
The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells
 , ,
, ,  and
and
Abstract
:1. Introduction
- Different ligands can induce dimerization or bind to different heterodimers of the GPR55 protein with CB1, CB2, or GPR18 receptors (CB1-GPR55, CB2-GPR55, and GPR18-GPR55) and thereby induce different intracellular signaling cascades.
- Different ligands can induce the coupling of the GPR55 receptor to different Gαα subunits (Gαq, Gα12, or Gα13), thus inducing opposite consequences for the cell (apoptosis or proliferation).
2. Results
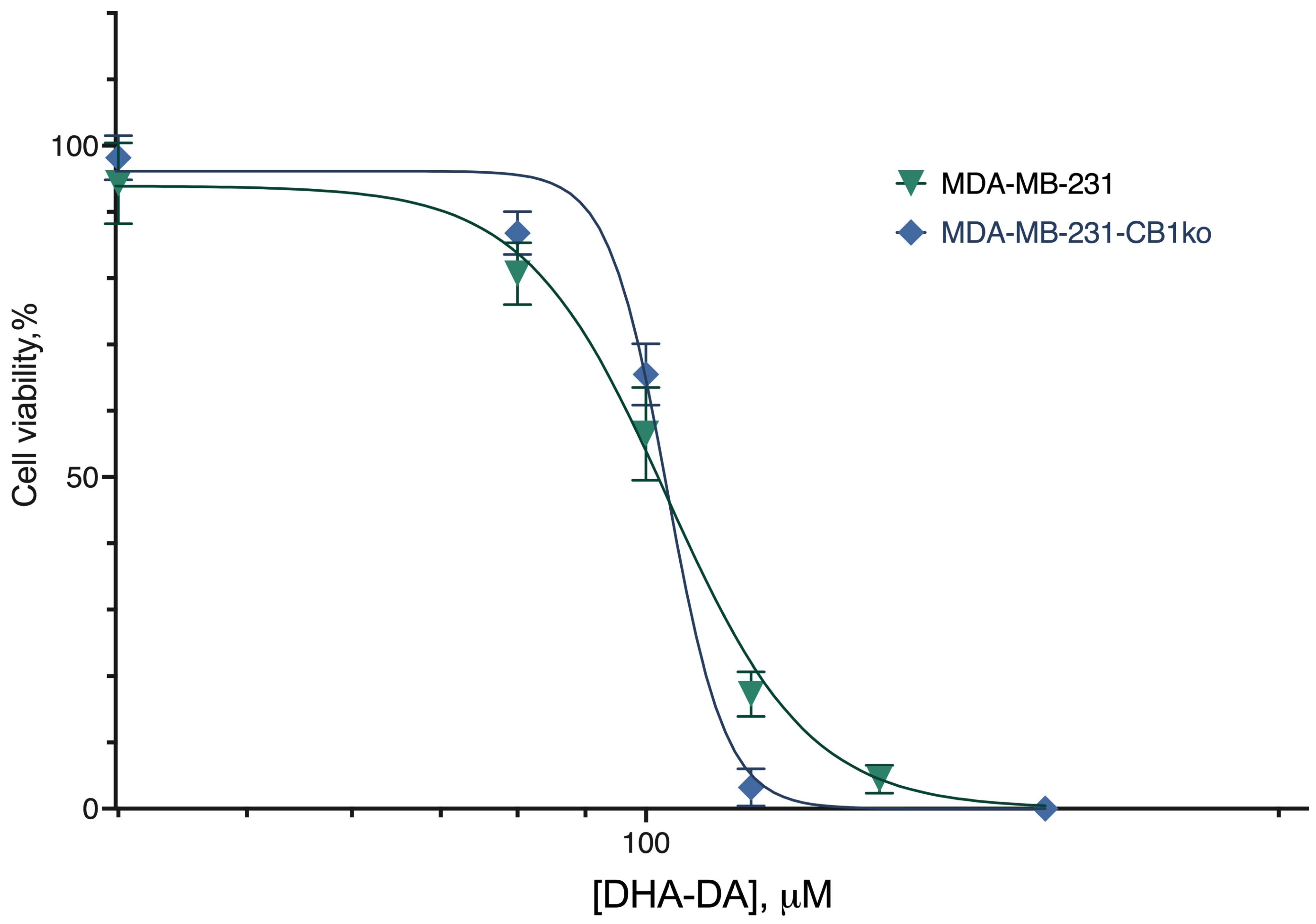
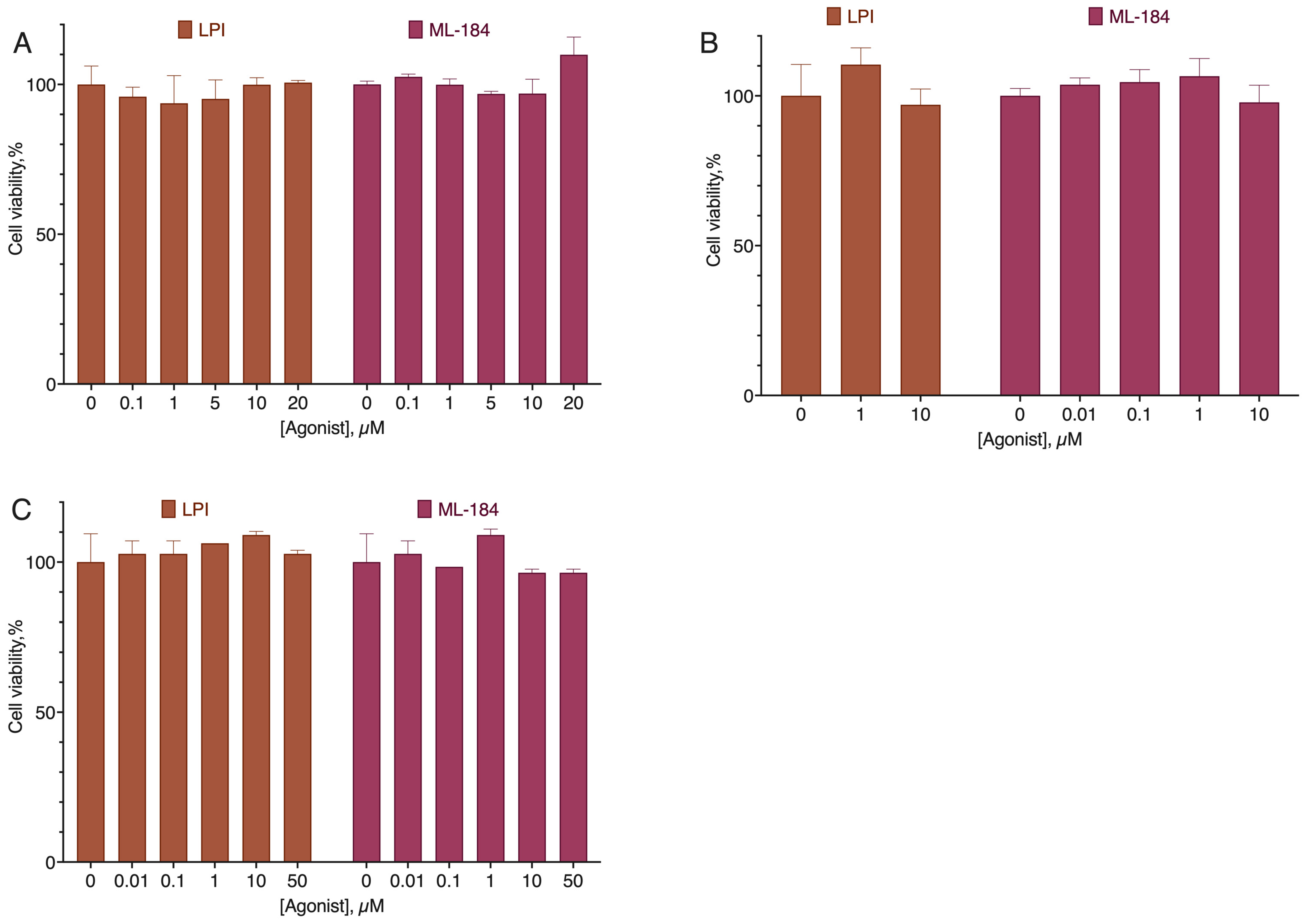
2.1. CB1-GPR55 Interaction
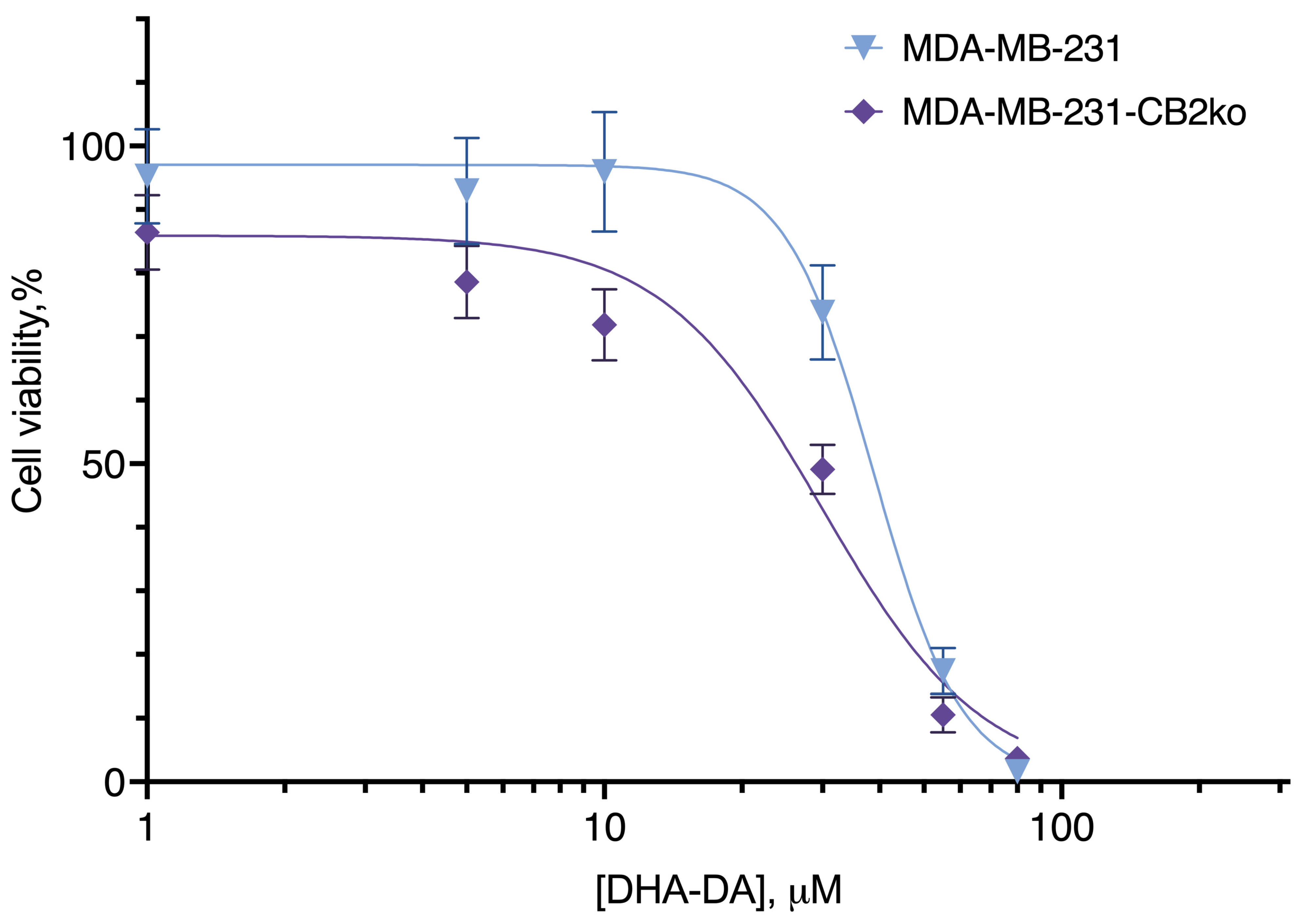
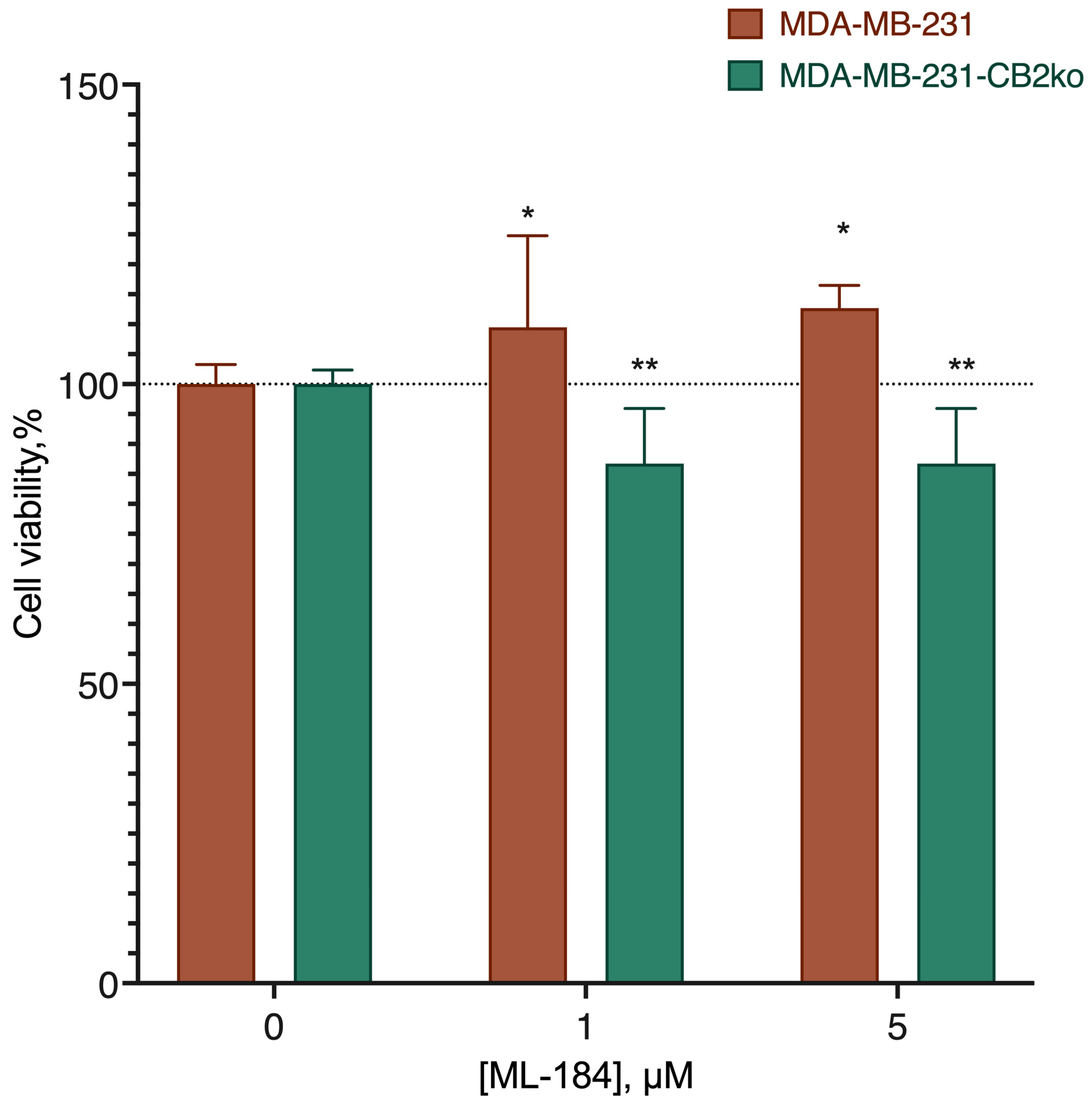
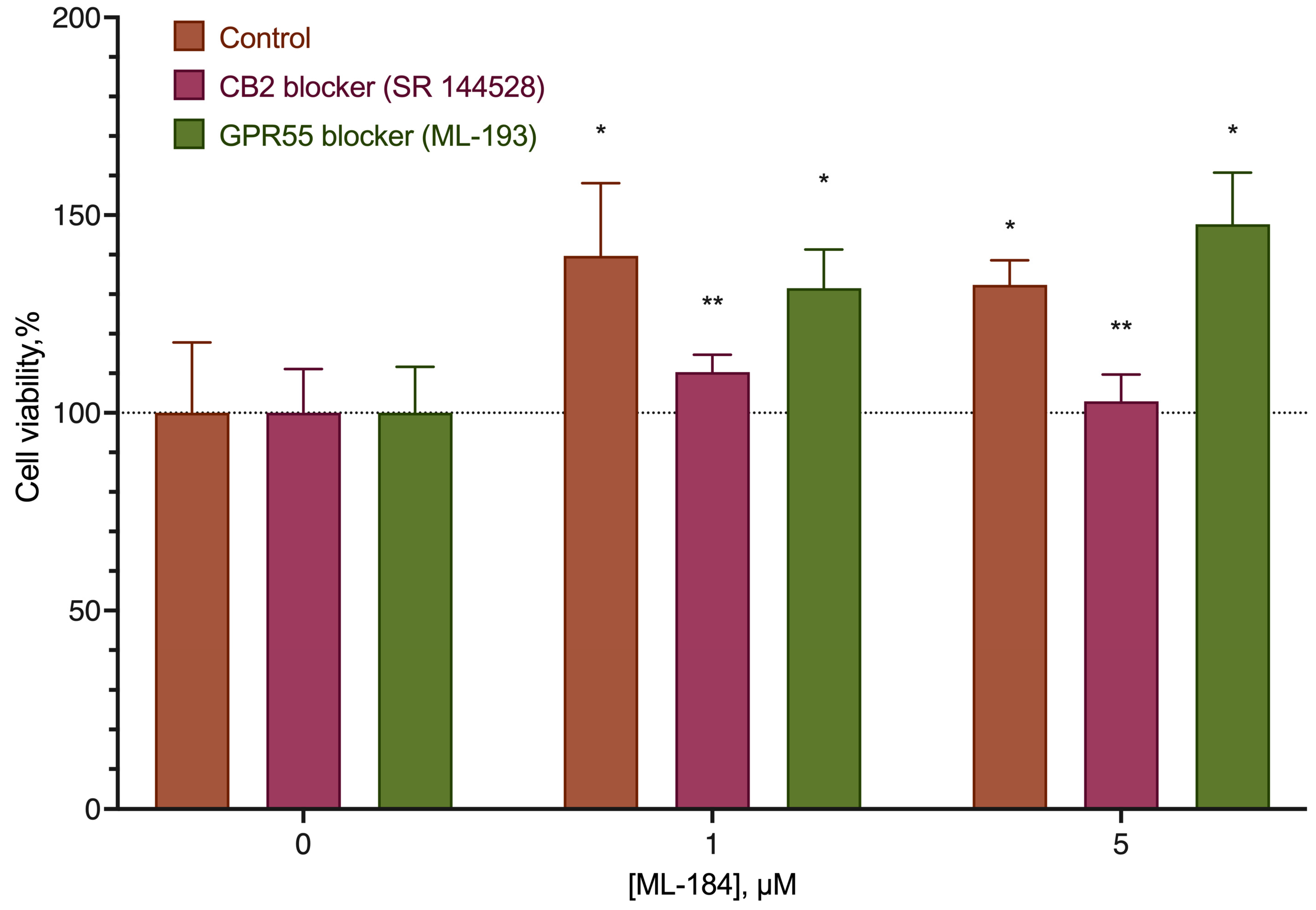
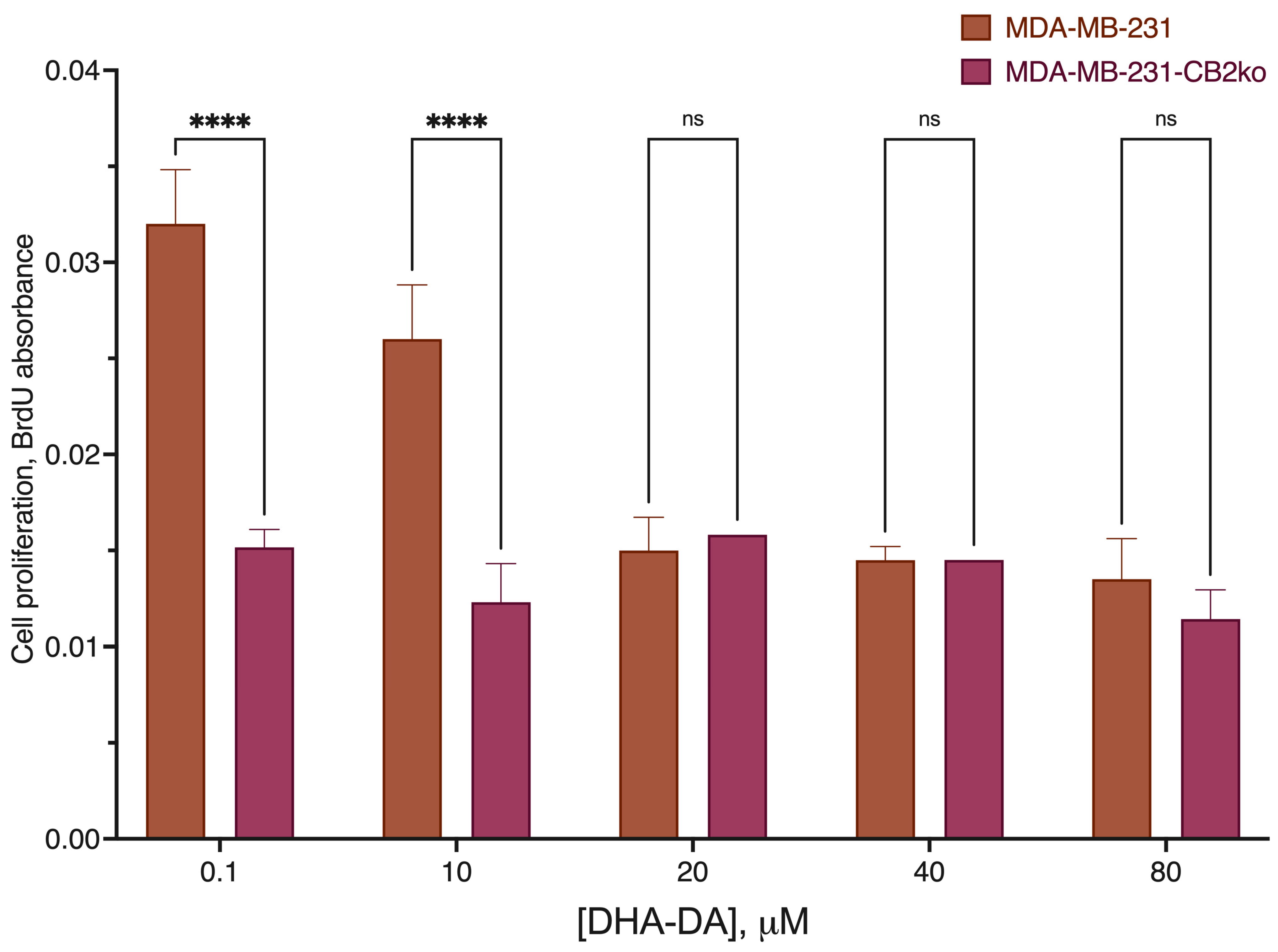
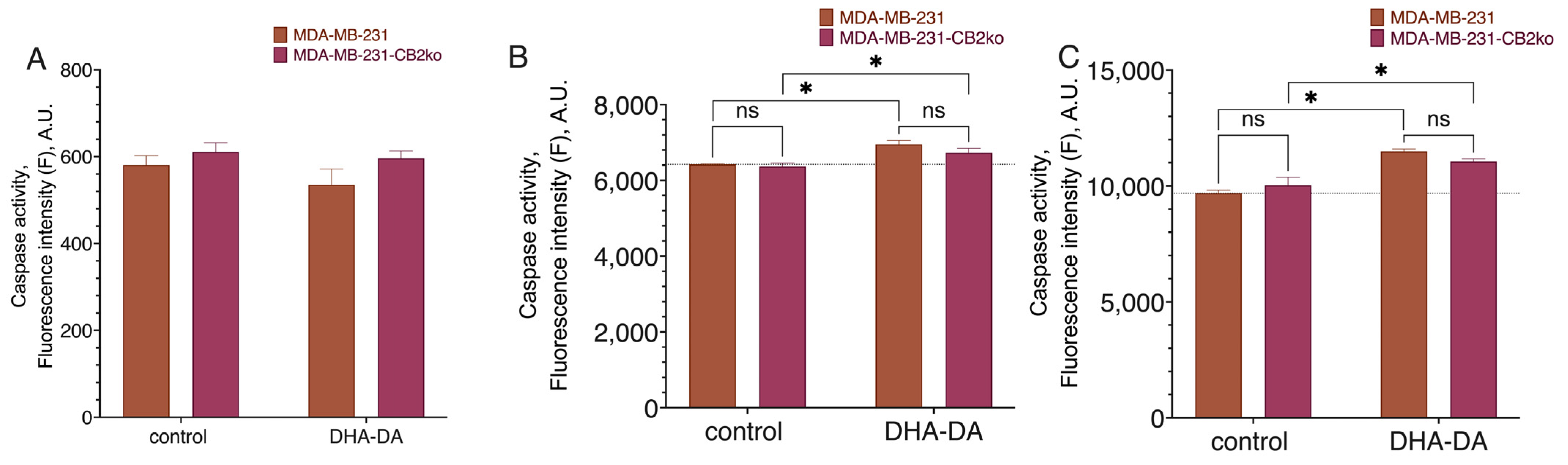
2.2. CB2-GPR55 Interaction
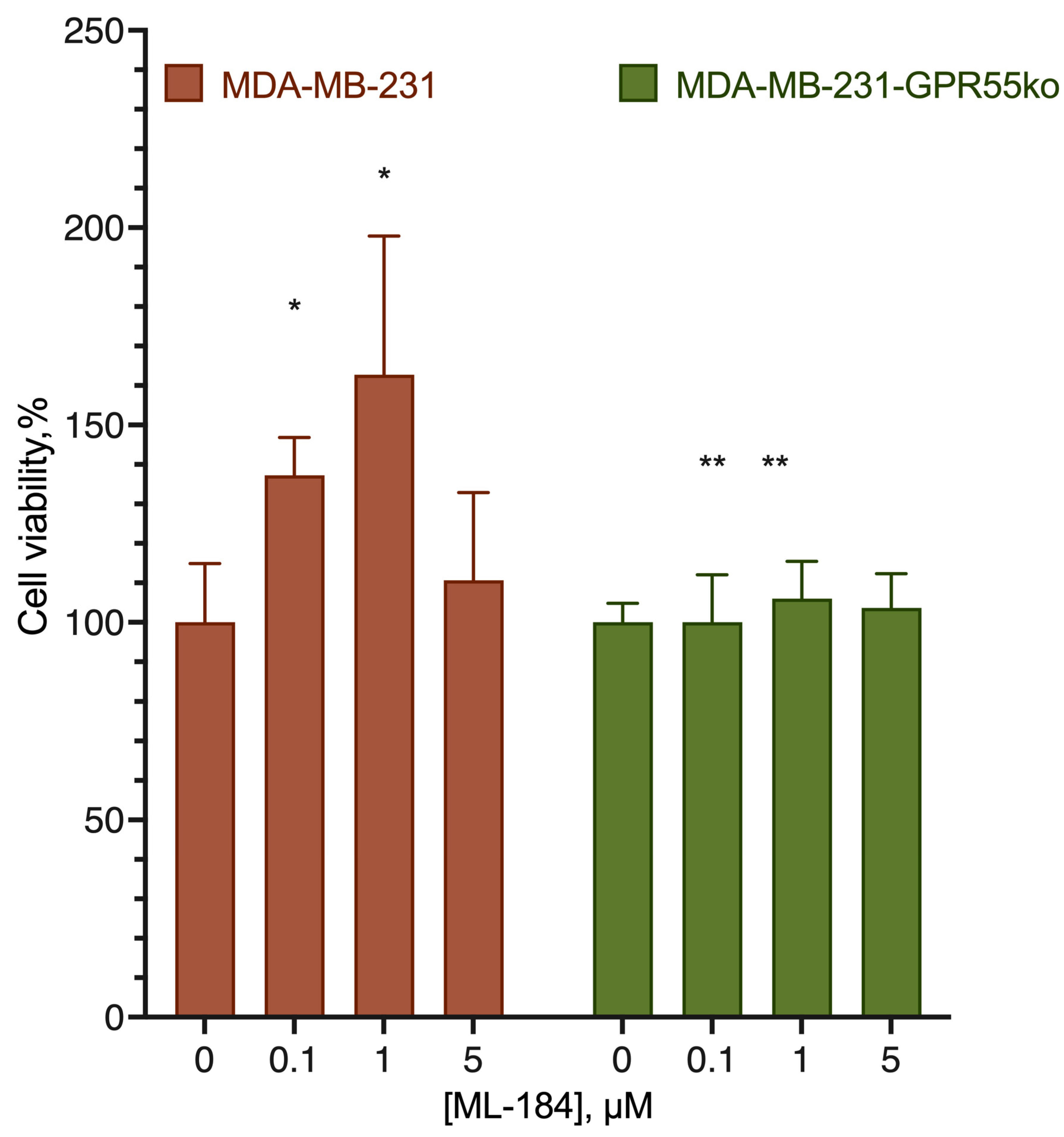
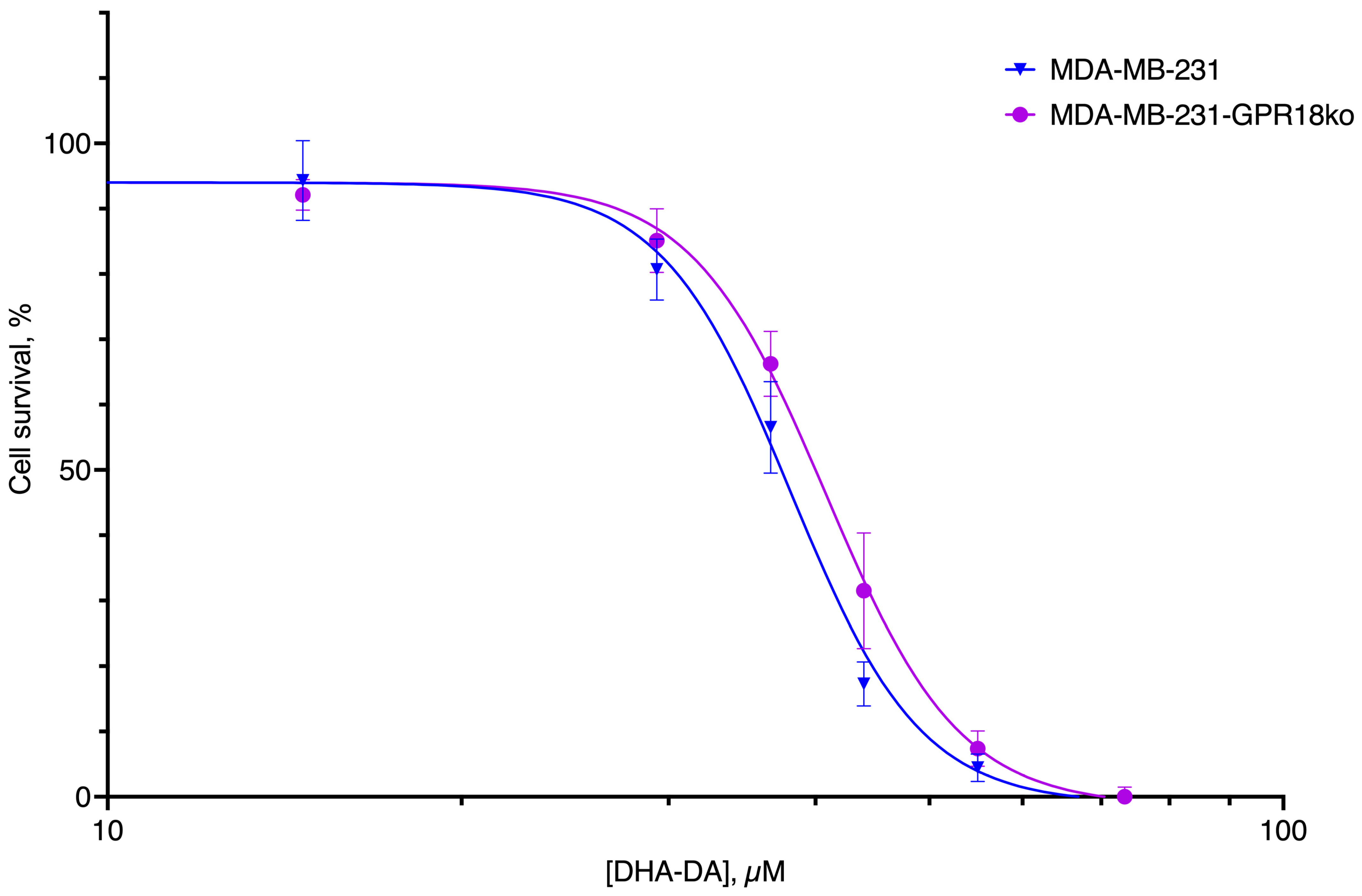
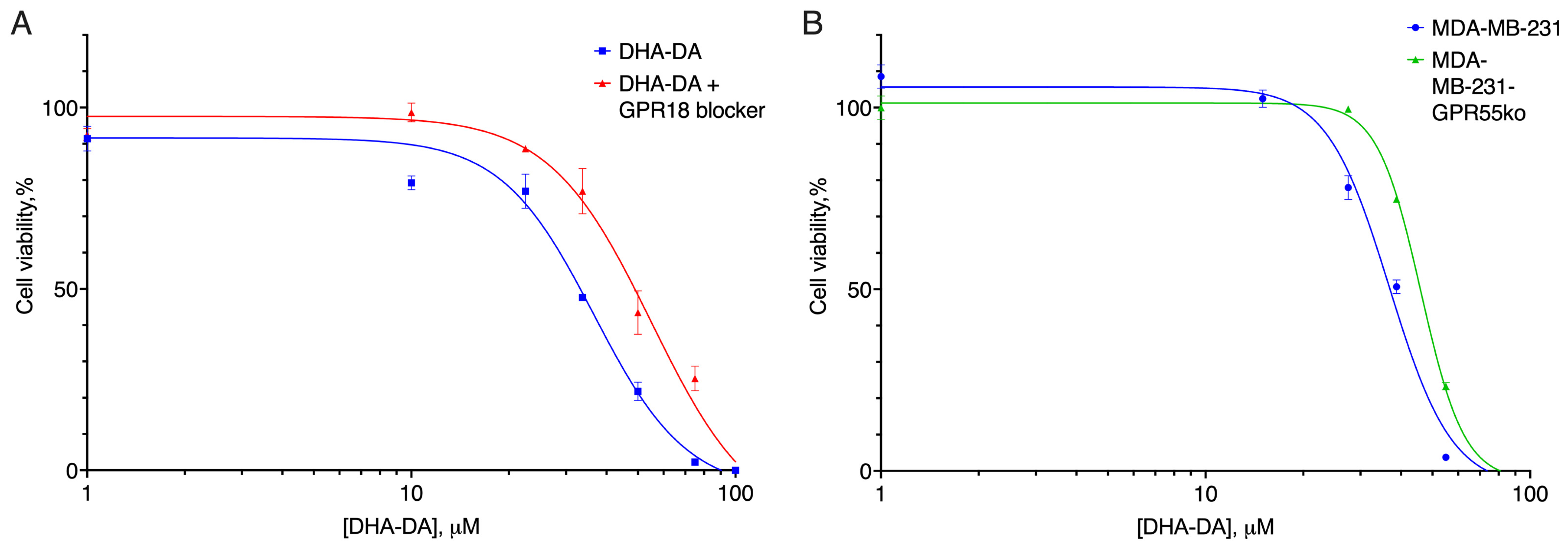
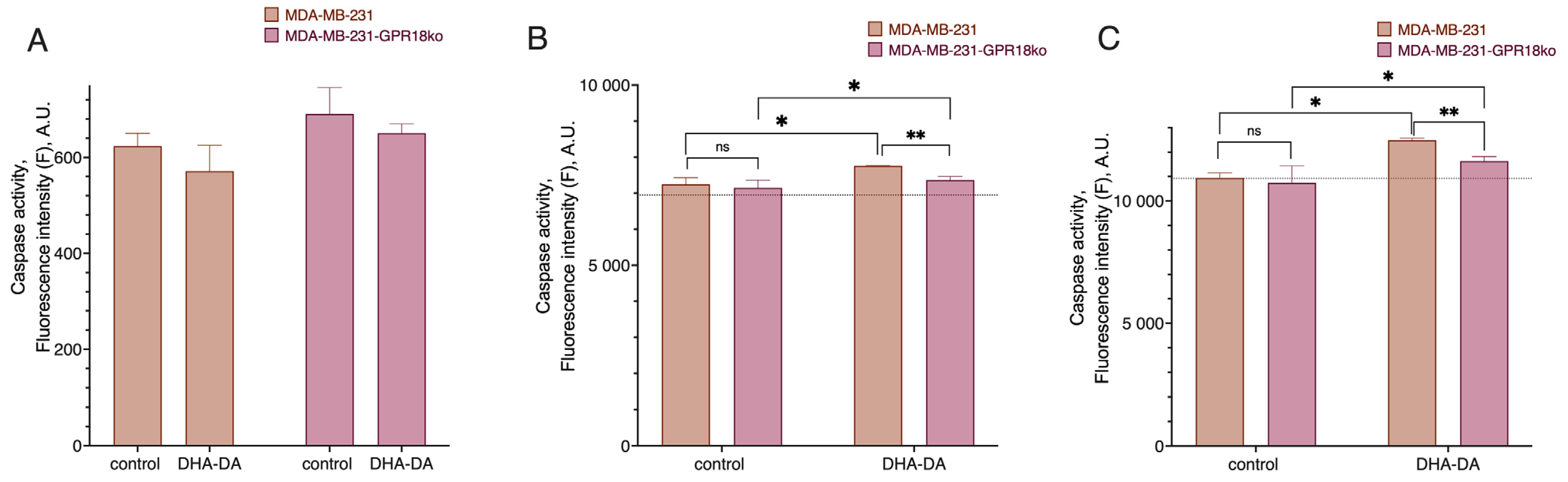
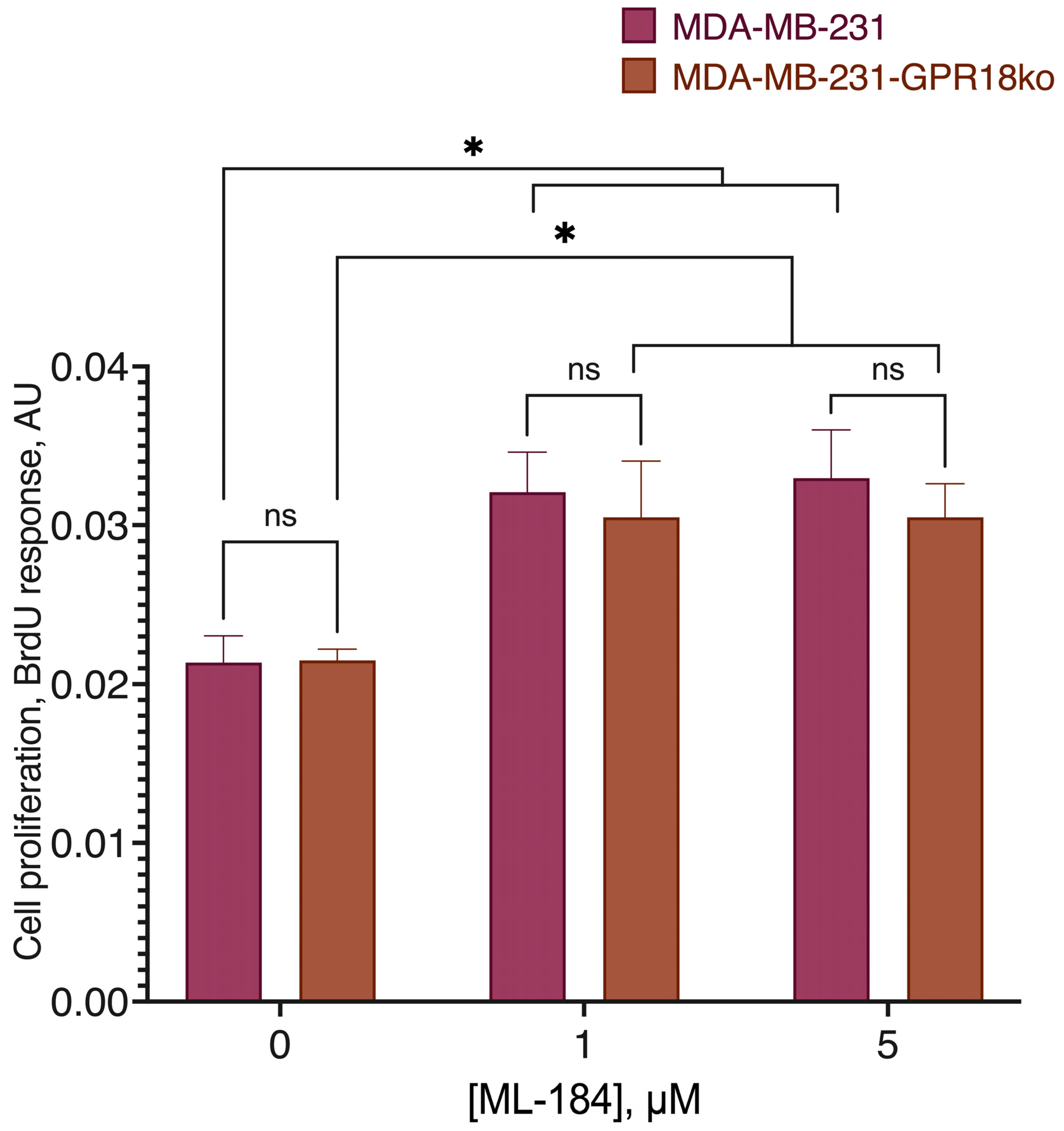
2.3. GPR18-GPR55 Interaction
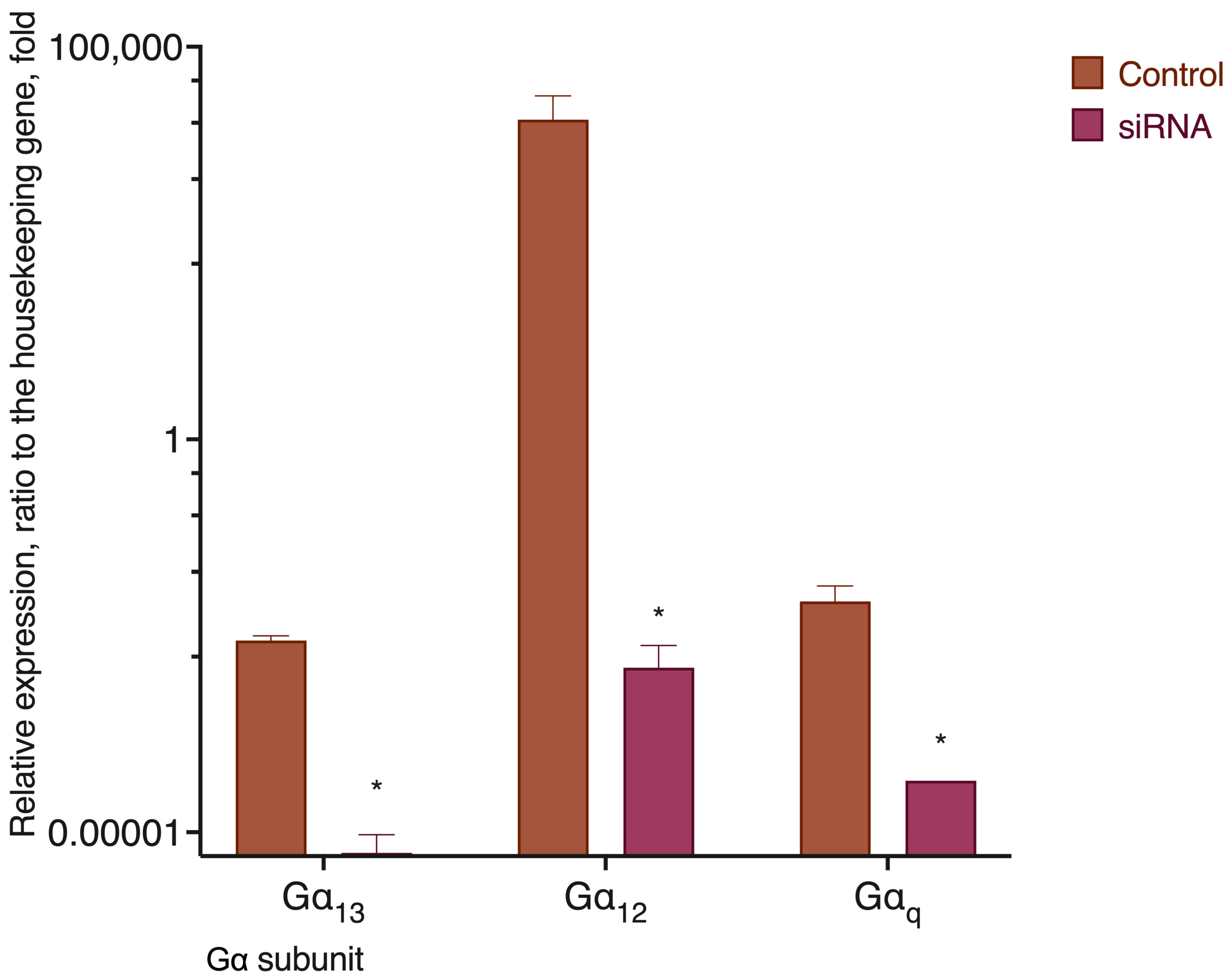
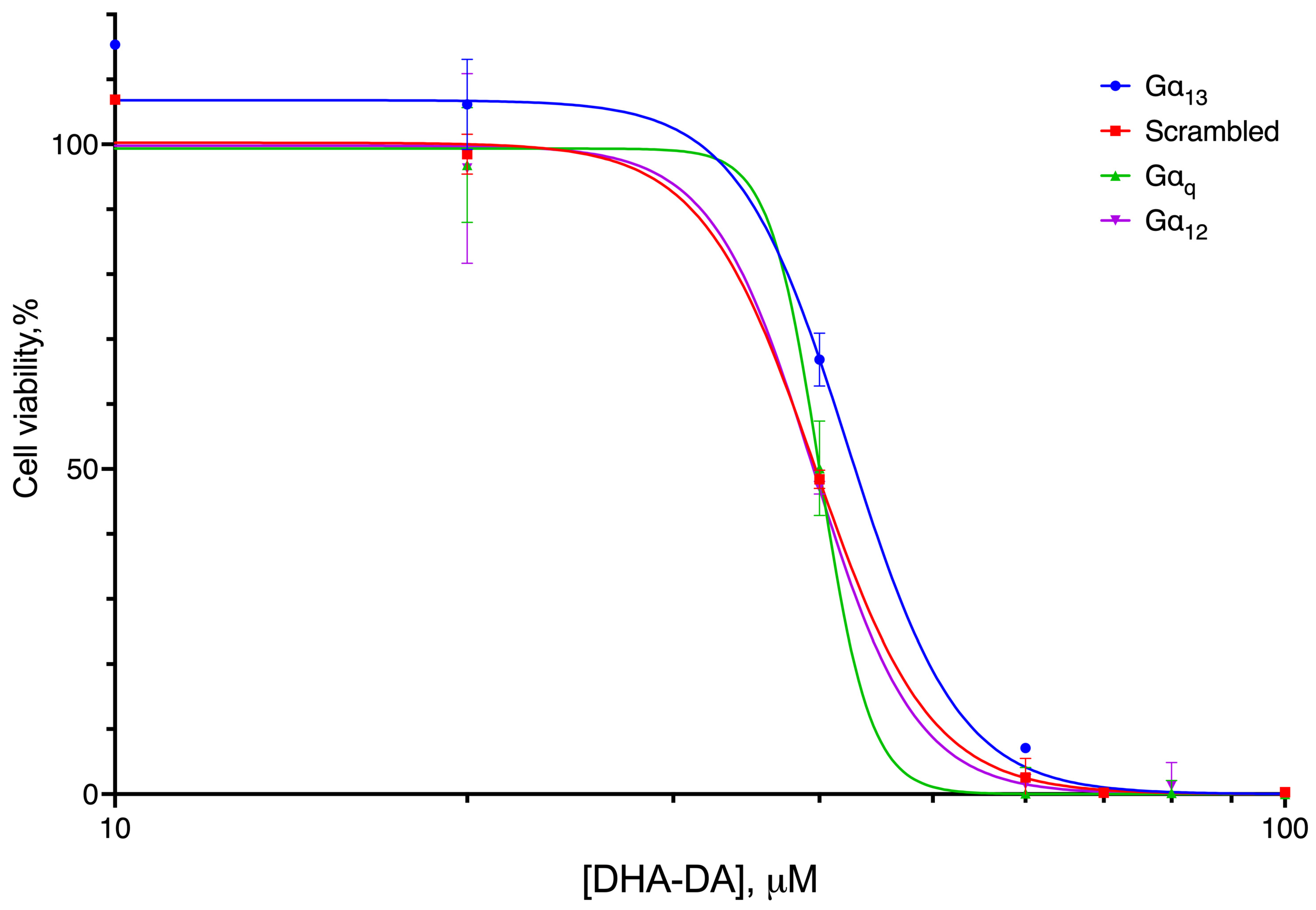
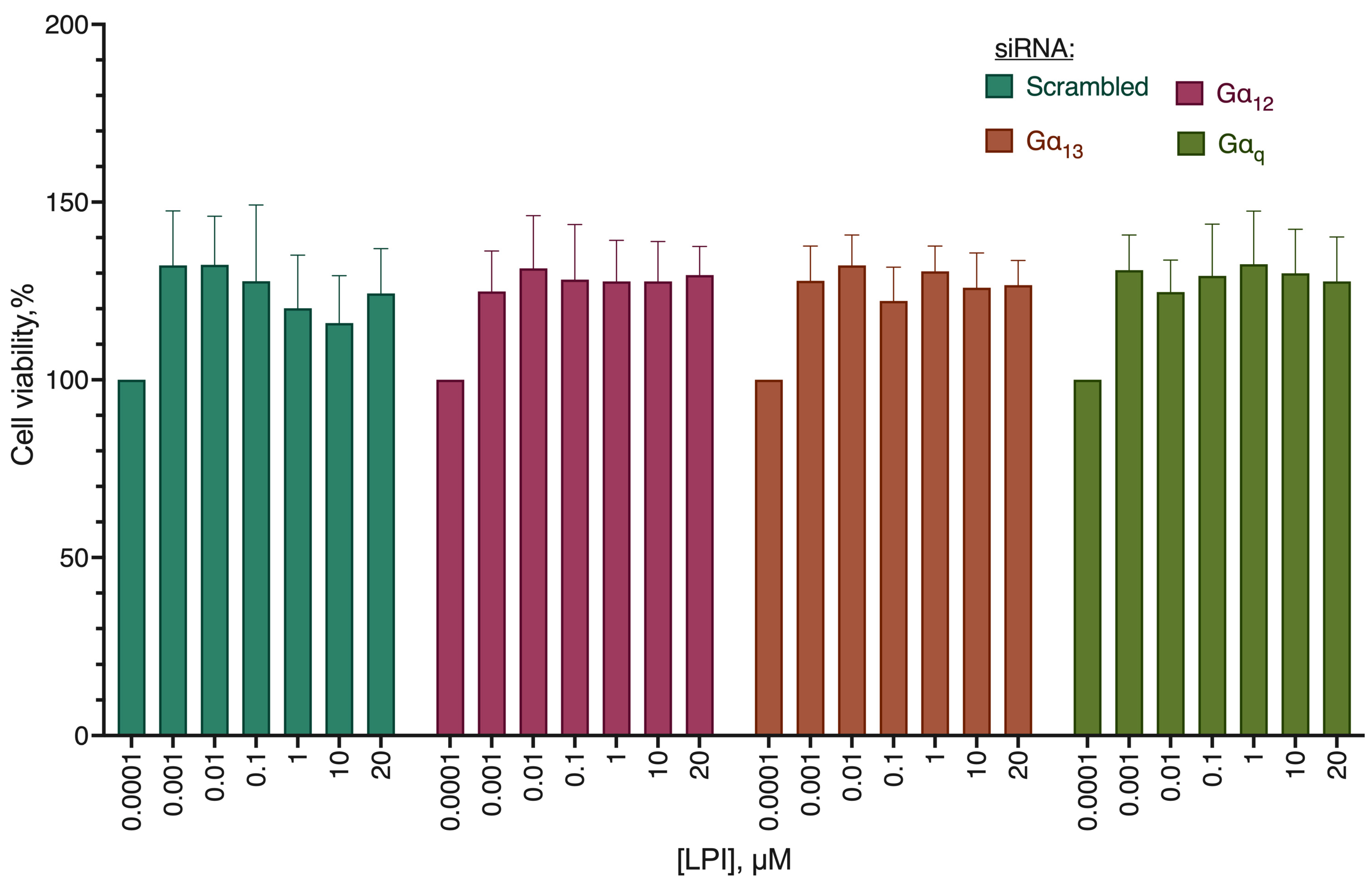
2.4. G Protein Knockdown Effect
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Chemical Synthesis
4.3. Cell Culture
4.4. siRNA Knockdown
4.5. CRISPR Knockdown
4.6. RT-qPCR
4.7. RNA Isolation and RT-PCR
4.8. Cytotoxicity and Proliferation Evaluation
4.9. Western Blot
4.10. BCA Protein Assay
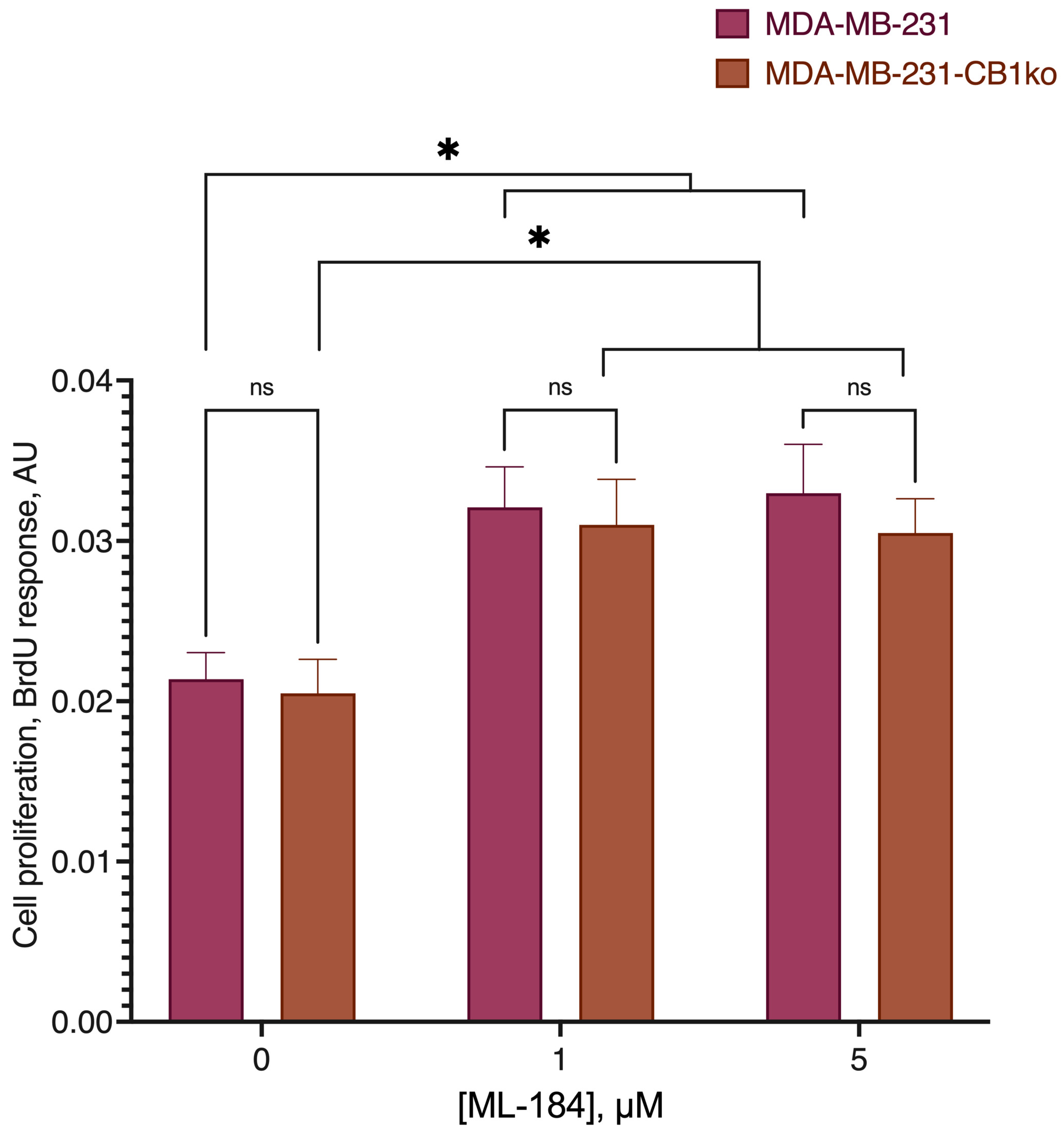
4.11. BrdU Proliferation Assay
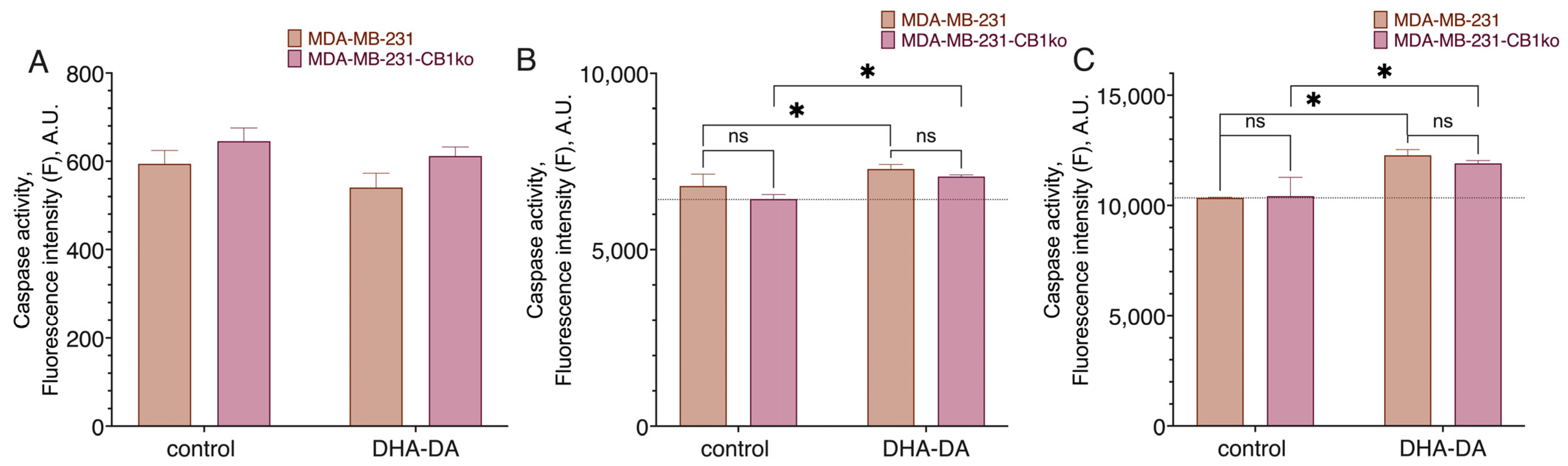
4.12. Caspase Activation Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DHA-DA | N-docosahexaenoyl dopamine |
| LPI | Lysophosphatidylinositol |
| ROS | Reactive oxygen species |
| BCIP | 5-Bromo-4-chloro-3-indolyl phosphate |
| NBT | Nitro blue tetrazolium |
| BCA | Bicinchoninic acid |
References
- Im, D.-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.B.; Kargl, J.; Andradas, C.; Brown, A.J.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent Developments in the Physiology and Pathology of the Lysophosphatidylinositol-Sensitive Receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Jagerovic, N. Advances towards the Discovery of GPR55 Ligands. Curr. Med. Chem. 2016, 23, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol. Sci. 2018, 39, 586–604. [Google Scholar] [CrossRef] [PubMed]
- Akimov, M.G.; Ashba, A.M.; Gretskaya, N.M.; Bezuglov, V.V. N-Acyl Dopamines Induce Apoptosis in PC12 Cell Line via the GPR55 Receptor Activation. Dokl. Biochem. Biophys. 2017, 474, 155–158. [Google Scholar] [CrossRef]
- Ashba, A.M.; Akimov, M.G.; Gretskaya, N.M.; Bezuglov, V.V. N-Acyl Dopamines Induce Cell Death in PC12 Cell Line via Induction of Nitric Oxide Generation and Oxidative Stress. Dokl. Biochem. Biophys. 2016, 467, 81–84. [Google Scholar] [CrossRef]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and Pharmacology of Endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef]
- Akimov, M.G.; Gretskaya, N.M.; Shevchenko, K.V.; Shevchenko, V.P.; Myasoedov, N.F.; Bobrov, M.Y.; Bezuglov, V.V. New Aspects of Biosynthesis and Metabolism of N-Acyldopamines in Rat Tissues. Russ. J. Bioorganic Chem. 2007, 33, 602–606. [Google Scholar] [CrossRef]
- Akimov, M.G.; Gamisonia, A.M.; Dudina, P.V.; Gretskaya, N.M.; Gaydaryova, A.A.; Kuznetsov, A.S.; Zinchenko, G.N.; Bezuglov, V.V. GPR55 Receptor Activation by the N-Acyl Dopamine Family Lipids Induces Apoptosis in Cancer Cells via the Nitric Oxide Synthase (NNOS) over-Stimulation. Int. J. Mol. Sci. 2021, 22, 622. [Google Scholar] [CrossRef]
- Tjalma, W.A.; Fiander, A.; Reich, O.; Powell, N.; Nowakowski, A.M.; Kirschner, B.; Koiss, R.; O’Leary, J.; Joura, E.A.; Rosenlund, M.; et al. Differences in Human Papillomavirus Type Distribution in High-Grade Cervical Intraepithelial Neoplasia and Invasive Cervical Cancer in Europe. Int. J. Cancer 2013, 132, 854–867. [Google Scholar] [CrossRef]
- Calvillo-Robledo, A.; Cervantes-Villagrana, R.D.; Morales, P.; Marichal-Cancino, B.A. The Oncogenic Lysophosphatidylinositol (LPI)/GPR55 Signaling. Life Sci. 2022, 301, 120596. [Google Scholar] [CrossRef] [PubMed]
- Kallendrusch, S.; Kremzow, S.; Nowicki, M.; Grabiec, U.; Winkelmann, R.; Benz, A.; Kraft, R.; Bechmann, I.; Dehghani, F.; Koch, M. The G Protein-Coupled Receptor 55 Ligand l-α-Lysophosphatidylinositol Exerts Microglia-Dependent Neuroprotection after Excitotoxic Lesion. Glia 2013, 61, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-Y.; Yu, R.; Li, W.; Liang, L.-F.; Han, Q.-Q.; Huang, H.-J.; Li, B.; Xu, S.-F.; Wu, G.-C.; Zhang, Y.-Q.; et al. The Neuroprotective Effects of GPR55 against Hippocampal Neuroinflammation and Impaired Adult Neurogenesis in CSDS Mice. Neurobiol. Dis. 2022, 169, 105743. [Google Scholar] [CrossRef] [PubMed]
- Cherif, H.; Argaw, A.; Cécyre, B.; Bouchard, A.; Gagnon, J.; Javadi, P.; Desgent, S.; Mackie, K.; Bouchard, J.-F. Role of GPR55 during Axon Growth and Target Innervation. eNeuro 2015, 2, ENEURO.0011-15.2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.A. The Enigmatic Pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Ren, G.; Shi, Y. The Putative Cannabinoid Receptor GPR55 Promotes Cancer Cell Proliferation. Oncogene 2011, 30, 139–141. [Google Scholar] [CrossRef]
- Zhou, X.-L.; Guo, X.; Song, Y.-P.; Zhu, C.-Y.; Zou, W. The LPI/GPR55 Axis Enhances Human Breast Cancer Cell Migration via HBXIP and p-MLC Signaling. Acta Pharmacol. Sin. 2018, 39, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.-J.; DeMorrow, S. Anandamide Exerts Its Antiproliferative Actions on Cholangiocarcinoma by Activation of the GPR55 Receptor. Lab. Investig. 2011, 91, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, C.M.; Balenga, N.A.; Schröder, R.; Kargl, J.K.; Platzer, W.; Martini, L.; Arthur, S.; Penman, J.; Whistler, J.L.; Kostenis, E.; et al. GPR55 Ligands Promote Receptor Coupling to Multiple Signalling Pathways. Br. J. Pharmacol. 2010, 160, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The Endocannabinoids Anandamide and Virodhamine Modulate the Activity of the Candidate Cannabinoid Receptor GPR55. J. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The Cannabinoid Receptor CB1 Modulates the Signaling Properties of the Lysophosphatidylinositol Receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Zhan, X.; Yu, X.; Jia, M.; Zhang, Y.; Yao, J.; Hu, X.; Bao, Z. Inhibiting CB1 Receptors Improves Lipogenesis in an in Vitro Non-Alcoholic Fatty Liver Disease Model. Lipids Health Dis. 2014, 13, 173. [Google Scholar] [CrossRef] [Green Version]
- Fogli, S.; Nieri, P.; Chicca, A.; Adinolfi, B.; Mariotti, V.; Iacopetti, P.; Breschi, M.C.; Pellegrini, S. Cannabinoid Derivatives Induce Cell Death in Pancreatic MIA PaCa-2 Cells via a Receptor-Independent Mechanism. FEBS Lett. 2006, 580, 1733–1739. [Google Scholar] [CrossRef] [Green Version]
- Hartel, M.; di Mola, F.F.; Selvaggi, F.; Mascetta, G.; Wente, M.N.; Felix, K.; Giese, N.A.; Hinz, U.; Di Sebastiano, P.; Büchler, M.W.; et al. Vanilloids in Pancreatic Cancer: Potential for Chemotherapy and Pain Management. Gut 2006, 55, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Paul, R.K.; Wnorowski, A.; Gonzalez-Mariscal, I.; Nayak, S.K.; Pajak, K.; Moaddel, R.; Indig, F.E.; Bernier, M.; Wainer, I.W. (R,R′)-4′-Methoxy-1-Naphthylfenoterol Targets GPR55-Mediated Ligand Internalization and Impairs Cancer Cell Motility. Biochem. Pharmacol. 2014, 87, 547–561. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pinilla, E.; Reyes-Resina, I.; Oñatibia-Astibia, A.; Zamarbide, M.; Ricobaraza, A.; Navarro, G.; Moreno, E.; Dopeso-Reyes, I.G.; Sierra, S.; Rico, A.J.; et al. CB1 and GPR55 Receptors Are Co-Expressed and Form Heteromers in Rat and Monkey Striatum. Exp. Neurol. 2014, 261, 44–52. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef] [PubMed]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and Cannabinoid CB2 Receptors Modulates Signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomko, A.; O’Leary, L.; Trask, H.; Achenbach, J.C.; Hall, S.R.; Goralski, K.B.; Ellis, L.D.; Dupré, D.J. Antitumor Activity of Abnormal Cannabidiol and Its Analog O-1602 in Taxol-Resistant Preclinical Models of Breast Cancer. Front. Pharmacol. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Reyes-Resina, I.; Navarro, G.; Aguinaga, D.; Canela, E.I.; Schoeder, C.T.; Załuski, M.; Kieć-Kononowicz, K.; Saura, C.A.; Müller, C.E.; Franco, R. Molecular and Functional Interaction between GPR18 and Cannabinoid CB2 G-Protein-Coupled Receptors. Relevance in Neurodegenerative Diseases. Biochem. Pharmacol. 2018, 157, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Yang, D.; Zhuang, Y.; Croll, T.I.; Cai, X.; Dai, A.; He, X.; Duan, J.; Yin, W.; Ye, C.; et al. Ligand Recognition and G-Protein Coupling Selectivity of Cholecystokinin A Receptor. Nat. Chem. Biol. 2021, 17, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- DeVree, B.T.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.J.; Edwald, E.; Fung, J.-J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric Coupling from G Protein to the Agonist-Binding Pocket in GPCRs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Bezuglov, V.; Bobrov, M.; Gretskaya, N.; Gonchar, A.; Zinchenko, G.; Melck, D.; Bisogno, T.; Di Marzo, V.; Kuklev, D.; Rossi, J.C.; et al. Synthesis and Biological Evaluation of Novel Amides of Polyunsaturated Fatty Acids with Dopamine. Bioorg. Med. Chem. Lett. 2001, 11, 447–449. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line Variant | |
|---|---|
| Source Cell Line | CB1ko |
| EC50, µM, mean (95% CI) | |
| 38.7 (34.62–43.0) | 38.8 (33.1–41.0) |
| Cell Line Variant | |
|---|---|
| MDA-MB-231 | MDA-MB-231-CB2ko |
| EC50, µM, mean (95% C.I.) | |
| 40.58 (38.13 to 43.12) | 33.57 * (31.35 to 35.88) |
| Cell Line Variant | |
|---|---|
| MDA-MB-231 | MDA-MB-231-GPR18ko |
| EC50 (95% CI), µM | |
| 38.7 (34.62–43.0) | 45.8 * (42.4–49.5) |
| Cell Line Variant | ||
|---|---|---|
| MDA-MB-231 | MDA-MB-231+PSB CB5 | MDA-MB-231-GPR55ko |
| EC50 (95% CI), µM | ||
| 36.83 (34.56 to 39.18) | 55.03 * (45.03 to 50.6) | 46.06 * (44.75 to 47.42) |
| Knockdown Variant | |||
|---|---|---|---|
| Scrambled | Gαq | Gα12 | Gα13 |
| EC50 (95% CI), µM | |||
| 39.69 (39.12 to 40.19) | 40.03 (39.41 to 41.05) | 39.55 (38.06 to 40.55) | 42.33 * (40.06 to 45.60) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akimov, M.G.; Gretskaya, N.M.; Dudina, P.V.; Sherstyanykh, G.D.; Zinchenko, G.N.; Serova, O.V.; Degtyaryova, K.O.; Deyev, I.E.; Bezuglov, V.V. The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells. Int. J. Mol. Sci. 2023, 24, 5524. https://doi.org/10.3390/ijms24065524
Akimov MG, Gretskaya NM, Dudina PV, Sherstyanykh GD, Zinchenko GN, Serova OV, Degtyaryova KO, Deyev IE, Bezuglov VV. The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells. International Journal of Molecular Sciences. 2023; 24(6):5524. https://doi.org/10.3390/ijms24065524
Chicago/Turabian StyleAkimov, Mikhail G., Natalia M. Gretskaya, Polina V. Dudina, Galina D. Sherstyanykh, Galina N. Zinchenko, Oksana V. Serova, Ksenia O. Degtyaryova, Igor E. Deyev, and Vladimir V. Bezuglov. 2023. "The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells" International Journal of Molecular Sciences 24, no. 6: 5524. https://doi.org/10.3390/ijms24065524
APA StyleAkimov, M. G., Gretskaya, N. M., Dudina, P. V., Sherstyanykh, G. D., Zinchenko, G. N., Serova, O. V., Degtyaryova, K. O., Deyev, I. E., & Bezuglov, V. V. (2023). The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells. International Journal of Molecular Sciences, 24(6), 5524. https://doi.org/10.3390/ijms24065524