
Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice

 ,
,
Abstract
:1. Introduction
2. Results
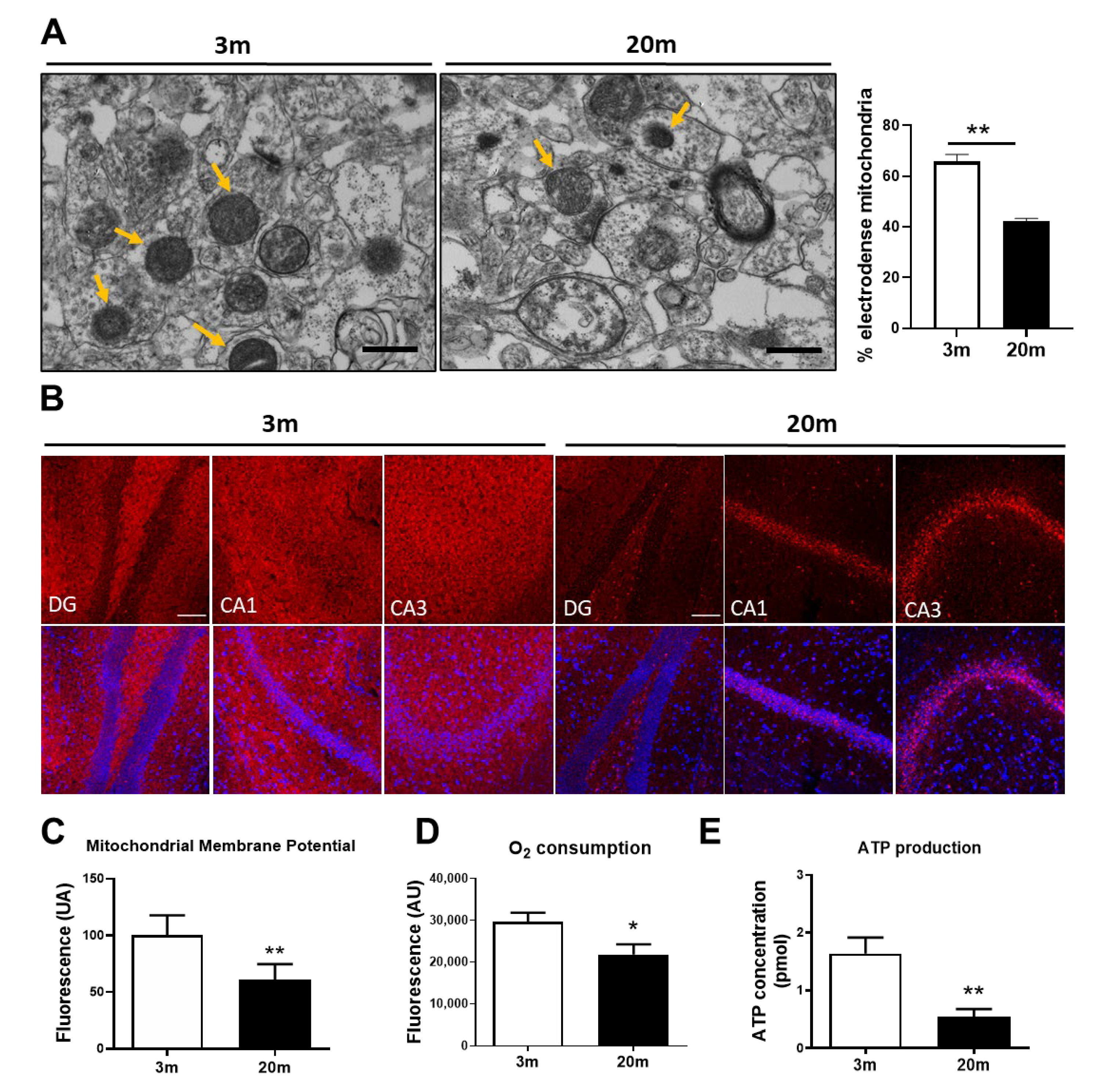
2.1. Mitochondrial Bioenergetic Dysfunction Is Observed in the Hippocampus of Aged Female C57BL/6J Mice
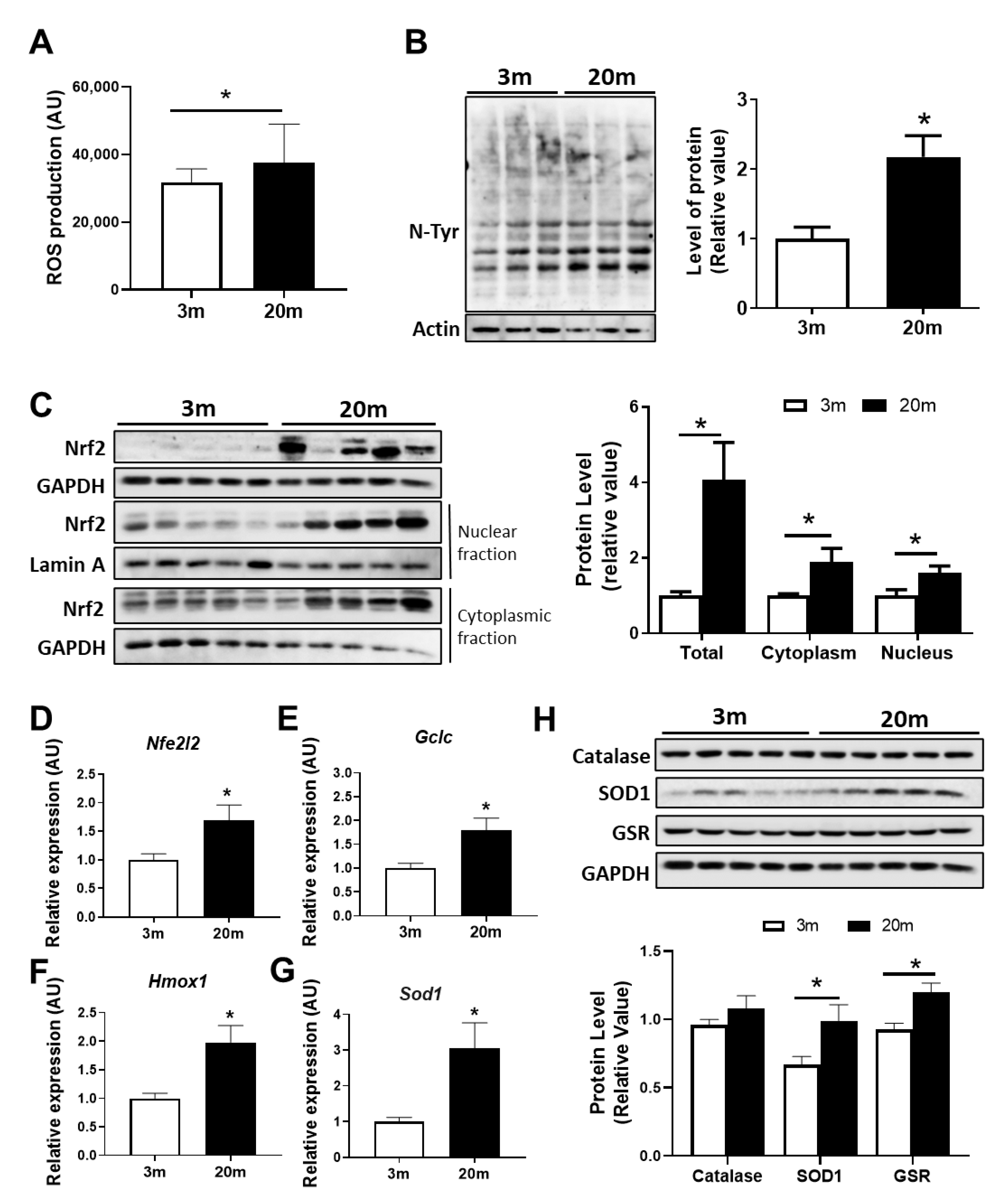
2.2. Redox Imbalance Is Observed in the Aged Hippocampus of Female C57BL/6J Mice
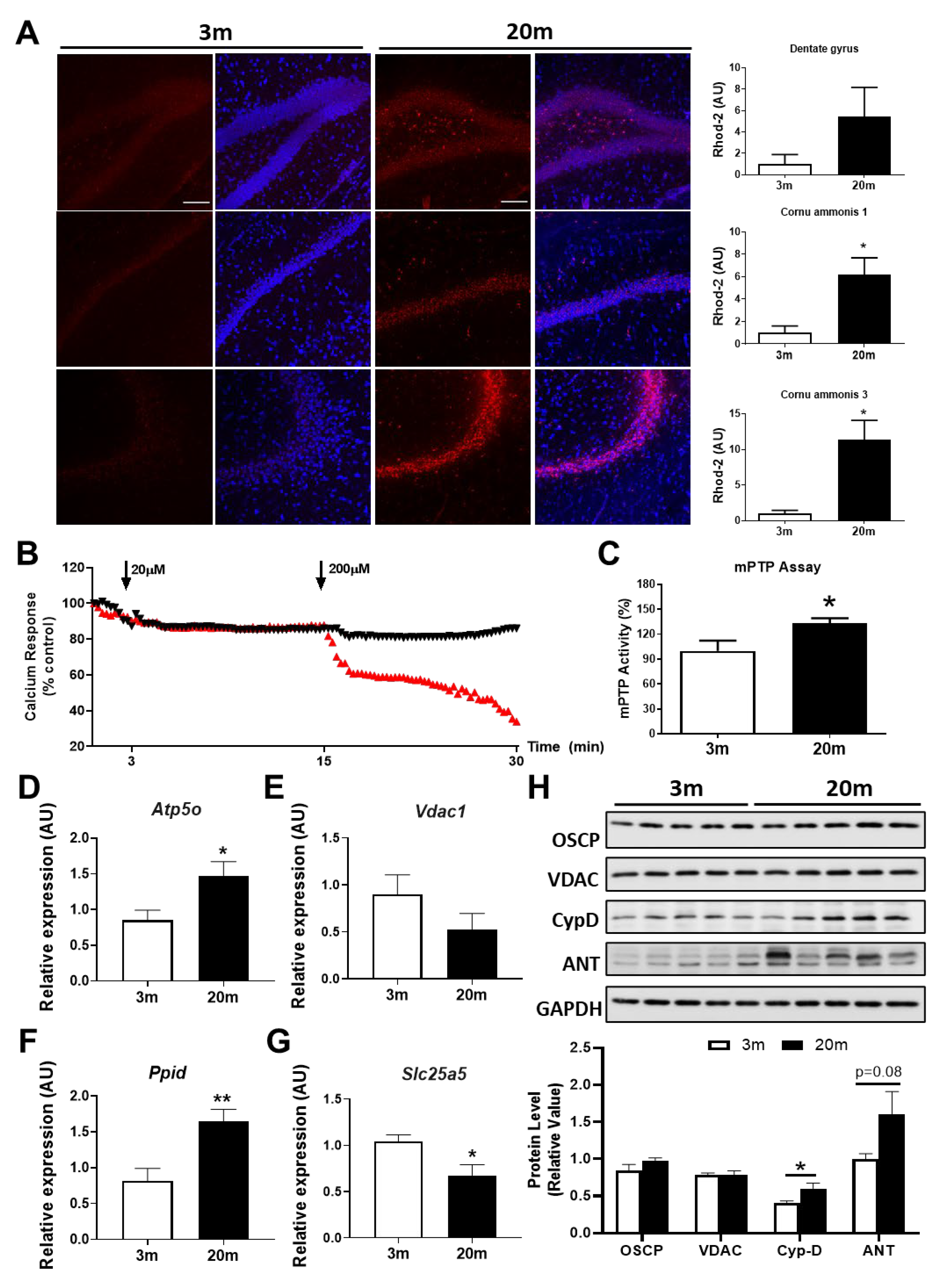
2.3. The Hippocampus of Aged Female Mice Shows Decreased Capacity for Mitochondrial Calcium Regulation
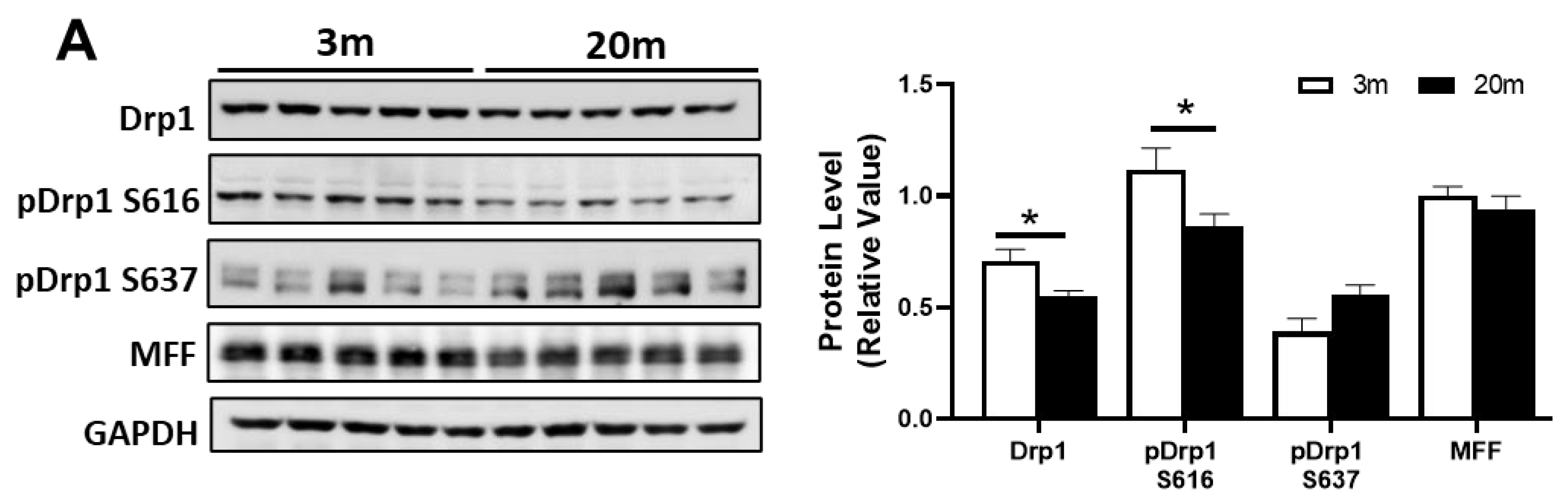
2.4. Altered Levels of Proteins Related to Fission and Fusion Processes Suggest an Imbalance of Mitochondrial Dynamics in the Hippocampus of Aged Mice
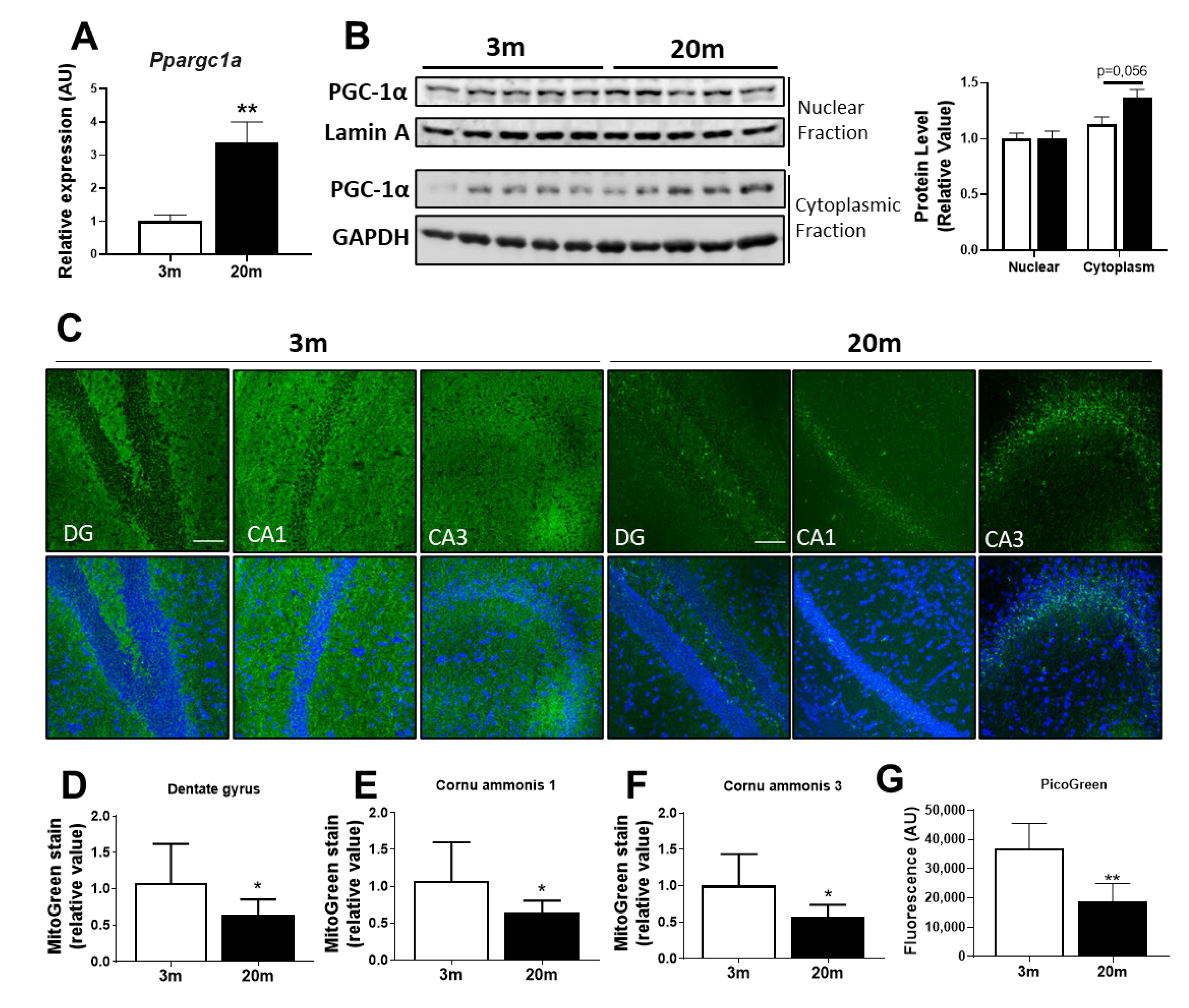
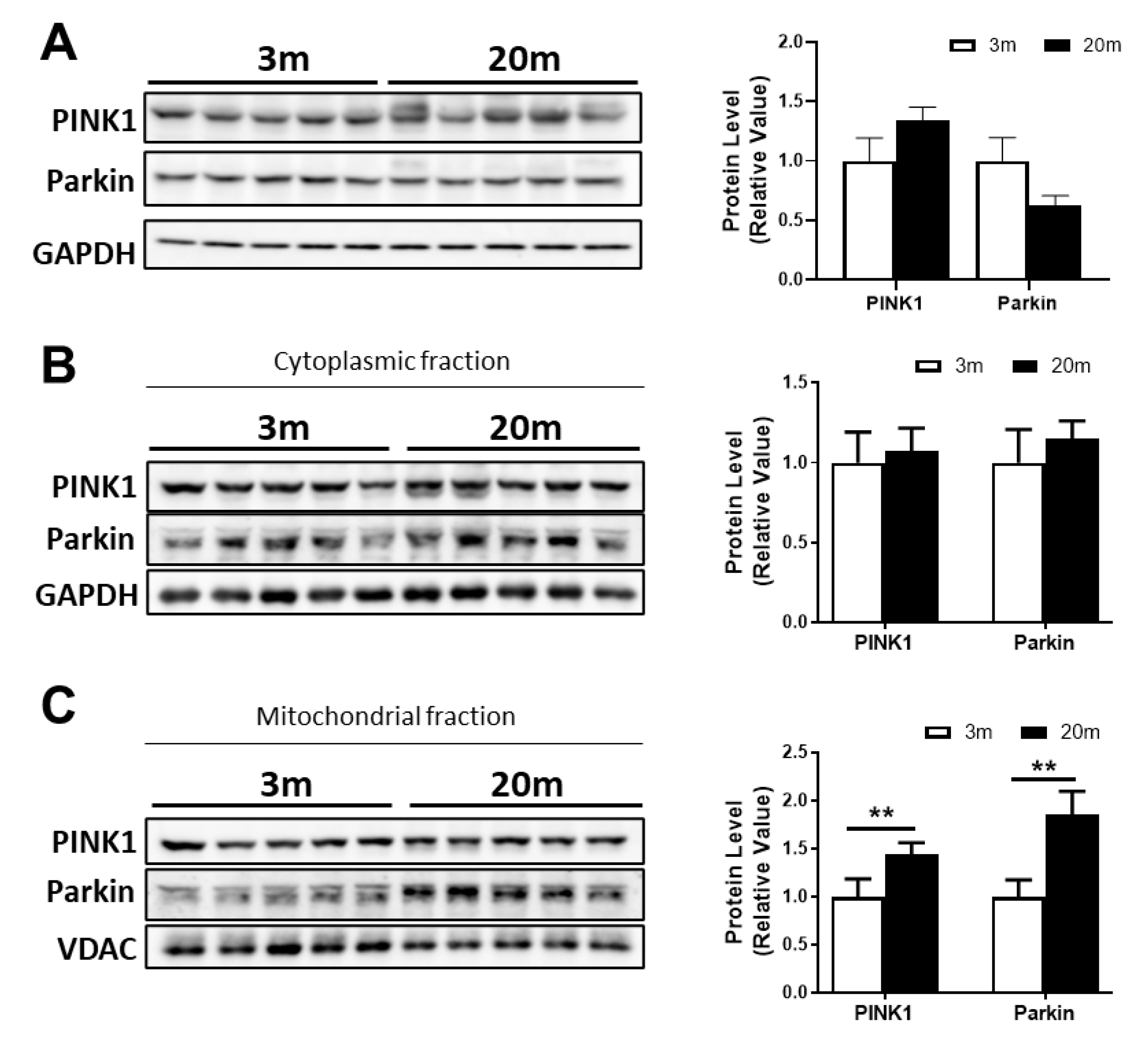
2.5. Reduced Mitochondrial Mass in the Aged Hippocampus, with Decreased Mitochondrial Biogenesis and Mitophagy
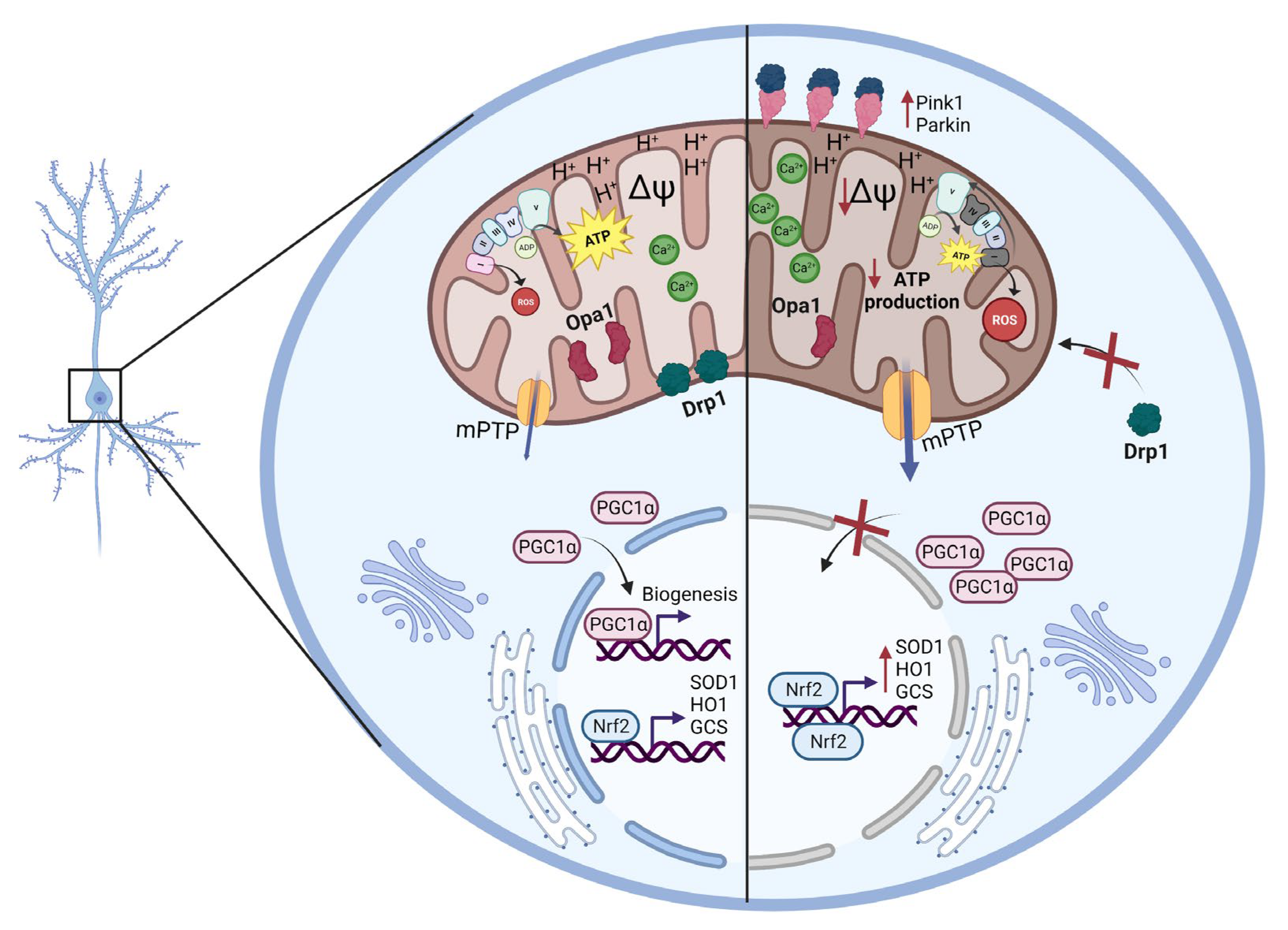
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jin, M.; Cai, S.Q. Mechanisms Underlying Brain Aging under Normal and Pathological Conditions. Neurosci. Bull. 2022, 39, 303–314. [Google Scholar] [CrossRef]
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Bettio, L.E.B.; Rajendran, L.; Gil-Mohapel, J. The effects of aging in the hippocampus and cognitive decline. Neurosci. Biobehav. Rev. 2017, 79, 66–86. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; McEwen, B.S. Mitochondria impact brain function and cognition. Proc. Natl. Acad. Sci. USA 2014, 111, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Palmeira, C.M.; Rolo, A.P. Mitochondrial Membrane Potential (DeltaPsi) Fluctuations Associated with the Metabolic States of Mitochondria. Methods Mol. Biol. 2018, 1782, 109–119. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Mira, R.G.; Cerpa, W. Building a Bridge between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2020, 41, 1413–1430. [Google Scholar] [CrossRef]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore: A potential drug target for neurodegeneration. Drug Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Cowell, R.M.; Blake, K.R.; Russell, J.W. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J. Comp. Neurol. 2007, 502, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Rybka, V.; Suzuki, Y.J.; Gavrish, A.S.; Dibrova, V.A.; Gychka, S.G.; Shults, N.V. Transmission Electron Microscopy Study of Mitochondria in Aging Brain Synapses. Antioxidants 2019, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Mishra, E.; Thakur, M.K. Alterations in hippocampal mitochondrial dynamics are associated with neurodegeneration and recognition memory decline in old male mice. Biogerontology 2022, 23, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Bandez, M.J.; Lopez-Cepero, J.M.; Gomez, C.; Boveris, A. High doses of vitamin E improve mitochondrial dysfunction in rat hippocampus and frontal cortex upon aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R827–R834. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Brown, M.R.; Geddes, J.W.; Sullivan, P.G. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J. Bioenerg. Biomembr. 2004, 36, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kumar Chaudhary, M.; Rizvi, S.I. Invertebrate and vertebrate models in aging research. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2019, 163, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A. Old and new models for the study of human ageing. Nat. Rev. Mol. Cell Biol. 2020, 21, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Brandvain, Y.; Klebanov, S.; Austad, S.N.; Miller, R.A.; Yuan, R.; Harrison, D.E. PohnB6F1: A cross of wild and domestic mice that is a new model of extended female reproductive life span. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Morales-Gonzalez, A.; Madrigal-Santillan, E.O.; Madrigal-Bujaidar, E.; Alvarez-Gonzalez, I.; Garcia-Melo, L.F.; Anguiano-Robledo, L.; Fregoso-Aguilar, T.; Morales-Gonzalez, J.A. Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise. Antioxidants 2019, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [Green Version]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Iosub, R.; Avitabile, D.; Grant, L.; Tsaneva-Atanasova, K.; Kennedy, H.J. Calcium-Induced calcium release during action potential firing in developing inner hair cells. Biophys. J. 2015, 108, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.L. A calcium-activated chloride current generates the after-depolarization of rat sensory neurones in culture. J. Physiol. 1985, 364, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [Green Version]
- Melachroinou, K.; Xilouri, M.; Emmanouilidou, E.; Masgrau, R.; Papazafiri, P.; Stefanis, L.; Vekrellis, K. Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiol. Aging 2013, 34, 2853–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino-Coppola, D. Regulation of the mitochondrial permeability transition pore and its effects on aging. Microb. Cell 2020, 7, 222–233. [Google Scholar] [CrossRef]
- Parks, R.J.; Menazza, S.; Holmstrom, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Li, M.; Xu, L.; Shen, Y.; Yang, H.; Ding, M.; Liu, X.; Xie, Z. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 2018, 29, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis in organismal senescence and neurodegeneration. Mech. Ageing Dev. 2020, 191, 111345. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taherzadeh-Fard, E.; Saft, C.; Akkad, D.A.; Wieczorek, S.; Haghikia, A.; Chan, A.; Epplen, J.T.; Arning, L. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol. Neurodegener. 2011, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Enriquez, S.; Kim, I.; Currin, R.T.; Lemasters, J.J. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006, 2, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Olesen, M.A.; Torres, A.K.; Jara, C.; Murphy, M.P.; Tapia-Rojas, C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020, 34, 101558. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Bovina, C.; Castelluccio, C.; Fato, R.; Formiggini, G.; Genova, M.L.; Marchetti, M.; Pich, M.M.; Pallotti, F.; Parenti Castelli, G.; et al. Mitochondrial complex I defects in aging. Mol. Cell. Biochem. 1997, 174, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Benson, T.; Albensi, B.C. Mitochondrial Transfusion Improves Mitochondrial Function through Up-regulation of Mitochondrial Complex II Protein Subunit SDHB in the Hippocampus of Aged Mice. Mol. Neurobiol. 2022, 59, 6009–6017. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Lopez-Cepero, J.M.; Bandez, M.J.; Sanchez-Pino, M.J.; Gomez, C.; Cadenas, E.; Boveris, A. Hippocampal mitochondrial dysfunction in rat aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R501–R509. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease–therapeutic aspects. Mol. Neurobiol. 2010, 41, 159–171. [Google Scholar] [CrossRef]
- Navarro, A.; Gomez, C.; Sanchez-Pino, M.J.; Gonzalez, H.; Bandez, M.J.; Boveris, A.D.; Boveris, A. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1392–R1399. [Google Scholar] [CrossRef] [Green Version]
- Leveille, C.F.; Mikhaeil, J.S.; Turner, K.D.; Silvera, S.; Wilkinson, J.; Fajardo, V.A. Mitochondrial cristae density: A dynamic entity that is critical for energy production and metabolic power in skeletal muscle. J. Physiol. 2017, 595, 2779–2780. [Google Scholar] [CrossRef]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ortenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Shen, T.; Huynh, H.; Fang, X.; Han, Z.; Ouyang, K. Cardiolipin Regulates Mitochondrial Ultrastructure and Function in Mammalian Cells. Genes 2022, 13, 1889. [Google Scholar] [CrossRef]
- Brandt, T.; Mourier, A.; Tain, L.S.; Partridge, L.; Larsson, N.G.; Kuhlbrandt, W. Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. Elife 2017, 6, e24662. [Google Scholar] [CrossRef]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The Functional Impact of Mitochondrial Structure across Subcellular Scales. Front. Physiol. 2020, 11, 541040. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, H.; Hu, W.; Wang, Y.; Wu, W.; Yin, Y.; Liang, Y.; Wang, C.; Huang, D.; Li, W. NADPH oxidase-derived production of reactive oxygen species is involved in learning and memory impairments in 16-month-old female rats. Mol. Med. Rep. 2015, 12, 4546–4553. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Serrano, F.; Oury, T.D.; Klann, E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J. Neurosci. 2006, 26, 3933–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. N. Y. Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangialasche, F.; Polidori, M.C.; Monastero, R.; Ercolani, S.; Camarda, C.; Cecchetti, R.; Mecocci, P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009, 8, 285–305. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Chen, B.H.; Shin, B.N.; Lee, T.K.; Cho, J.H.; Kim, I.H.; Park, J.H.; Lee, J.C.; Tae, H.J.; Lee, C.H.; et al. Comparison of catalase immunoreactivity in the hippocampus between young, adult and aged mice and rats. Mol. Med. Rep. 2016, 14, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Leutner, S.; Eckert, A.; Muller, W.E. ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J. Neural Transm. 2001, 108, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Borras, C.; Gambini, J.; Sastre, J.; Pallardo, F.V. Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett. 2005, 579, 2541–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo-Pelaez, F.; Song, S.; Parthasarathy, A.; Hazzi, C.; Naidu, K.; Sanchez-Ramos, J. Oxidative DNA damage in the aging mouse brain. Mov. Disord. 1999, 14, 972–980. [Google Scholar] [CrossRef]
- Navarro, A.; Sanchez Del Pino, M.J.; Gomez, C.; Peralta, J.L.; Boveris, A. Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R985–R992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, R.; Santandreu, F.M.; Valle, A.; Gianotti, M.; Oliver, J.; Roca, P. Sex-dependent differences in aged rat brain mitochondrial function and oxidative stress. Free Radic Biol. Med. 2009, 46, 169–175. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Aranguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88 Pt B, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Khorolskaya, V.G.; Sadovnikova, I.S.; Shaforostova, E.A.; Cherednichenko, V.R.; Burakova, I.Y.; Plotnikov, E.Y.; Popov, V.N. Age-Related Decline in Nrf2/ARE Signaling Is Associated with the Mitochondrial DNA Damage and Cognitive Impairments. Int. J. Mol. Sci. 2022, 23, 15197. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflug. Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Lopez-Vazquez, S.; Nunez, J.; Villalobos, C.; Nunez, L. Remodeling of Intracellular Ca2+ Homeostasis in Rat Hippocampal Neurons Aged In Vitro. Int. J. Mol. Sci. 2020, 21, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 2016, 1863, 2637–2649. [Google Scholar] [CrossRef]
- Jara, C.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Tau Deletion Prevents Cognitive Impairment and Mitochondrial Dysfunction Age Associated by a Mechanism Dependent on Cyclophilin-D. Front. Neurosci. 2020, 14, 586710. [Google Scholar] [CrossRef]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimer’s Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging 2014, 6, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.W.; Zhou, W.; Zhang, B.Y.; Jia, H.; Xu, L.J.; Liu, A.L.; Du, G.H. DL0410 ameliorates cognitive disorder in SAMP8 mice by promoting mitochondrial dynamics and the NMDAR-CREB-BDNF pathway. Acta Pharmacol. Sin. 2021, 42, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, K.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Archer, S.L. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: Implications for human disease. Clin. Sci. 2016, 130, 1861–1874. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Sirago, G.; Fracasso, F.; Leeuwenburgh, C.; Lezza, A.M.S. “What makes some rats live so long?” The mitochondrial contribution to longevity through balance of mitochondrial dynamics and mtDNA content. Exp. Gerontol. 2016, 85, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenz, T. Mitochondria and PGC-1alpha in Aging and Age-Associated Diseases. J. Aging Res. 2011, 2011, 810619. [Google Scholar] [CrossRef] [Green Version]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARgamma/PGC1alpha signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef]
- El Hayek, L.; Khalifeh, M.; Zibara, V.; Abi Assaad, R.; Emmanuel, N.; Karnib, N.; El-Ghandour, R.; Nasrallah, P.; Bilen, M.; Ibrahim, P.; et al. Lactate Mediates the Effects of Exercise on Learning and Memory through SIRT1-Dependent Activation of Hippocampal Brain-Derived Neurotrophic Factor (BDNF). J. Neurosci. 2019, 39, 2369–2382. [Google Scholar] [CrossRef] [Green Version]
- Kalfalah, F.; Sobek, S.; Bornholz, B.; Gotz-Rosch, C.; Tigges, J.; Fritsche, E.; Krutmann, J.; Kohrer, K.; Deenen, R.; Ohse, S.; et al. Inadequate mito-biogenesis in primary dermal fibroblasts from old humans is associated with impairment of PGC1A-independent stimulation. Exp. Gerontol. 2014, 56, 59–68. [Google Scholar] [CrossRef]
- Baek, J.H.; Son, H.; Jeong, Y.H.; Park, S.W.; Kim, H.J. Chronological Aging Standard Curves of Telomere Length and Mitochondrial DNA Copy Number in Twelve Tissues of C57BL/6 Male Mouse. Cells 2019, 8, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chiang, W.C. Mitophagy in aging and longevity. IUBMB Life 2022, 74, 296–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia-Rojas, C.; Inestrosa, N.C. Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.J.; Ait-Aissa, K.; Levchenko, V.; Staruschenko, A.; Gutterman, D.D.; Beyer, A.M. Visualization and quantification of mitochondrial structure in the endothelium of intact arteries. Cardiovasc. Res. 2019, 115, 1546–1556. [Google Scholar] [CrossRef]
- Koopman, W.J.; Verkaart, S.; Visch, H.J.; van der Westhuizen, F.H.; Murphy, M.P.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Inhibition of complex I of the electron transport chain causes O2−. -mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005, 288, C1440–C1450. [Google Scholar] [CrossRef]
- Torres, A.K.; Tapia-Rojas, C.; Cerpa, W.; Quintanilla, R.A. Stimulation of Melanocortin Receptor-4 (MC4R) Prevents Mitochondrial Damage Induced by Binge Ethanol Protocol in Adolescent Rat Hippocampus. Neuroscience 2020, 438, 70–85. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Torres, A.K.; Quintanilla, R.A. Adolescence binge alcohol consumption induces hippocampal mitochondrial impairment that persists during the adulthood. Neuroscience 2019, 406, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Karadayian, A.G.; Bustamante, J.; Czerniczyniec, A.; Lombardi, P.; Cutrera, R.A.; Lores-Arnaiz, S. Alcohol hangover induces mitochondrial dysfunction and free radical production in mouse cerebellum. Neuroscience 2015, 304, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Hmox1 | 5′-CACAGCACTATGTAAAGCGTCT-3′ | 5′-TGTGCAATCTTCTTCAGGACC-3′ |
| Gclc | 5′-GGGGTGACGAGGTGGAGTA-3′ | 5′-GTTGGGGTTTGTCCTCTCCC-3′ |
| Nfe2l2 | 5′-ACCCGAAGCACGCTGAAGGC-3′ | 5′-GTCACTGAACCCAGGCGGTGG-3′ |
| Ppid | 5′-AGGAGATAGCCCCAGGAGAT-3′ | 5′-TTGCATACACGGCCTTCTCTT-3′ |
| Slc25a5 | 5′-CCACCCAGGCTCTCAACTTT-3′ | 5′-AAGCACAAGGATGTAGCCCC-3′ |
| Atp5o | 5′-CAAGCGCACCGTCAAAGTG-3′ | 5′-GCACCGTCTTTAACTCAGAGAG-3′ |
| Vdac1 | 5′-AGTAACACTCGCTTCGGAATAG-3′ | 5′-TGGTTTTAGGGTCTGAGTGTAC-3′ |
| Ppargc1a | 5′-ACAGAGACACTGGACAGTCT-3′ | 5′-CATTGTAGCTGAGCTGAGTG-3′ |
| Cycs | 5′-GTTCAGAAGTGTGCCCAGTG-3′ | 5′-TACTCCATCAGGGTATCCTC-3′ |
| Sod1 | 5′-ACTTCGAGCAGAAGGCAAGC-3′ | 5′-AACATGCCTCTCTTCATCCG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, A.K.; Jara, C.; Llanquinao, J.; Lira, M.; Cortés-Díaz, D.; Tapia-Rojas, C. Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice. Int. J. Mol. Sci. 2023, 24, 5476. https://doi.org/10.3390/ijms24065476
Torres AK, Jara C, Llanquinao J, Lira M, Cortés-Díaz D, Tapia-Rojas C. Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice. International Journal of Molecular Sciences. 2023; 24(6):5476. https://doi.org/10.3390/ijms24065476
Chicago/Turabian StyleTorres, Angie K., Claudia Jara, Jesús Llanquinao, Matías Lira, Daniela Cortés-Díaz, and Cheril Tapia-Rojas. 2023. "Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice" International Journal of Molecular Sciences 24, no. 6: 5476. https://doi.org/10.3390/ijms24065476
APA StyleTorres, A. K., Jara, C., Llanquinao, J., Lira, M., Cortés-Díaz, D., & Tapia-Rojas, C. (2023). Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice. International Journal of Molecular Sciences, 24(6), 5476. https://doi.org/10.3390/ijms24065476