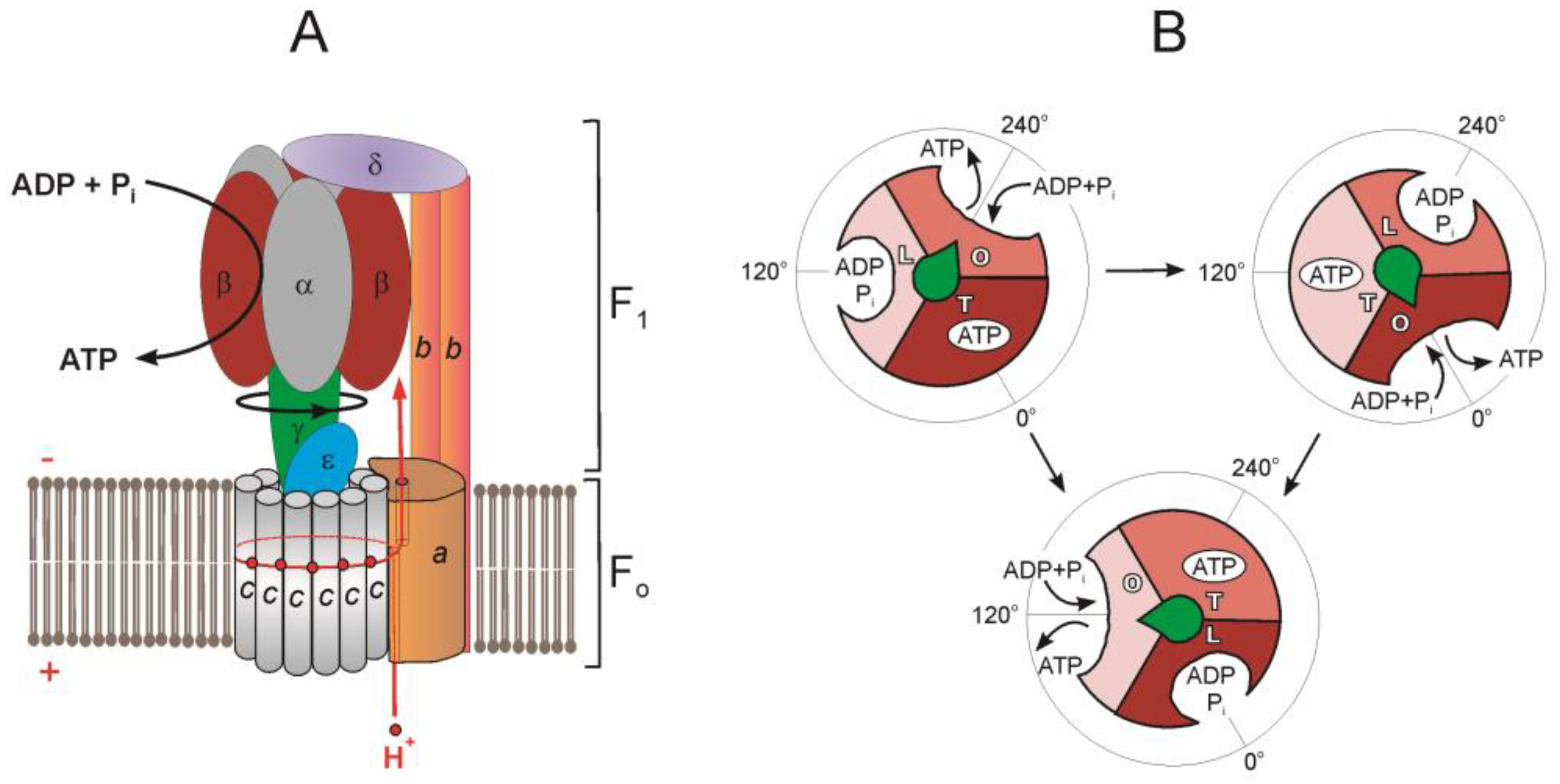
2. Common Subunit Composition and Function of F1·Fo
3. F1·Fo Rotary Catalysis
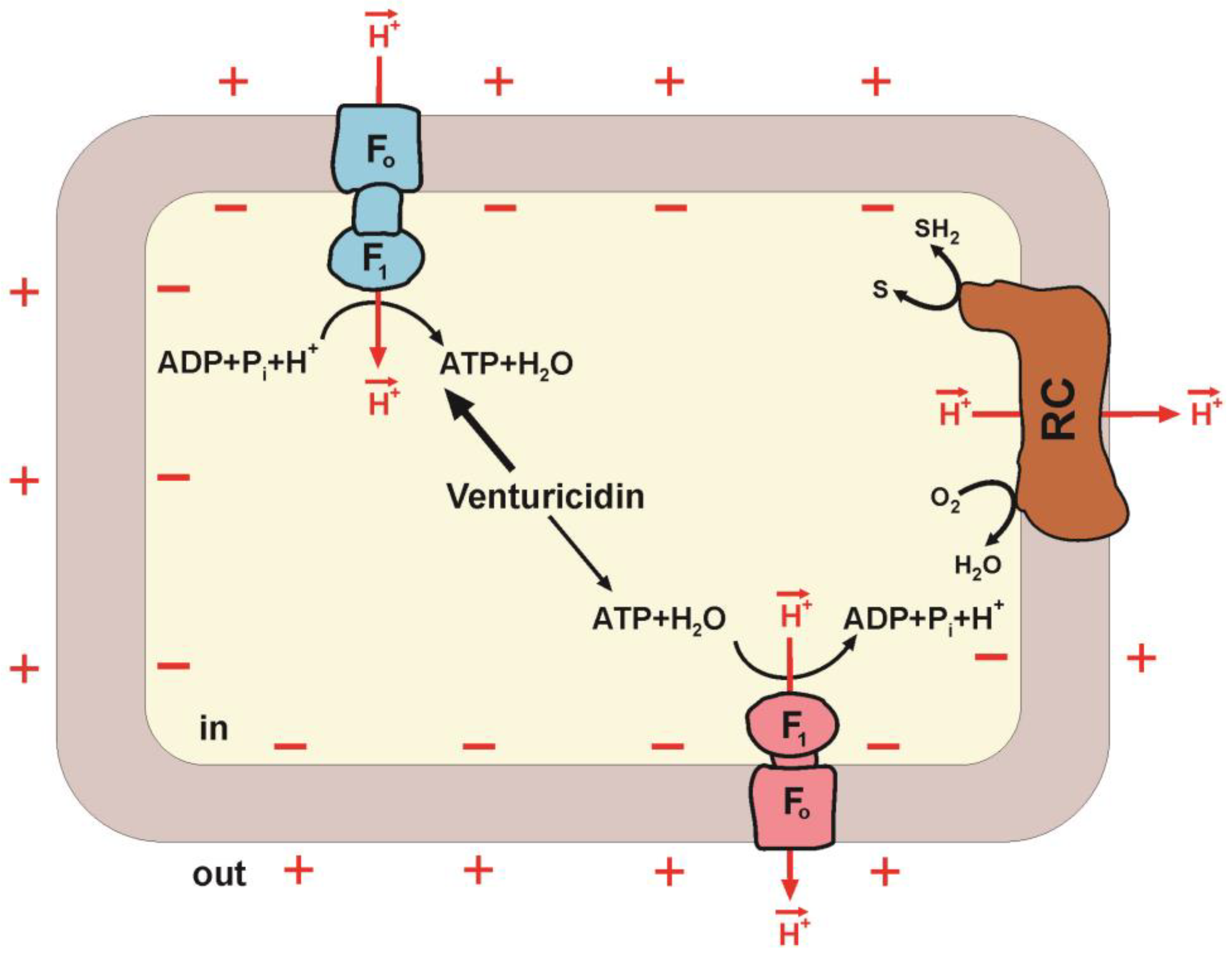
4. Reversibility of F1 Fo ATP Synthase Reaction and the Problem of Preventing Wasteful ATP Hydrolysis
4.1. ADP(Mg2+)-Inhibition
4.2. Natural Inhibitor Proteins
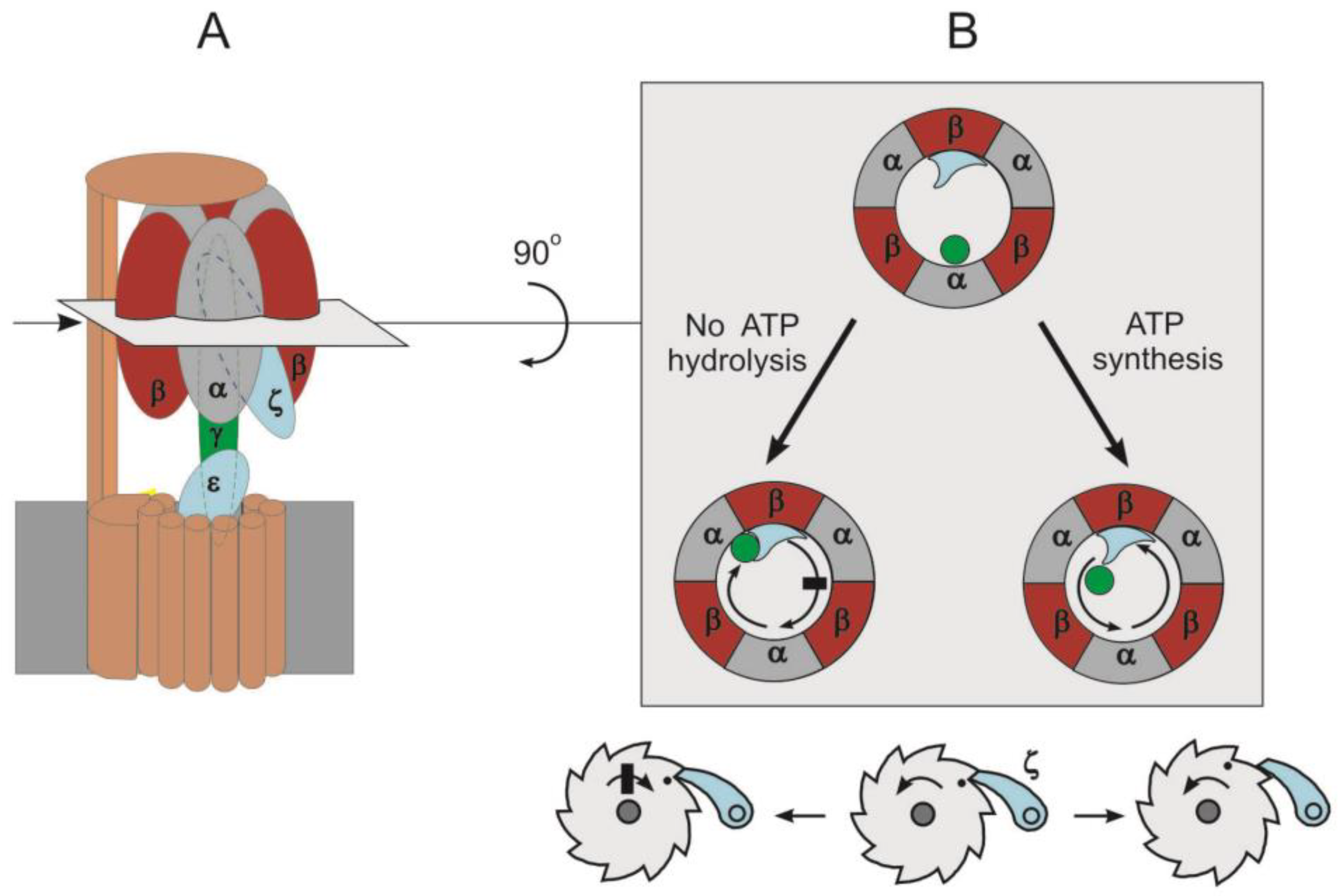
4.3. «Ratchet and Pawl» Mechanism of F1∙Fo
5. Paracoccus denitrificans as a Unidirectional F1∙Fo Model
Hypothesis of Two Forms of F1∙Fo: ATP Synthase and ATP Hydrolase
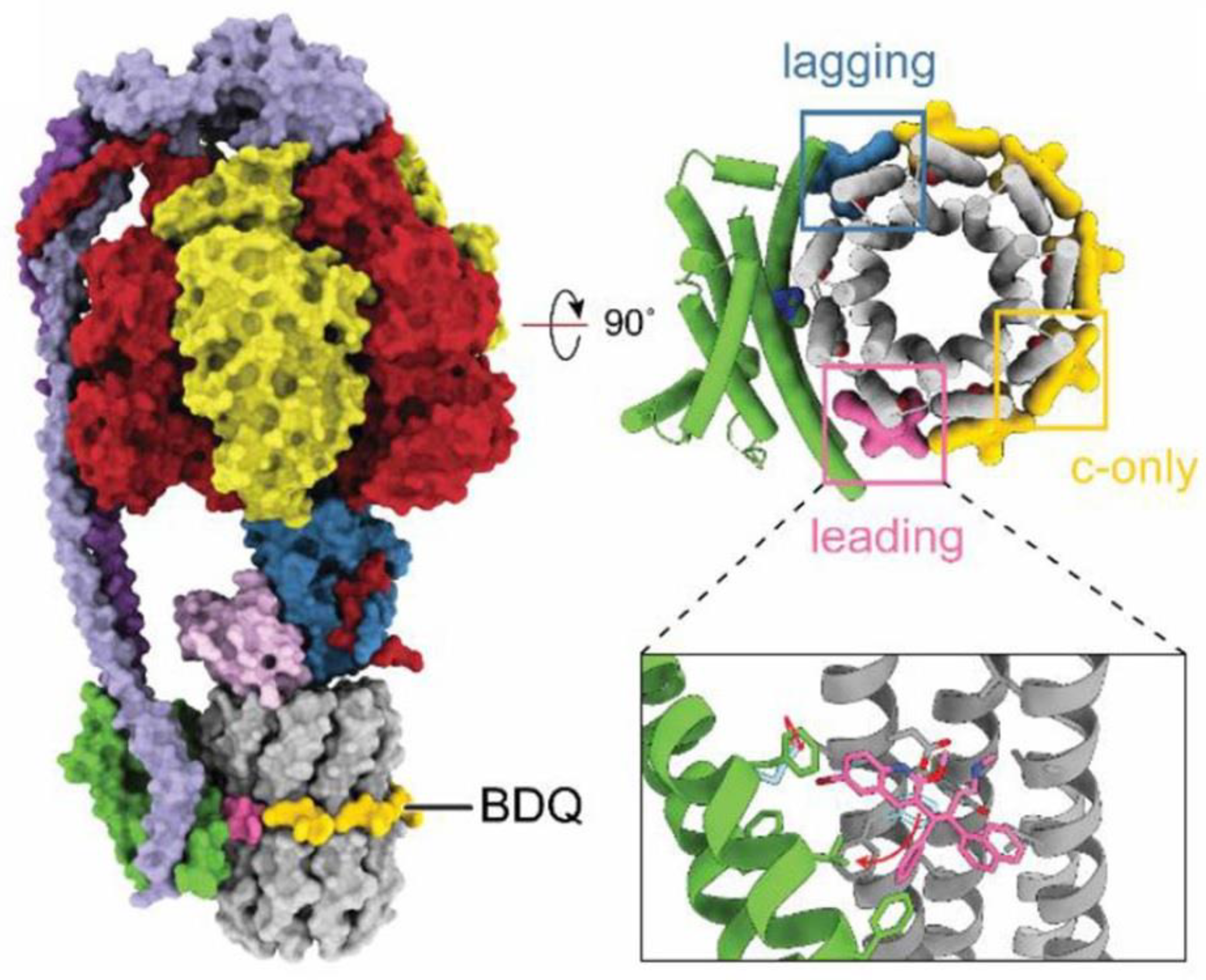
6. Mycobacterium tuberculosis F1∙Fo as a Promising Drug Target
Bedaquiline Is Effective in Curing Highly Drug-Resistant Tuberculosis via Targeting M. tuberculosis Fo∙F1
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Ríos, E.; Montgomery, M.G.; Leslie, A.G.; García-Trejo, J.J.; Walker, J.E. Structure of a catalytic dimer of the α- and β-subunits of the F-ATPase from Paracoccus denitrificans at 2.3Å resolution. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1309–1317. [Google Scholar] [CrossRef] [Green Version]
- Petri, J.; Nakatani, Y.; Montgomery, M.G.; Ferguson, S.A.; Aragão, D.; Leslie, A.G.W.; Heikal, A.; Walker, J.E.; Cook, G.M. Structure of F1-ATPase from the obligate anaerobe Fusobacterium nucleatum. Open Biol. 2019, 9, 190066. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Rubinstein, J.L. Cryo-EM of ATP synthases. Curr. Opin. Struct. Biol. 2018, 52, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.M.; Rubinstein, J.L. CryoEM Reveals the Complexity and Diversity of ATP Synthases. Front. Microbiol. 2022, 13, 864006. [Google Scholar] [CrossRef] [PubMed]
- Noji, H.; Ueno, H.; McMillan, D.G.G. Catalytic robustness and torque generation of the F1-ATPase. Biophys. Rev. 2017, 9, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Noji, H.; Ueno, H. How Does F1-ATPase Generate Torque?: Analysis From Cryo-Electron Microscopy and Rotational Catalysis of Thermophilic F1. Front. Microbiol. 2022, 13, 904084. [Google Scholar] [CrossRef]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Cook, G.M.; Greening, C.; Hards, K.; Berney, M. Energetics of pathogenic bacteria and opportunities for drug development. Adv. Microb. Physiol. 2014, 65, 1–62. [Google Scholar] [CrossRef]
- Narang, R.; Kumar, R.; Kalra, S.; Nayak, S.K.; Khatik, G.L.; Kumar, G.N.; Sudhaka, K.; Singh, S.K. Recent advancements in mechanistic studies and structure activity relationship of FoF1 ATP synthase inhibitor as antimicrobial agent. Eur. J. Med. Chem. 2019, 182, 111644. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.E.; Bertrand, B.P.; Heim, C.E.; Alqarzaee, A.A.; Chaudhari, S.S.; Aldrich, A.L.; Fey, P.D.; Thomas, V.C.; Kielian, T. Staphylococcus aureus ATP synthase promotes biofilm persistence by influencing innate immunity. MBio 2020, 11, e01581-20. [Google Scholar] [CrossRef] [PubMed]
- Demmer, J.K.; Phillips, B.P.; Uhrig, O.L.; Filloux, A.; Allsopp, L.P.; Bublitz, M.; Meier, T. Structure of ATP synthase from ESKAPE pathogen Acinetobacter baumannii. Sci. Adv. 2022, 8, eabl5966. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Ragunathan, P.; Shin, J.; Saw, W.-G.; Wong, C.-F.; Dick, T.; Grüber, G. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: Implications for drug design. Prog. Biophys. Mol. Biol. 2020, 152, 64–73. [Google Scholar] [CrossRef]
- Guo, H.; Suzuki, T.; Rubinstein, J.L. Structure of a bacterial ATP synthase. eLife 2019, 8, e43128. [Google Scholar] [CrossRef]
- Morales-Ríos, E.; de la Rosa-Morales, F.; Mendoza-Hernández, G.; Rodríguez-Zavala, J.S.; Celis, H.; Zarco-Zavala, M.; García-Trejo, J.J. A novel 11-kDa inhibitory subunit in the F1FO ATP synthase of Paracoccus denitrificans and related alpha-proteobacteria. FASEB J. 2010, 24, 599–608. [Google Scholar] [CrossRef]
- Zarco-Zavala, M.; Morales-Ríos, E.; Mendoza-Hernández, G.; Ramírez-Silva, L.; Pérez-Hernández, G.; García-Trejo, J.J. The ζ subunit of the F1FO-ATP synthase of α-proteobacteria controls rotation of the nanomotor with a different structure. FASEB J. 2014, 28, 2146–2157. [Google Scholar] [CrossRef]
- Hahn, A.; Vonck, J.; Mills, D.J.; Meier, T.; Kühlbrandt, W. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 2018, 360, eaat4318. [Google Scholar] [CrossRef] [Green Version]
- Fillingame, R.H.; Steed, P.R. Half channels mediating H(+) transport and the mechanism of gating in the Fo sector of Escherichia coli F1Fo ATP synthase. Biochim. Biophys. Acta 2014, 1837, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, C.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat. Struct. Biol. 2000, 7, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Sobti, M.; Smits, C.; Wong, A.S.; Ishmukhametov, R.; Stock, D.; Sandin, S.; Stewart, A.G. Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. eLife 2016, 5, e21598. [Google Scholar] [CrossRef]
- Morales-Ríos, E.; Watt, I.N.; Zhang, Q.; Ding, S.; Fearnley, I.M.; Montgomery, M.G.; Wakelam, M.J.O.; Walker, J.E. Purification, characterization and crystallization of the F-ATPase from Paracoccus denitrificans. Open Biol. 2015, 5, 150119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobti, M.; Ishmukhametov, R.; Bouwer, J.C.; Ayer, A.; Suarna, C.; Smith, N.J.; Christie, M.; Stocker, R.; Duncan, T.M.; Stewart, A.G. Cryo-EM reveals distinct conformations of E. coli ATP synthase on exposure to ATP. eLife 2019, 8, e43864. [Google Scholar] [CrossRef]
- Sobti, M.; Walshe, J.L.; Wu, D.; Ishmukhametov, R.; Zeng, Y.C.; Robinson, C.V.; Berry, R.M.; Stewart, A.G. Cryo-EM structures provide insight into how E. coli F1Fo ATP synthase accommodates symmetry mismatch. Nat. Commun. 2020, 11, 2615. [Google Scholar] [CrossRef]
- Sobti, M.; Zeng, Y.C.; Walshe, J.L.; Brown, S.H.J.; Ishmukhametov, R.; Stewart, A.G. Changes within the central stalk of E. coli F1Fo ATP synthase observed after addition of ATP. Commun. Biol. 2023, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gómez, J.D.; Liao, M.; Mueller, D.M. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 2018, 360, eaas9699. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.J.; Klusch, N.; Langer, J.D.; Mills, D.J.; Yildiz, Ö.; Kühlbrandt, W. Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-Fo coupling. Science 2019, 364, eaaw9128. [Google Scholar] [CrossRef]
- Junge, W.; Hendrik, S.; Engelbrecht, S. Torque generation and elastic power transmission in the rotary F(O)F(1)-ATPase. Nature 2009, 459, 364–370. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sobti, M.; Ueno, H.; Noji, H.; Stewart, A.G. The six steps of the complete F1-ATPase rotary catalytic cycle. Nat. Commun. 2021, 12, 4690. [Google Scholar] [CrossRef] [PubMed]
- Zarco-Zavala, M.; Watanabe, R.; McMillan, D.G.G.; Suzuki, T.; Ueno, H.; Mendoza-Hoffmann, F.; García-Trejo, J.J.; Noji, H. The 3 × 120° rotary mechanism of Paracoccus denitrificans F1-ATPase is different from that of the bacterial and mitochondrial F1-ATPases. Proc. Natl. Acad. Sci. USA 2020, 117, 29647–29657. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanaka, K.; Wakabayashi, C.; Saita, E.; Yoshida, M. Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat. Chem. Biol. 2014, 10, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Rondelez, Y.; Tresset, G.; Nakashima, T.; Kato-Yamada, Y.; Fujita, H.; Takeuchi, S.; Noji, H. Highly coupled ATP synthesis by F1-ATPase single molecules. Nature 2005, 433, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.D. Steady-state and pre-steady-state kinetics of the mitochondrial F(1)F(o) ATPase: Is ATP synthase a reversible molecular machine? J. Exp. Biol. 2000, 203, 41–49. [Google Scholar] [CrossRef]
- Lapashina, A.S.; Feniouk, B.A. ADP-Inhibition of H+-FOF1-ATP Synthase. Biochemistry 2018, 83, 1141–1160. [Google Scholar] [CrossRef]
- Murakami, S.; Kondo, K.; Katayama, S.; Hara, S.; Sunamura, E.I.; Yamashita, E.; Groth, G.; Hisabori, T. Structure of the γ-ε complex of cyanobacterial F1-ATPase reveals a suppression mechanism of the γ subunit on ATP hydrolysis in phototrophs. Biochem J. 2018, 475, 2925–2939. [Google Scholar] [CrossRef]
- Pullman, M.E.; Monroy, G.C. A naturally occurring inhibitor of mitochondrial adenosine triphosphatase. J. Biol. Chem. 1963, 238, 3762–3769. [Google Scholar] [CrossRef]
- Bason, J.V.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F1-ATPase. Proc. Natl. Acad. Sci. USA 2014, 111, 11305–11310. [Google Scholar] [CrossRef] [Green Version]
- Junge, W.; Nelson, N. ATP synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Williams, D.; Kandiah, E.; Fromme, P.; Chiu, P.L. Structural basis of redox modulation on chloroplast ATP synthase. Commun. Biol. 2020, 3, 482. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, G.; Duncan, T.M. Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 2011, 18, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Trejo, J.J.; Zarco-Zavala, M.; Mendoza-Hoffmann, F.; Hernández-Luna, E.; Ortega, R.; Mendoza-Hernández, G. The Inhibitory Mechanism of the ζ Subunit of the F1FO-ATPase Nanomotor of Paracoccus denitrificans and Related α-Proteobacteria. J. Biol. Chem. 2016, 291, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Varghese, F.; Blaza, J.N.; Jones, A.J.Y.; Jarman, O.D.; Hirst, J. Deleting the IF1-like zeta subunit from Paracoccus denitrificans ATP synthase is not sufficient to activate ATP hydrolysis. Open. Biol. 2018, 8, 170206. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.D. New Perspective on the Reversibility of ATP Synthesis and Hydrolysis by Fo∙F1-ATP Synthase (Hydrolase). Biochemistry 2019, 84, 1247–1255. [Google Scholar] [CrossRef]
- Hyndman, D.J.; Milgrom, Y.M.; Bramhall, E.A.; Cross, R.L. Nucleotide-binding sites on Escherichia coli F1-ATPase. Specificity of noncatalytic sites and inhibition at catalytic sites by MgADP. J. Biol. Chem. 1994, 269, 28871–28877. [Google Scholar] [CrossRef] [PubMed]
- Jault, J.M.; Matsui, T.; Jault, F.M.; Kaibara, C.; Muneyuki, E.; Yoshida, M.; Kagawa, Y.; Allison, W.S. The alpha 3 beta 3 gamma complex of the F1-ATPase from thermophilic Bacillus PS3 containing the alpha D261N substitution fails to dissociate inhibitory MgADP from a catalytic site when ATP binds to noncatalytic sites. Biochemistry 1995, 34, 16412–16418. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Moises, F.; García, J.J.; Rodríguez-Zavala, J.S.; Moreno-Sánchez, R. Sulfite and membrane energization induce two different active states of the Paracoccus denitrificans F0F1-ATPase. Eur. J. Biochem. 2000, 267, 993–1000. [Google Scholar] [CrossRef]
- Milgrom, Y.M.; Duncan, T.M. F-ATP-ase of Escherichia coli membranes: The ubiquitous MgADP-inhibited state and the inhibited state induced by the ε-subunit’s C-terminal domain are mutually exclusive. Biochim. Biophys. Acta Bioenerg. 2020, 186, 148189. [Google Scholar] [CrossRef]
- Bowler, M.W.; Montgomery, M.G.; Leslie, A.; Walker, J.E. How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. USA 2006, 103, 8646–8649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkin, M.A.; Vinogradov, A.D. Energy-dependent transformation of the catalytic activities of the mitochondrial F0∙F1-ATP synthase. FEBS Lett. 1999, 448, 123–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, S.; Graber, P.; Turina, P. The activity of the ATP synthase from Escherichia coli is regulated by the transmembrane proton motive force. J. Biol. Chem. 2000, 275, 30157–30162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zharova, T.V.; Vinogradov, A.D. Energy-dependent transformation of F0.F1-ATPase in Paracoccus denitrificans plasma membranes. J. Biol. Chem. 2004, 279, 12319–12324. [Google Scholar] [CrossRef] [Green Version]
- Feniouk, B.A.; Suzuki, T.; Yoshida, M. Regulatory interplay between proton motive force, ADP, phosphate, and subunit epsilon in bacterial ATP synthase. J. Biol. Chem. 2007, 282, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.; Muneyuki, E.; Allison, W.S. The characteristics of the (alpha V371C)3(beta R337C)3 gamma double mutant subcomplex of the TF1-ATPase indicate that the catalytic site at the alpha TP-beta TP interface with bound MgADP in crystal structures of MF1 represents a catalytic site containing inhibitory MgADP. Biochemistry 2005, 44, 2441–2448. [Google Scholar] [CrossRef]
- Hirono-Hara, Y.; Ishizuka, K.; Kinosita, K., Jr.; Yoshida, M.; Noji, H. Activation of pausing F1 motor by external force. Proc. Natl. Acad. Sci. USA 2005, 102, 4288–4293. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Kato-Yamada, Y. Severe MgADP inhibition of Bacillus subtilis F1-ATPase is not due to the absence of nucleotide binding to the noncatalytic nucleotide binding sites. PLoS ONE 2014, 9, e107197. [Google Scholar] [CrossRef] [Green Version]
- Mizumoto, J.; Kikuchi, Y.; Nakanishi, Y.H.; Mouri, N.; Cai, A.; Ohta, T.; Haruyama, T.; Kato-Yamada, Y. ε subunit of Bacillus subtilis F1-ATPase relieves MgADP inhibition. PLoS ONE 2013, 8, e73888. [Google Scholar] [CrossRef] [Green Version]
- Krah, A.; Zarco-Zaval, M.; McMillan, D.G.G. Insights into the regulatory function of the epsilon subunit from bacterial F-type ATP synthases: A comparison of structural, biochemical and biophysical data. Open Biol. 2018, 8, 170275. [Google Scholar] [CrossRef] [Green Version]
- Lucero, R.A.; Mercedes, E.P.; Thorsten, L.; Giovanni, G.C.; Michael, F.; Guadalupe, Z.; Pablo, P.J.; Federico, M.; Oscar, F.H. Deletion of the natural inhibitory protein Inh1 in Ustilagomaydis has no effect on the dimeric state of the F1FO-ATP synthase but increases the ATPase activity and reduces the stability. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148429. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Hoffmann, F.; Pérez-Oseguera, Á.; Cevallos, M.Á.; Zarco-Zavala, M.; Ortega, R.; Peña-Segura, C.; Espinoza-Simón, E.; Uribe-Carvajal, S.; García-Trejo, J.J. The biological role of the ζ subunit as uni-directional inhibitor of the F1FO-ATPase of Paracoccus denitrificans. Cell. Rep. 2018, 22, 1067–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krah, A.; Kato-Yamada, Y.; Takada, S. The structural basis of a high affinity ATP binding ε subunit from a bacterial ATP synthase. PLoS ONE 2017, 12, e0177907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akanuma, G.; Tagana, T.; Sawada, M.; Suzuki, S.; Shimada, T.; Tanaka, K.; Kawamura, F.; Kato-Yamada, Y. C-terminal regulatory domain of the ε subunit of FoF1 ATP synthase enhances the ATP-dependent H+ pumping that is involved in the maintenance of cellular membrane potential in Bacillus subtilis. Microbiol. Open 2019, 8, e00815. [Google Scholar] [CrossRef] [Green Version]
- Kato-Yamada, Y.; Yoshida, M. Isolated epsilon subunit of thermophilic F1-ATPase binds ATP. J. Biol. Chem. 2003, 278, 36013–36016. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Kajiwara, N.; Tanaka, H.; Tsukihara, T.; Kato-Yamada, Y.; Yoshida, M.; Akutsu, H. Structures of the thermophilic F1-ATPase ε subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1. Proc. Natl. Acad. Sci. USA 2007, 104, 11233–11238. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.A.; Cook, G.M.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. Regulation of the thermoalkaliphilic F1-ATPase from Caldalkalibacillus thermarum. Proc. Natl. Acad. Sci. USA 2016, 113, 10860–10865. [Google Scholar] [CrossRef] [Green Version]
- Sielaff, H.; Duncan, T.M.; Börsch, M. The regulatory subunit ε in Escherichia coli FOF1-ATP synthase. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 775–788. [Google Scholar] [CrossRef]
- Zhang, A.T.; Montgomery, M.G.; Leslie, A.G.W.; Cook, G.M.; Walker, J.E. The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs. Proc. Natl. Acad. Sci. USA 2019, 116, 4206–4211. [Google Scholar] [CrossRef] [Green Version]
- Biukovic, G.; Basak, S.; Manimekalai, M.S.S.; Rishikesan, S.; Roessle, M.; Dick, T.; Rao, S.P.S.; Hunke, C.; Grüber, G. Variations of subunit epsilon of the Mycobacterium tuberculosis F1Fo ATP synthase and a novel model for mechanism of action of the tuberculosis drug TMC207. Antimicrob. Agents Chemother. 2013, 57, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, R.; Mori, S.; Ueno, H.; Noji, H. Kinetic analysis of the inhibition mechanism of bovine mitochondrial F1-ATPase inhibitory protein using biochemical assay. J. Biochem. 2021, 170, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Hoffmann, F.; Zarco-Zavala, M.; Ortega, R.; García-Trejo, J.J. Control of rotation of the F1FO-ATP synthase nanomotor by an inhibitory α-helix from unfolded ε or intrinsically disordered ζ and IF1 proteins. J. Bioenerg. Biomembr. 2018, 50, 403–424. [Google Scholar] [CrossRef]
- Tsunoda, S.P.; Rodgers, A.J.; Aggeler, R.; Wilce, M.C.; Yoshida, M.; Capaldi, R.A. Large conformational changes of the epsilon subunit in the bacterial F1F0 ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 6560–6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.J. Paracoccus denitrificans Oxidative Phosphorylation: Retentions, Gains, Losses, and Lessons En Route to Mitochondria. IUBMB Life 2018, 70, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Hoffmann, F.; Zarco-Zavala, M.; Ortega, R.; Celis-Sandoval, H.; Torres-Larios, A.; García-Trejo, J.J. Evolution of the Inhibitory and Non-Inhibitory ε, ζ, and IF1 Subunits of the F1FO-ATPase as Related to the Endosymbiotic Origin of Mitochondria. Microorganisms 2022, 10, 1372. [Google Scholar] [CrossRef]
- Zarco-Zavala, M.; Mendoza-Hoffmann, F.; García-Trejo, J.J. Unidirectional regulation of the F1FO-ATP synthase nanomotor by the ζ pawl-ratchet inhibitor protein of Paracoccus denitrificans and related αproteobacteria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Astudillo, H.; Zarco-Zavala, M.; García-Trejo, J.J.; González-Halphen, D. Regulation of bacterial ATP synthase activity: A gear-shifting or a pawl-ratchet mechanism? FEBS J. 2021, 288, 3159–3163. [Google Scholar] [CrossRef]
- Cook, G.M.; Keis, S.; Morgan, H.W.; von Ballmoos, C.; Matthey, U.; Kaim, G.; Dimroth, P. Purification and biochemical characterization of the F1Fo-ATP synthase from thermoalkaliphilic Bacillus sp. strain TA2.A1. J. Bacteriol. 2003, 185, 4442–4449. [Google Scholar] [CrossRef] [Green Version]
- Haagsma, A.C.; Driessen, N.N.; Hahn, M.M.; Lill, H.; Bald, D. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 2010, 313, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Hotra, A.; Suter, M.; Biukovic, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Gruber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase γ subunit sheds light on its inhibitory role in ATP hydrolysis driven H+ pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [Green Version]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biukovic, G.; Manimekalai, M.S.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit α of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, J.A.; Ferguson, S.J. Kinetics of oxidative phosphorylation in Paracoccus denitrificans. 1. Mechanism of ATP synthesis at the active site(s) of F0F1-ATPase. Biochemistry 1990, 29, 10503–10518. [Google Scholar] [CrossRef]
- Fujiwara, M.; Kato-Yamada, Y. ATP-binding affinity of the ε subunit of thermophilic F1-ATPase under label-free conditions. Biochem. Biophys. Rep. 2020, 21, 100725. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014, 4, 6522. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Beahm, D.R.; Ionov, S.; Sarpeshkar, R. Measuring and modeling energy and power consumption in living microbial cells with a synthetic ATP reporter. BMC Biol. 2021, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Suzuki, T.; Berney, M.; Yoshida, M.; Cook, G.M. The regulatory C-terminal domain of subunit ε of F₀F₁ ATP synthase is dispensable for growth and survival of Escherichia coli. J. Bacteriol. 2011, 193, 2046–2052. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.B.; Duncan, T.M. Aerobic Growth of Escherichia coli Is Reduced, and ATP Synthesis Is Selectively Inhibited when Five C-terminal Residues Are Deleted from the ϵ Subunit of ATP Synthase. J. Biol. Chem. 2015, 290, 21032–21041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iino, R.; Hasegawa, R.; Tabata, K.V.; Noji, H. Mechanism of inhibition by C-terminal alpha-helices of the epsilon subunit of Escherichia coli FoF1-ATP synthase. J. Biol. Chem. 2009, 284, 17457–17464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Ernster, L. Inactive to active transitions of the mitochondrial ATPase complex as controlled by the ATPase inhibitor. Biochim. Biophys. Acta 1979, 547, 252–257. [Google Scholar] [CrossRef]
- Syroeshkin, A.V.; Vasilyeva, E.A.; Vinogradov, A.D. ATP synthesis catalyzed by the mitochondrial F1-F0 ATP synthase is not a reversal of its ATPase activity. FEBS Lett. 1995, 366, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Jarman, O.D.; Biner, O.; Hirst, J. Regulation of ATP hydrolysis by the ε subunit, ζ subunit and Mg-ADP in the ATP synthase of Paracoccus denitrificans. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148355. [Google Scholar] [CrossRef]
- Galkina, K.V.; Zubareva, V.M.; Kashko, N.D.; Lapashina, A.S.; Markova, O.V.; Feniouk, B.A.; Knorre, D.A. Heterogeneity of Starved Yeast Cells in IF1 Levels Suggests the Role of This Protein in vivo. Front. Microbiol. 2022, 13, 816622. [Google Scholar] [CrossRef]
- Saita, E.; Iino, R.; Suzuki, T.; Feniouk, B.A.; Kinosita, K., Jr.; Yoshida, M. Activation and stiffness of the inhibited states of F1-ATPase probed by single-molecule manipulation. J. Biol. Chem. 2010, 285, 11411–11417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Murakami, T.; Iino, R.; Suzuki, J.; Ono, S.; Shirakihara, Y.; Yoshida, M. F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of epsilon subunit in response to proton motive force and ADP/ATP balance. J. Biol. Chem. 2003, 278, 46840–46846. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yu, J.; Wang, M.; Zeng, Q.; Fu, X.; Chang, Z. A high-throughput genetically directed protein crosslinking analysis reveals the physiological relevance of the ATP synthase ‘inserted’ state. FEBS J. 2021, 288, 2989–3009. [Google Scholar] [CrossRef] [PubMed]
- John, P.; Whatley, F.R. Oxidative phosphorylation coupled to oxygen uptake and nitrate reduction in Micrococcus denitrificans. Biochim. Biophys. Acta 1970, 216, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Covian, R.; Edwards, L.; He, Y.; Kim, G.; Houghton, C.; Levine, R.L.; Balaban, R.S. Energy homeostasis is a conserved process: Evidence from Paracoccus denitrificans’ response to acute changes in energy demand. PLoS ONE 2021, 16, e0259636. [Google Scholar] [CrossRef]
- Zharova, T.V.; Vinogradov, A.D. ATPase/synthase activity of Paracoccus denitrificans Fo·F1 as related to the respiratory control phenomenon. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1322–1329. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.Q.; Yang, W.; Karplus, M. A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase. Cell 2005, 23, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Zharova, T.V.; Vinogradov, A.D. Functional heterogeneity of Fo·F1H+-ATPase/synthase in coupled Paracoccus denitrificans plasma membranes. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 939–944. [Google Scholar] [CrossRef]
- Johnson, K.M.; Swenson, L.; Opipari, A.W.; Reuter, R.; Zarrabi, N.; Fierke, C.A.; Börsch, M.; Glick, G.D. Mechanistic basis for differential inhibition of the F1 Fo-ATPase by aurovertin. Biopolymers 2009, 91, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco-Moises, F.; Minauro-Sanmiguel, F.; Bravo, C.; García, J.J. Sulfite inhibits the F1F0-ATP synthase and activates the F1F0-ATPase of Paracoccus denitrificans. J. Bioenerg. Biomembr. 2002, 34, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Zharova, T.V.; Kozlovsky, V.S.; Grivennikova, V.G. Interaction of Venturicidin and Fo·F1-ATPase/ATP Synthase of Tightly Coupled Subbacterial Particles of Paracoccus denitrificans in Energized Membranes. Biochemistry 2022, 87, 742–751. [Google Scholar] [CrossRef]
- Milgrom, Y.M.; Duncan, T.M. Complex effects of macrolide venturicidins on bacterial F-ATPases likely contribute to their action as antibiotic adjuvants. Sci. Rep. 2021, 11, 13631. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, M.; Turina, P.; Melandri, B.A.; Dunn, S.D. Modulation of coupling in the Escherichia coli ATP synthase by ADP and Pi: Role of the ε subunit C-terminal domain. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 34–44. [Google Scholar] [CrossRef]
- Turina, P. Modulation of the H+/ ATP coupling ratio by ADP and ATP as a possible regulatory feature in the F-type ATP synthases. Front. Mol. Biosci. 2022, 9, 1023031. [Google Scholar] [CrossRef]
- Vestergaard, M.; Dirk, B.; Ingmer, H. Targeting the ATP synthase in bacterial and fungal pathogens: Beyond Mycobacterium tuberculosis. J. Glob. Antimicrob. Resist. 2022, 29, 29–41. [Google Scholar] [CrossRef]
- Cofas-Vargas, L.F.; Mendoza-Espinosa, P.; Avila-Barrientos, L.P.; Prada-Gracia, D.; Riveros-Rosas, H.; García-Hernández, E. Exploring the druggability of the binding site of aurovertin, an exogenous allosteric inhibitor of FOF1-ATP synthase. Front. Pharmacol. 2022, 13, 1012008. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Roshanak, S.; Ingmer, H. Targeting the ATP synthase in Staphylococcus aureus small colony Variants, Streptococcus pyogenes and pathogenic fungi. Antibiotics 2021, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Hards, K.; Cook, G.M. Targeting bacterial energetics to produce new antimicrobials. Drug. Resist. Updat. 2018, 36, 1–12. [Google Scholar] [CrossRef]
- Saw, W.G.; Wu, M.L.; Ragunathan, P.; Biukovic, G.; Lau, A.M.; Shin, J.; Harikishore, A.; Cheung, C.Y.; Hards, K.; Sarathy, J.P.; et al. Disrupting coupling within mycobacterial F-ATP synthases subunit ϵ causes dysregulated energy production and cell wall biosynthesis. Sci. Rep. 2019, 9, 16759. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Akhter, Y. A review on enzyme complexes of electron transport chain from Mycobacterium tuberculosis as promising drug targets. Int. J. Biol. Macromol. 2022, 212, 474–494. [Google Scholar] [CrossRef]
- McNeil, M.B.; Cheung, C.Y.; Waller, N.J.E.; Adolph, C.; Chapman, C.L.; Seeto, N.E.J.; Jowsey, W.; Li, Z.; Hameed, H.M.A.; Zhang, T.; et al. Uncovering interactions between mycobacterial respiratory complexes to target drug-resistant Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 2022, 12, 980844. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Lamprecht, D.A.; Asmal, R.; Adamson, J.H.; Borah, K.; Beste, D.J.V.; Lee, B.S.; Pethe, K.; Rousseau, S.; Krieger, I.; et al. Bedaquiline reprograms central metabolism to reveal glycolytic vulnerability in Mycobacterium tuberculosis. Nat. Commun. 2020, 11, 6092. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.G.; Wu, S.Q.; He, J.Q. Efficacy of bedaquiline in the treatment of drug-resistant tuberculosis: A systematic review and meta-analysis. BMC Infect. Dis. 2021, 21, 970. [Google Scholar] [CrossRef]
- Deshkar, A.T.; Shirure, P.A. Bedaquiline: A novel diarylquinoline for multidrug-resistant pulmonary tuberculosis. Cureus 2022, 14, e28519. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.A. Pharmacodynamics and bactericidal activity of bedaquiline in pulmonary tuberculosis. Antimicrob. Agents Chemother. 2022, 66, e0163621. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Preiss, L.; Langer, J.D.; Yildiz, O.; Eckhardt-Strelau, L.; Guillemont, J.E.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [Green Version]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Gohlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [Green Version]
- Sarathy, J.P.; Ragunathan, P.; Cooper, C.B.; Upton, A.M.; Gruber, G.; Dick, T. TBAJ-876 displays bedaquiline-like mycobactericidal potency without retaining the parental drug’s uncoupler activity. Antimicrob. Agents Chemother. 2020, 64, e01540-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamariah, N.; Huber, R.G.; Nartey, W.; Bhushan, S.; Bond, P.J.; Gruber, G. Structure and subunit arrangement of Mycobacterial F1FO ATP synthase and novel features of the unique mycobacterial subunit delta. J. Struct. Biol. 2019, 207, 199–208. [Google Scholar] [CrossRef]
- Guo, H.; Courbon, G.M.; Bueler, S.A.; Mai, J.; Liu, J.; Rubinstein, J.L. Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature 2021, 589, 143–147. [Google Scholar] [CrossRef]
- Montgomery, M.G.; Petri, J.; Spikes, T.E.; Walker, J.E. Structure of the ATP synthase from Mycobacterium smegmatis provides targets for treating tuberculosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2111899118. [Google Scholar] [CrossRef]
- Kubo, S.; Niina, T.; Takada, S. Molecular dynamics simulation of proton-transfer coupled rotations in ATP synthase FO motor. Sci. Rep. 2020, 10, 8225. [Google Scholar] [CrossRef] [PubMed]
- Krah, A.; Gruber, G.; Bond, P.J. Binding properties of the anti-TB drugs bedaquiline and TBAJ-876 to a mycobacterial F-ATP synthase. Curr. Res. Struct. Biol. 2022, 4, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-F.; Grüber, G. The unique C-terminal extension of mycobacterial F-ATP synthase subunit α is the major contributor to its latent ATP hydrolysis activity. Antimicrob. Agents Chemother. 2020, 64, e01568. [Google Scholar] [CrossRef]
- Wong, C.F.; Lau, A.M.; Harikishore, A.; Saw, W.G.; Shin, J.; Ragunathan, P.; Bhushan, S.; Ngan, S.C.; Sze, S.K.; Bates, R.W.; et al. A systematic assessment of mycobacterial F1-ATPase subunit ε’s role in latent ATPase hydrolysis. FEBS J. 2021, 288, 818–836. [Google Scholar] [CrossRef]
- Hards, K.; McMillan, D.G.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, T.; Takaki, A.; Aono, A.; Mitarai, S.; Okumura, M.; Ohta, K.; Kato, S. Multidrug resistant tuberculosis with simultaneously acquired drug resistance to bedaquiline and delamanid. Clin. Infect. Dis. 2021, 73, 2329–2331. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhou, W.; Patel, H.; Srivastava, A.P.; Symersky, J.; Bonar, M.M.; Faraldo-Gomez, J.D.; Liao, M.; Mueller, D.M. Bedaquiline inhibits the yeast and human mitochondrial ATP synthases. Commun. Biol. 2020, 3, 452. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Abdillahi-Ibrahim, R.; Wagner, M.J.; Krab, K.; Vergauwen, K.; Guillemont, J.; Andries, K.; Lill, H.; Koul, A.; Bald, D. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.U.; Denny, W.A.; et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorg. Med. Chem. 2018, 26, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.S.; Lu, G.L.; Tong, A.S.T.; Conole, D.; Franzblau, S.G.; Upton, A.M.; Lotlikar, M.U.; Cooper, C.B.; Palmer, B.D.; Choi, P.J.; et al. Synthesis and structure-activity relationships for a new class of tetrahydronaphthalene amide inhibitors of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2022, 229, 114059. [Google Scholar] [CrossRef]
- Yarlagadda, V.; Medina, R.; Wright, G.D. Venturicidin A, A Membrane-active Natural Product Inhibitor of ATP synthase Potentiates Aminoglycoside Antibiotics. Sci. Rep. 2020, 10, 8134. [Google Scholar] [CrossRef]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020, 152, 45–54. [Google Scholar] [CrossRef]
- Mascolo, L.; Bald, D. Cytochrome bd in Mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol. 2020, 152, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Siletsky, S.A.; Paiardini, A.; Hoogewijs, D.; Forte, E.; Giuffre, A.; Poole, R.K. Bacterial oxidases of the cytochrome bd family: Redox enzymes of unique structure, function and utility as drug targets. Antioxid. Redox Signal. 2021, 34, 1280–1318. [Google Scholar] [CrossRef]
- Friedrich, T.; Wohlwend, D.; Borisov, V.B. Recent advances in structural studies of cytochrome bd and its potential application as a drug target. Int. J. Mol. Sci. 2022, 23, 3166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shao, M.; Wang, W.; Cheung, C.Y.; Wu, Y.; Yu, H.; Hu, X.; Cook, G.M.; Gong, H.; Lu, X. Discovery of 1-hydroxy-2-methylquinolin-4(1H)-one derivatives as new cytochrome bd oxidase inhibitors for tuberculosis therapy. Eur. J. Med. Chem. 2023, 245, 114896. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zharova, T.V.; Grivennikova, V.G.; Borisov, V.B. F1·Fo ATP Synthase/ATPase: Contemporary View on Unidirectional Catalysis. Int. J. Mol. Sci. 2023, 24, 5417. https://doi.org/10.3390/ijms24065417
Zharova TV, Grivennikova VG, Borisov VB. F1·Fo ATP Synthase/ATPase: Contemporary View on Unidirectional Catalysis. International Journal of Molecular Sciences. 2023; 24(6):5417. https://doi.org/10.3390/ijms24065417
Chicago/Turabian StyleZharova, Tatyana V., Vera G. Grivennikova, and Vitaliy B. Borisov. 2023. "F1·Fo ATP Synthase/ATPase: Contemporary View on Unidirectional Catalysis" International Journal of Molecular Sciences 24, no. 6: 5417. https://doi.org/10.3390/ijms24065417
APA StyleZharova, T. V., Grivennikova, V. G., & Borisov, V. B. (2023). F1·Fo ATP Synthase/ATPase: Contemporary View on Unidirectional Catalysis. International Journal of Molecular Sciences, 24(6), 5417. https://doi.org/10.3390/ijms24065417