

Targeting Tumor Microenvironment Akt Signaling Represents a Potential Therapeutic Strategy for Aggressive Thyroid Cancer

Abstract
1. Introduction
2. Results
2.1. Tumor Microenvironment Stromal Cells Demonstrated the Capability of Promoting CSC Growth and Invasion
2.2. Tumor Stromal Cells Were Capable of Enhancing JAK/STAT3 and PI3K/Akt Signaling in CSCs
2.3. PI3K/Akt and JAK/STAT3 Signaling Engaged in the Promoting Effect of Tumor Stromal Cells on CSC Sphere Formation and Clonal Growth
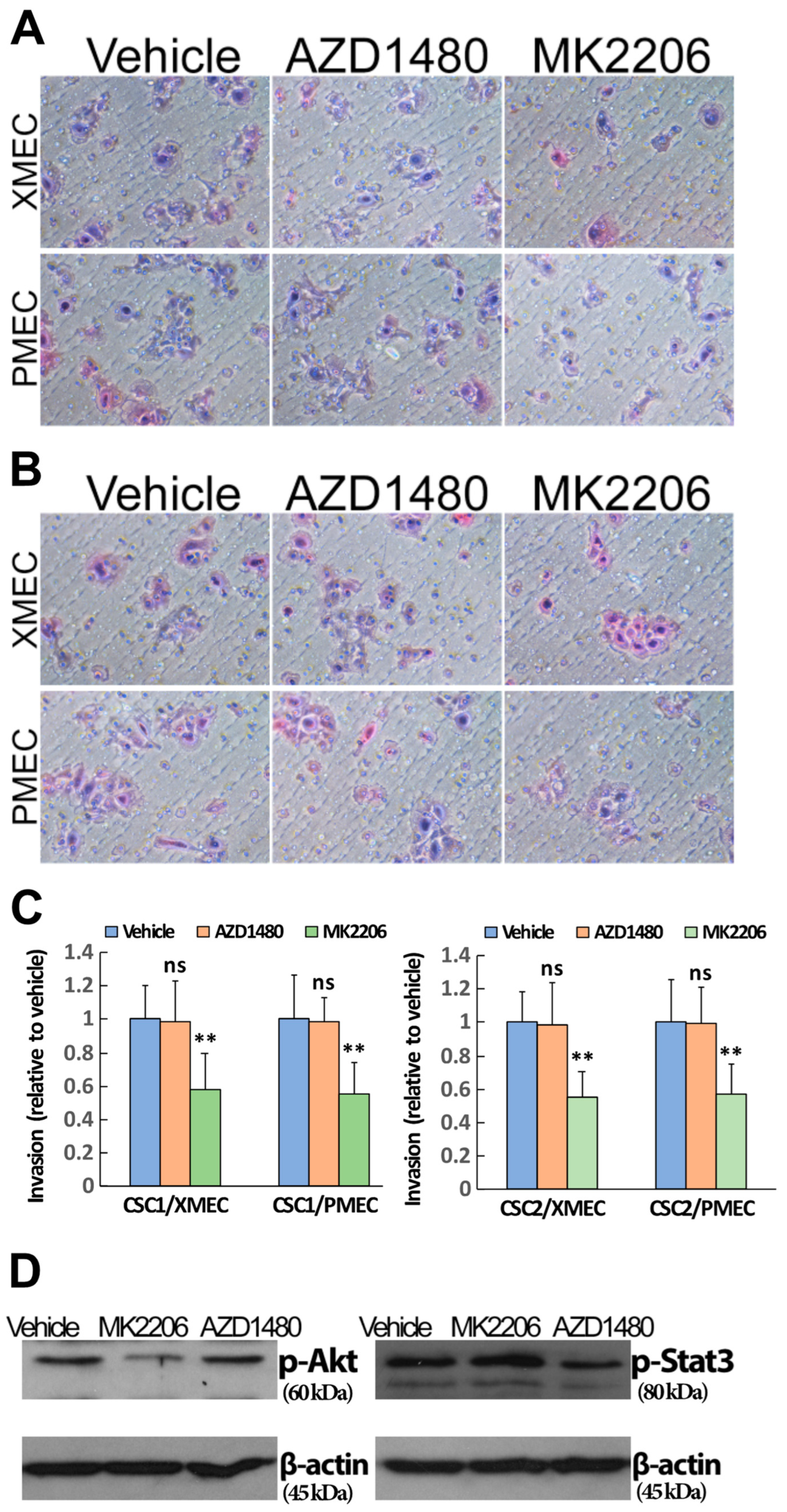
2.4. PI3K/Akt Signaling Pathway Contributed to the Enhanced CSC Invasion Induced by Tumor Stromal Cells
2.5. Inhibiting Akt Signaling Activity Represents a Therapeutic Strategy to Target PTC Tumor Progression
3. Discussion
4. Materials and Methods
4.1. Preparation of Primary Cells from Patient PTC and Non-Tumor Thyroid Tissues
4.2. PTC and Non-Tumor Thyroid Tissue-Derived Cells In Vitro Cultures
4.3. Patient Derived Xenograft Tumors
4.4. Sphere Formation and Clonal Growth
4.5. Tumor Cell Invasion Assay
4.6. Western Blotting
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Elisei, R. A Narrative Review of Genetic Alterations in Primary Thyroid Epithelial Cancer. Int. J. Mol. Sci. 2021, 22, 1726. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chen, J.; Zhou, J.; Xiao, S.; Zeng, W.; Xia, J.; Zeng, X. Epigenetic modification and BRAF gene mutation in thyroid carcinoma. Cancer Cell Int. 2021, 21, 687. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jeon, M.J.; Oh, H.S.; Park, S.; Kim, T.Y.; Shong, Y.K.; Kim, W.B.; Kim, K.; Kim, W.G.; Song, D.E. BRAF and RAS Mutational Status in Noninvasive Follicular Thyroid Neoplasm with Papillary-Like Nuclear Features and Invasive Subtype of Encapsulated Follicular Variant of Papillary Thyroid Carcinoma in Korea. Thyroid 2018, 28, 504–510. [Google Scholar] [CrossRef]
- Agarwal, S.; Bychkov, A.; Jung, C.K. Emerging Biomarkers in Thyroid Practice and Research. Cancers 2021, 14, 204. [Google Scholar] [CrossRef]
- Elia, G.; Patrizio, A.; Ragusa, F.; Paparo, S.R.; Mazzi, V.; Balestri, E.; Botrini, C.; Rugani, L.; Benvenga, S.; Materazzi, G.; et al. Molecular features of aggressive thyroid cancer. Front. Oncol. 2022, 12, 1099280. [Google Scholar] [CrossRef]
- Are, C.; Shaha, A.R. Anaplastic thyroid carcinoma: Biology, pathogenesis, prognostic factors, and treatment approaches. Ann. Surg. Oncol. 2006, 13, 453–464. [Google Scholar] [CrossRef]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef]
- Harper, C.; Michael, J.; Rahmeh, T.; Munro, V. Rare distant metastases to pancreas, liver, and lung as initial presentation of mixed tall cell and columnar cell variants of papillary thyroid cancer. Endocrinol. Diabetes Metab. Case Rep. 2022, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.R.; Montierth, M.; Xu, L.; Goswami, M.; Zhao, X.; Cote, G.; Wang, W.; Iyer, P.; Dadu, R.; Busaidy, N.L.; et al. Impact of Somatic Mutations on Survival Outcomes in Patients With Anaplastic Thyroid Carcinoma. JCO Precis Oncol. 2022, 6, e2100504. [Google Scholar] [CrossRef] [PubMed]
- Soe, M.H.; Chiang, J.M.; Flavell, R.R.; Khanafshar, E.; Mendoza, L.; Kang, H.; Liu, C. Non-Iodine-Avid Disease Is Highly Prevalent in Distant Metastatic Differentiated Thyroid Cancer With Papillary Histology. J. Clin. Endocrinol. Metab. 2022, 107, e3206–e3216. [Google Scholar] [CrossRef]
- Franco, A.T.; Ricarte-Filho, J.C.; Isaza, A.; Jones, Z.; Jain, N.; Mostoufi-Moab, S.; Surrey, L.; Laetsch, T.W.; Li, M.M.; DeHart, J.C.; et al. Fusion Oncogenes Are Associated With Increased Metastatic Capacity and Persistent Disease in Pediatric Thyroid Cancers. J. Clin. Oncol. 2022, 40, 1081–1090. [Google Scholar] [CrossRef]
- Tchekmedyian, V.; Dunn, L.; Sherman, E.; Baxi, S.S.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Enhancing Radioiodine Incorporation in BRAF-Mutant, Radioiodine-Refractory Thyroid Cancers with Vemurafenib and the Anti-ErbB3 Monoclonal Antibody CDX-3379: Results of a Pilot Clinical Trial. Thyroid 2022, 32, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, I.; Shinozaki, E.; Matsushima, T.; Wakatsuki, T.; Ogura, M.; Ichimura, T.; Ozaka, M.; Takahari, D.; Suenaga, M.; Chin, K.; et al. Retrospective study of RAS/PIK3CA/BRAF tumor mutations as predictors of response to first-line chemotherapy with bevacizumab in metastatic colorectal cancer patients. BMC Cancer 2017, 17, 38. [Google Scholar] [CrossRef]
- Sherman, S.I. Targeted therapy of thyroid cancer. Biochem. Pharmacol. 2010, 80, 592–601. [Google Scholar] [CrossRef]
- Krajewska, J.; Handkiewicz-Junak, D.; Jarzab, B. Sorafenib for the treatment of thyroid cancer: An updated review. Expert Opin. Pharmacother. 2015, 16, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Boucai, L. Editorial: Targeted therapy in advanced thyroid cancer. Front. Endocrinol. 2022, 13, 1022698. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.C.; Kunnimalaiyaan, M.; Wang, J.R.; Busaidy, N.L.; Sherman, S.I.; Lai, S.Y.; Zafereo, M.; Cabanillas, M.E. Molecular mechanisms of resistance to kinase inhibitors and redifferentiation in thyroid cancers. Endocr. Relat. Cancer 2022, 29, R173–R190. [Google Scholar]
- Hescheler, D.A.; Hartmann, M.J.M.; Riemann, B.; Michel, M.; Bruns, C.J.; Alakus, H.; Chiapponi, C. Anaplastic thyroid cancer: Genome-based search for new targeted therapy options. Endocr. Connect. 2022, 11, e210624. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Laha, D.; Grant, R.; Nilubol, N. Advances in Biomarker-Driven Targeted Therapies in Thyroid Cancer. Cancers 2021, 13, 6194. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Ricarte-Filho, J.C.; Laetsch, T.W.; Bauer, A.J. Oncogene-specific inhibition in the treatment of advanced pediatric thyroid cancer. J. Clin. Investig. 2021, 131, e152696. [Google Scholar] [CrossRef]
- Fullmer, T.; Cabanillas, M.E.; Zafereo, M. Novel Therapeutics in Radioactive Iodine-Resistant Thyroid Cancer. Front. Endocrinol. 2021, 12, 720723. [Google Scholar] [CrossRef]
- Weitzman, S.P.; Sherman, S.I. Novel Drug Treatments of Progressive Radioiodine-Refractory Differentiated Thyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.J.; Ruan, D.T.; Paulson, V.A.; Barletta, J.A.; Hanna, G.J.; Kraft, S.; Calles, A.; Nehs, M.A.; Moore, F.D., Jr.; Taylor-Weiner, A.; et al. Genomic Heterogeneity and Exceptional Response to Dual Pathway Inhibition in Anaplastic Thyroid Cancer. Clin. Cancer Res. 2017, 23, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Peix, F.; Casanovas, O. Promalignant effects of antiangiogenics in the tumor microenvironment. Semin. Cancer Biol. 2022, 86 Pt 3, 199–206. [Google Scholar] [CrossRef]
- Yuzhalin, A.E. Parallels between the extracellular matrix roles in developmental biology and cancer biology. Semin. Cell Dev. Biol. 2022, 128, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Dey, P. Co-dependencies in the tumor immune microenvironment. Oncogene 2022, 41, 3821–3829. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Das, A.; Sagi, I. Emerging roles of ECM remodeling processes in cancer. Semin. Cancer Biol. 2020, 62, 192–200. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Laplagne, C.; Domagala, M.; Le Naour, A.; Quemerais, C.; Hamel, D.; Fournie, J.J.; Couderc, B.; Bousquet, C.; Ferrand, A.; Poupot, M. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int. J. Mol. Sci. 2019, 20, 4719. [Google Scholar] [CrossRef]
- Wels, J.; Kaplan, R.N.; Rafii, S.; Lyden, D. Migratory neighbors and distant invaders: Tumor-associated niche cells. Genes Dev. 2008, 22, 559–574. [Google Scholar] [CrossRef]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef]
- Cheng, H.C.; Huang, L.T. Tumor Progression, Microenvironments, and Therapeutics. Life 2022, 12, 1599. [Google Scholar] [CrossRef] [PubMed]
- Bozyk, A.; Wojas-Krawczyk, K.; Krawczyk, P.; Milanowski, J. Tumor Microenvironment—A Short Review of Cellular and Interaction Diversity. Biology 2022, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Rodrigues, F.S.; Ciccarelli, F.D.; Malanchi, I. Reflected stemness as a potential driver of the tumour microenvironment. Trends Cell Biol. 2022, 32, 979–987. [Google Scholar] [CrossRef]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2022, 78, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Bayik, D.; Lathia, J.D. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer 2021, 21, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, A.M.; Aguirre, S.; Hao, X.; Waldvogel, S.M.; Zhang, X.H. Exploiting bone niches: Progression of disseminated tumor cells to metastasis. J. Clin. Investig. 2021, 131, e143764. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Mirshahidi, S.; Simental, A.; Lee, S.C.; De Andrade Filho, P.A.; Peterson, N.R.; Cao, W.; Necochea-Campion, R.; Yang, H.; Duerksen-Hughes, P.; Yuan, X. Subpopulations of cancer stem cells found in papillary thyroid carcinoma. Exp. Cell Res. 2018, 362, 515–524. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Ham, I.H.; Wang, L.; Lee, D.; Woo, J.; Kim, T.H.; Jeong, H.Y.; Oh, H.J.; Choi, K.S.; Kim, T.M.; Hur, H. Curcumin inhibits the cancer-associated fibroblast-derived chemoresistance of gastric cancer through the suppression of the JAK/STAT3 signaling pathway. Int. J. Oncol. 2022, 61, 1–12. [Google Scholar] [CrossRef]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de Los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to Durvalumab and Durvalumab plus Tremelimumab Is Associated with Functional STK11 Mutations in Patients with Non-Small Cell Lung Cancer and Is Reversed by STAT3 Knockdown. Cancer Discov. 2021, 11, 2828–2845. [Google Scholar] [CrossRef]
- Hu, X.; Xiang, F.; Feng, Y.; Gao, F.; Ge, S.; Wang, C.; Zhang, X.; Wang, N. Neutrophils Promote Tumor Progression in Oral Squamous Cell Carcinoma by Regulating EMT and JAK2/STAT3 Signaling Through Chemerin. Front. Oncol. 2022, 12, 812044. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Klein, K.; Zdarsky, B.; Stoiber, D. The Multifaceted Role of STAT3 in NK-Cell Tumor Surveillance. Front. Immunol. 2022, 13, 947568. [Google Scholar] [CrossRef]
- Dosch, A.R.; Singh, S.; Dai, X.; Mehra, S.; Silva, I.C.; Bianchi, A.; Srinivasan, S.; Gao, Z.; Ban, Y.; Chen, X.; et al. Targeting Tumor-Stromal IL6/STAT3 Signaling through IL1 Receptor Inhibition in Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 2280–2290. [Google Scholar] [CrossRef]
- Sun, L.; Ke, M.; Wang, X.; Yin, M.; Wei, J.; Xu, L.; Tian, X.; Wang, F.; Zhang, H.; Fu, S.; et al. FAP(high) alpha-SMA(low) cancer-associated fibroblast-derived SLPI protein encapsulated in extracellular vesicles promotes ovarian cancer development via activation of PI3K/AKT and downstream signaling pathways. Mol. Carcinog. 2022, 61, 910–923. [Google Scholar] [CrossRef]
- Chen, X.; Yang, M.; Yin, J.; Li, P.; Zeng, S.; Zheng, G.; He, Z.; Liu, H.; Wang, Q.; Zhang, F.; et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/beta-catenin signaling. Cell Commun. Signal 2022, 20, 92. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zhang, P.; Yu, H.; Xi, J. Extracellular Vesicles-Encapsulated miR-153-3p Potentiate the Survival and Invasion of Lung Adenocarcinoma. Mol. Cells 2022, 45, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.X.; Zhang, Y.Q.; Sun, H.; Chen, Z.Q.; Chang, J.J.; Wang, X.; Wang, X.; Tan, C.; Ni, S.J.; Weng, W.W.; et al. Calcipotriol abrogates cancer-associated fibroblast-derived IL-8-mediated oxaliplatin resistance in gastric cancer cells via blocking PI3K/Akt signaling. Acta Pharmacol. Sin. 2022, 44, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Gul, D.; Schweitzer, A.; Khamis, A.; Knauer, S.K.; Ding, G.B.; Freudelsperger, L.; Karampinis, I.; Strieth, S.; Hagemann, J.; Stauber, R.H. Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer. Cancers 2022, 14, 2337. [Google Scholar] [CrossRef]
- Tey, S.K.; Wong, S.W.K.; Chan, J.Y.T.; Mao, X.; Ng, T.H.; Yeung, C.L.S.; Leung, Z.; Fung, H.L.; Tang, A.H.N.; Wong, D.K.H.; et al. Patient pIgR-enriched extracellular vesicles drive cancer stemness, tumorigenesis and metastasis in hepatocellular carcinoma. J. Hepatol. 2022, 76, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.J.; Li, T.W.; Zhou, L.; Wong, T.L.; Liu, X.; Ma, V.W.S.; Lo, C.M.; Man, K.; Lee, T.K.; Ning, W.; et al. FSTL1 Secreted by Activated Fibroblasts Promotes Hepatocellular Carcinoma Metastasis and Stemness. Cancer Res. 2021, 81, 5692–5705. [Google Scholar] [CrossRef]
- Brockmueller, A.; Shayan, P.; Shakibaei, M. Evidence That beta1-Integrin Is Required for the Anti-Viability and Anti-Proliferative Effect of Resveratrol in CRC Cells. Int. J. Mol. Sci. 2022, 23, 4714. [Google Scholar] [CrossRef]
- Liu, D.; Shi, K.; Fu, M.; Chen, F. Melatonin indirectly decreases gastric cancer cell proliferation and invasion via effects on cancer-associated fibroblasts. Life Sci. 2021, 277, 119497. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-a signaling. Exp. Cell Res. 2020, 396, 112315. [Google Scholar] [CrossRef]
- Lazarian, G.; Friedrich, C.; Quinquenel, A.; Tran, J.; Ouriemmi, S.; Dondi, E.; Martin, A.; Mihoub, I.; Chiron, D.; Bellanger, C.; et al. Stabilization of beta-catenin upon B-cell receptor signaling promotes NF-kB target genes transcription in mantle cell lymphoma. Oncogene 2020, 39, 2934–2947. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, Y.; Tan, X.; Ke, K.; Zheng, X.; Wang, F.; Lan, S.; Liao, N.; Cai, Z.; Shi, Y.; et al. Inflammatory Micro-environment Contributes to Stemness Properties and Metastatic Potential of HCC via the NF-kappaB/miR-497/SALL4 Axis. Mol. Ther. Oncolytics 2019, 15, 79–90. [Google Scholar] [CrossRef]
- Zheng, S.; Hu, C.; Lin, H.; Li, G.; Xia, R.; Zhang, X.; Su, D.; Li, Z.; Zhou, Q.; Chen, R. circCUL2 induces an inflammatory CAF phenotype in pancreatic ductal adenocarcinoma via the activation of the MyD88-dependent NF-kappaB signaling pathway. J. Exp. Clin. Cancer Res. 2022, 41, 71. [Google Scholar] [CrossRef]
- Lin, H.Y.; Ko, C.J.; Lo, T.Y.; Wu, S.R.; Lan, S.W.; Huang, C.A.; Lin, Y.C.; Lin, H.H.; Tu, H.F.; Lee, C.F.; et al. Matriptase-2/NR4A3 axis switches TGF-beta action toward suppression of prostate cancer cell invasion, tumor growth, and metastasis. Oncogene 2022, 41, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; He, J.; Pan, Q.Z.; Yang, J.; Zhao, J.; Zhang, Y.J.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735. [Google Scholar] [CrossRef]
- Yang, M.; Li, D.; Jiang, Z.; Li, C.; Ji, S.; Sun, J.; Chang, Y.; Ruan, S.; Wang, Z.; Liang, R.; et al. TGF-beta-Induced FLRT3 Attenuation Is Essential for Cancer-Associated Fibroblast-Mediated Epithelial-Mesenchymal Transition in Colorectal Cancer. Mol. Cancer Res. 2022, 20, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Z.; Wang, Y.; Zhao, H.; Du, Y. The Role of Cancer-Associated Fibroblasts in Ovarian Cancer. Cancers 2022, 14, 2637. [Google Scholar] [CrossRef] [PubMed]
- Dhainaut, M.; Rose, S.A.; Akturk, G.; Wroblewska, A.; Nielsen, S.R.; Park, E.S.; Buckup, M.; Roudko, V.; Pia, L.; Sweeney, R.; et al. Spatial CRISPR genomics identifies regulators of the tumor microenvironment. Cell 2022, 185, 1223–1239.e20. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, M.U.; Bansal, S.; Kaur, P.; Jain, S.K.; Altadil, T.; Hinzman, C.P.; Li, Y.; Moulton, J.; Singh, B.; Bansal, S.; et al. TGFbeta Drives Metabolic Perturbations during Epithelial Mesenchymal Transition in Pancreatic Cancer: TGFbeta Induced EMT in PDAC. Cancers 2021, 13, 6204. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, G.; Huo, X.; Wang, Y.; Tigyi, G.; Zhu, B.M.; Yue, J.; Zhang, W. Adipose-Derived Stem Cells Facilitate Ovarian Tumor Growth and Metastasis by Promoting Epithelial to Mesenchymal Transition Through Activating the TGF-beta Pathway. Front. Oncol. 2021, 11, 756011. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Mbeunkui, F.; Johann, D.J., Jr. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.A.; Novitskiy, S.; Owens, P.; Massoll, N.; Cheng, N.; Fang, W.; Moses, H.L.; Franco, A.T. Fibroblast-Mediated Collagen Remodeling Within the Tumor Microenvironment Facilitates Progression of Thyroid Cancers Driven by BrafV600E and Pten Loss. Cancer Res. 2016, 76, 1804–1813. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Ng, M.R.; Brugge, J.S. A stiff blow from the stroma: Collagen crosslinking drives tumor progression. Cancer Cell 2009, 16, 455–457. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Yarchoan, M.; LiVolsi, V.A.; Brose, M.S. BRAF mutation and thyroid cancer recurrence. J. Clin. Oncol. 2015, 33, 7–8. [Google Scholar] [CrossRef]
- Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 2007, 28, 742–762. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Zhang, Y.; Patel, R.; Sharma, A.; Wu, H.; Robertson, G.P.; Yan, L.; Rubin, E.; Yang, J.M. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011, 71, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Leong, S.W.; Wang, J.; Wu, Q.; Ghauri, M.A.; Sarwar, A.; Su, Q.; Zhang, Y. Cephalomannine inhibits hypoxia-induced cellular function via the suppression of APEX1/HIF-1alpha interaction in lung cancer. Cell Death Dis. 2021, 12, 490. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metastasis Status | PTC Tested (Cases) | pAkt Expression ↑ (Cases) | pAkt Expression ↑ and BRAFV600E (Cases) |
|---|---|---|---|
| Lymph node + | 26 | 25 | 25 |
| Lymph node − | 45 | 11 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirshahidi, S.; Yuan, I.J.; Simental, A.; Lee, S.C.; Peterson, N.R.; Andrade Filho, P.A.; Murry, T.; Duerksen-Hughes, P.; Yuan, X. Targeting Tumor Microenvironment Akt Signaling Represents a Potential Therapeutic Strategy for Aggressive Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 5471. https://doi.org/10.3390/ijms24065471
Mirshahidi S, Yuan IJ, Simental A, Lee SC, Peterson NR, Andrade Filho PA, Murry T, Duerksen-Hughes P, Yuan X. Targeting Tumor Microenvironment Akt Signaling Represents a Potential Therapeutic Strategy for Aggressive Thyroid Cancer. International Journal of Molecular Sciences. 2023; 24(6):5471. https://doi.org/10.3390/ijms24065471
Chicago/Turabian StyleMirshahidi, Saied, Isabella J. Yuan, Alfred Simental, Steve C. Lee, Nathaniel R. Peterson, Pedro A. Andrade Filho, Thomas Murry, Penelope Duerksen-Hughes, and Xiangpeng Yuan. 2023. "Targeting Tumor Microenvironment Akt Signaling Represents a Potential Therapeutic Strategy for Aggressive Thyroid Cancer" International Journal of Molecular Sciences 24, no. 6: 5471. https://doi.org/10.3390/ijms24065471
APA StyleMirshahidi, S., Yuan, I. J., Simental, A., Lee, S. C., Peterson, N. R., Andrade Filho, P. A., Murry, T., Duerksen-Hughes, P., & Yuan, X. (2023). Targeting Tumor Microenvironment Akt Signaling Represents a Potential Therapeutic Strategy for Aggressive Thyroid Cancer. International Journal of Molecular Sciences, 24(6), 5471. https://doi.org/10.3390/ijms24065471