
Valproic Acid Inhibits Progressive Hereditary Hearing Loss in a KCNQ4 Variant Model through HDAC1 Suppression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
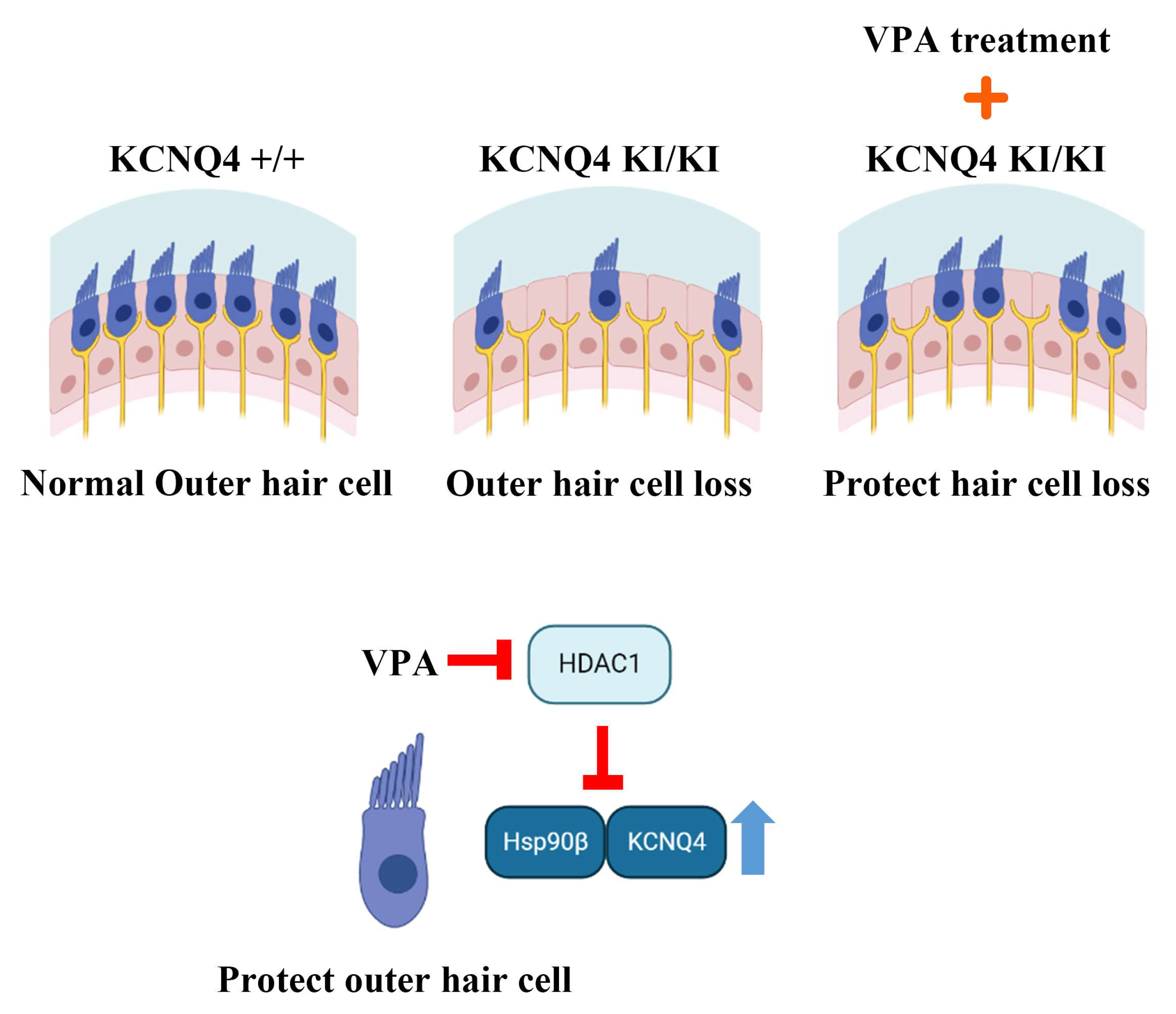
Abstract
:1. Introduction
2. Results
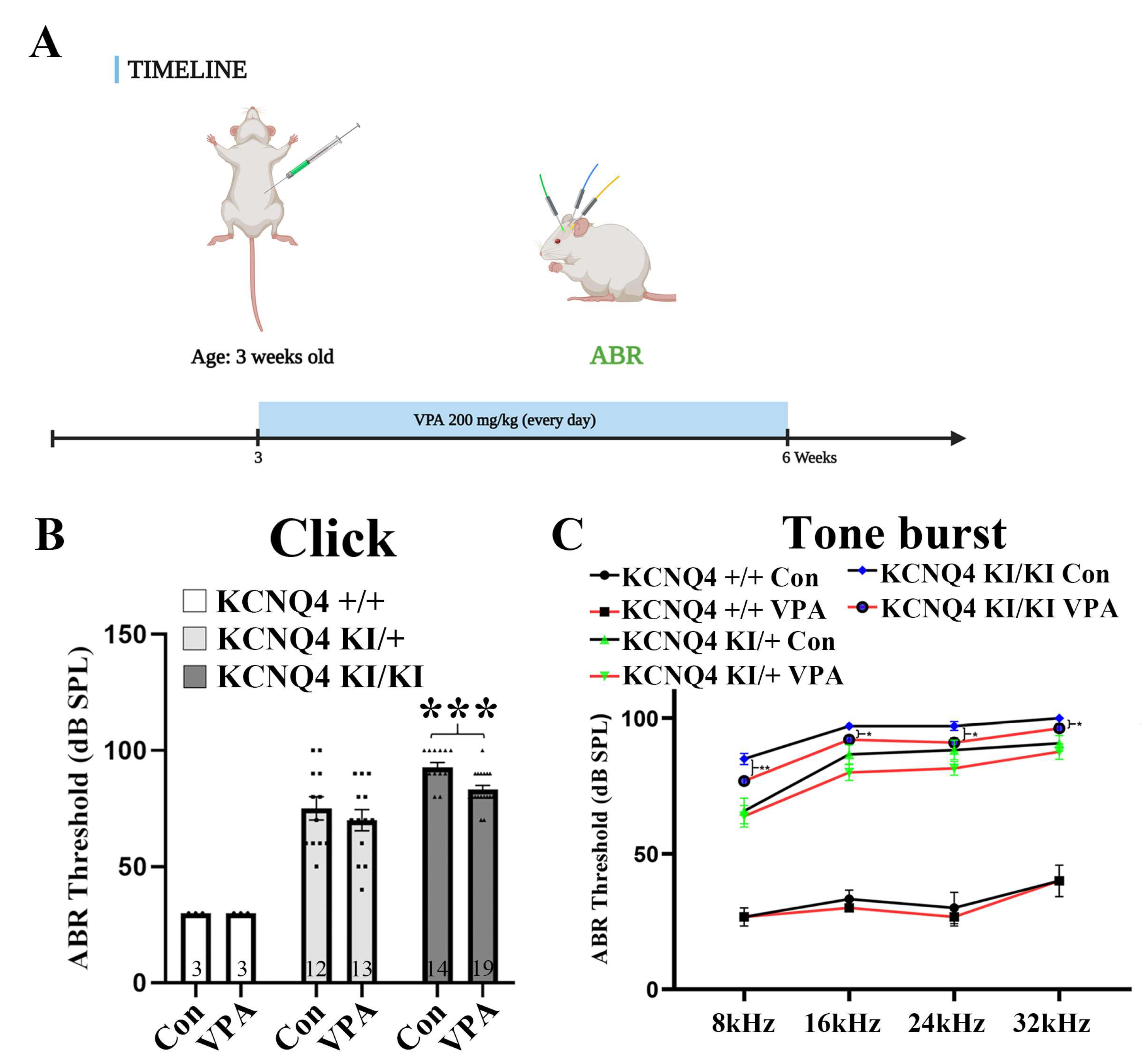
2.1. Valproic Acid (VPA) Inhibits Progressive Hearing Deterioration in a KCNQ4 p.W276S Variant Mouse Model
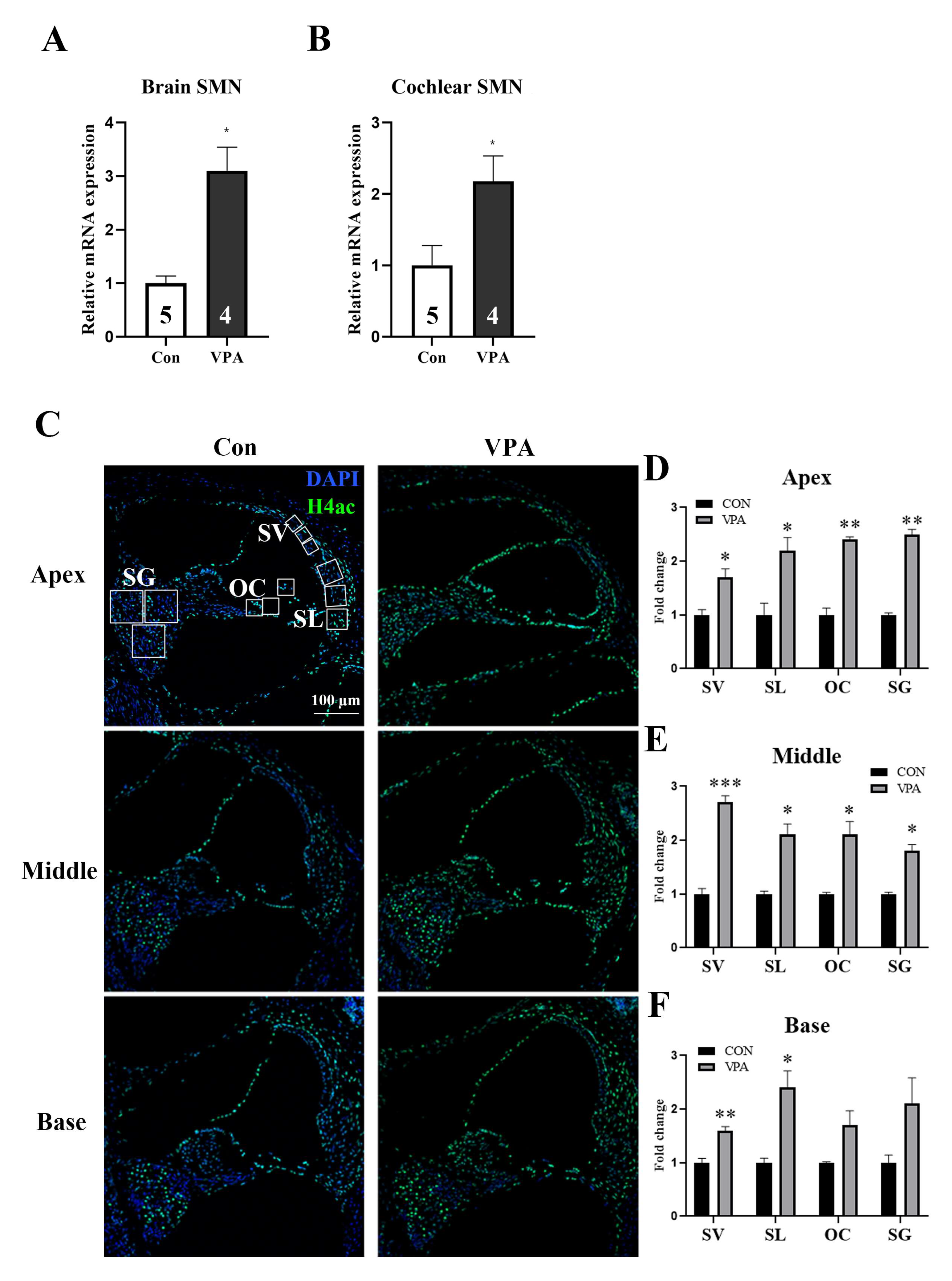
2.2. Systemic Injection of VPA Directly Affects the Cochlea
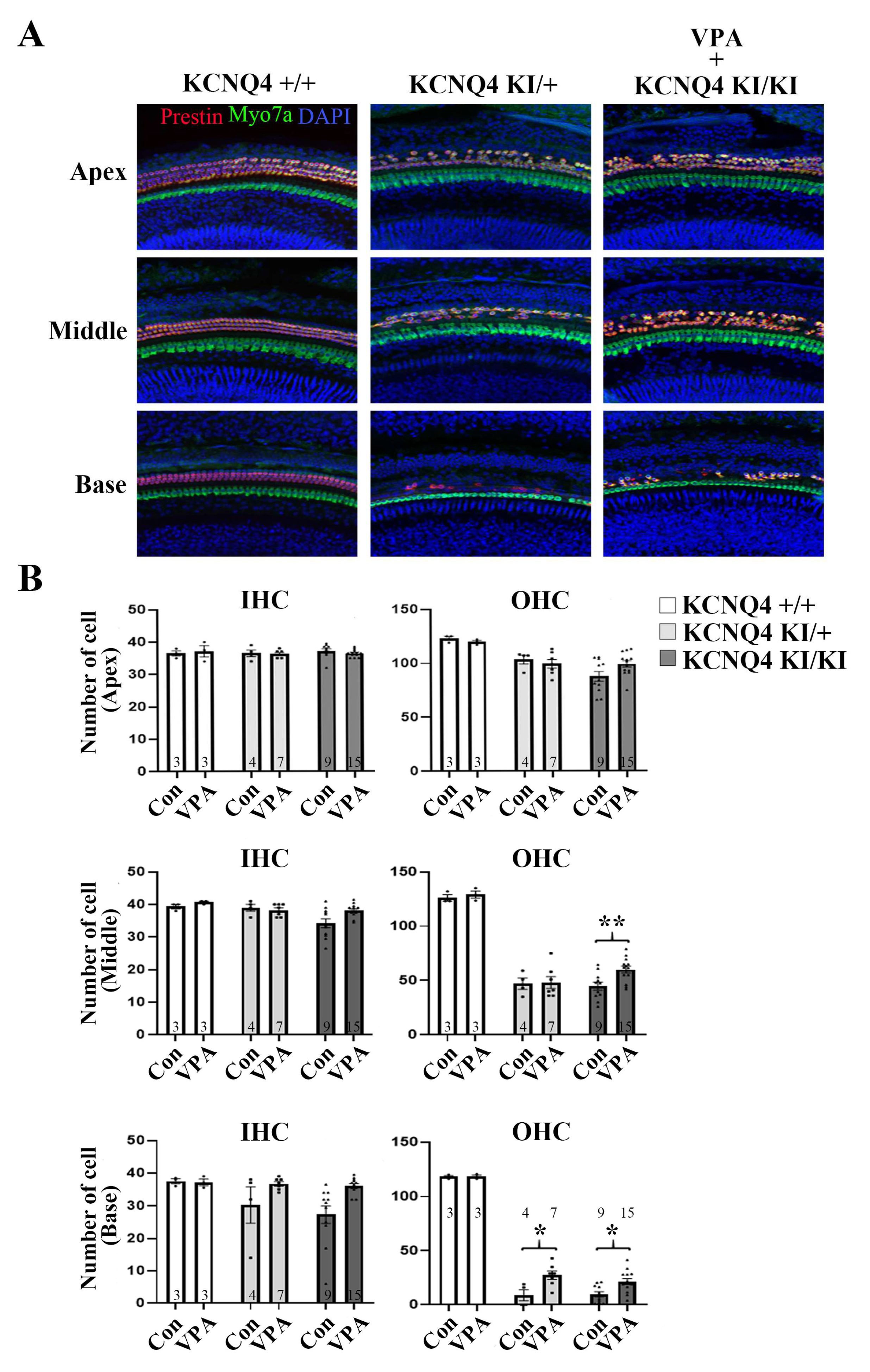
2.3. VPA Rescues the OHC from Cell Death in the KCNQ4 p.W276S Variant
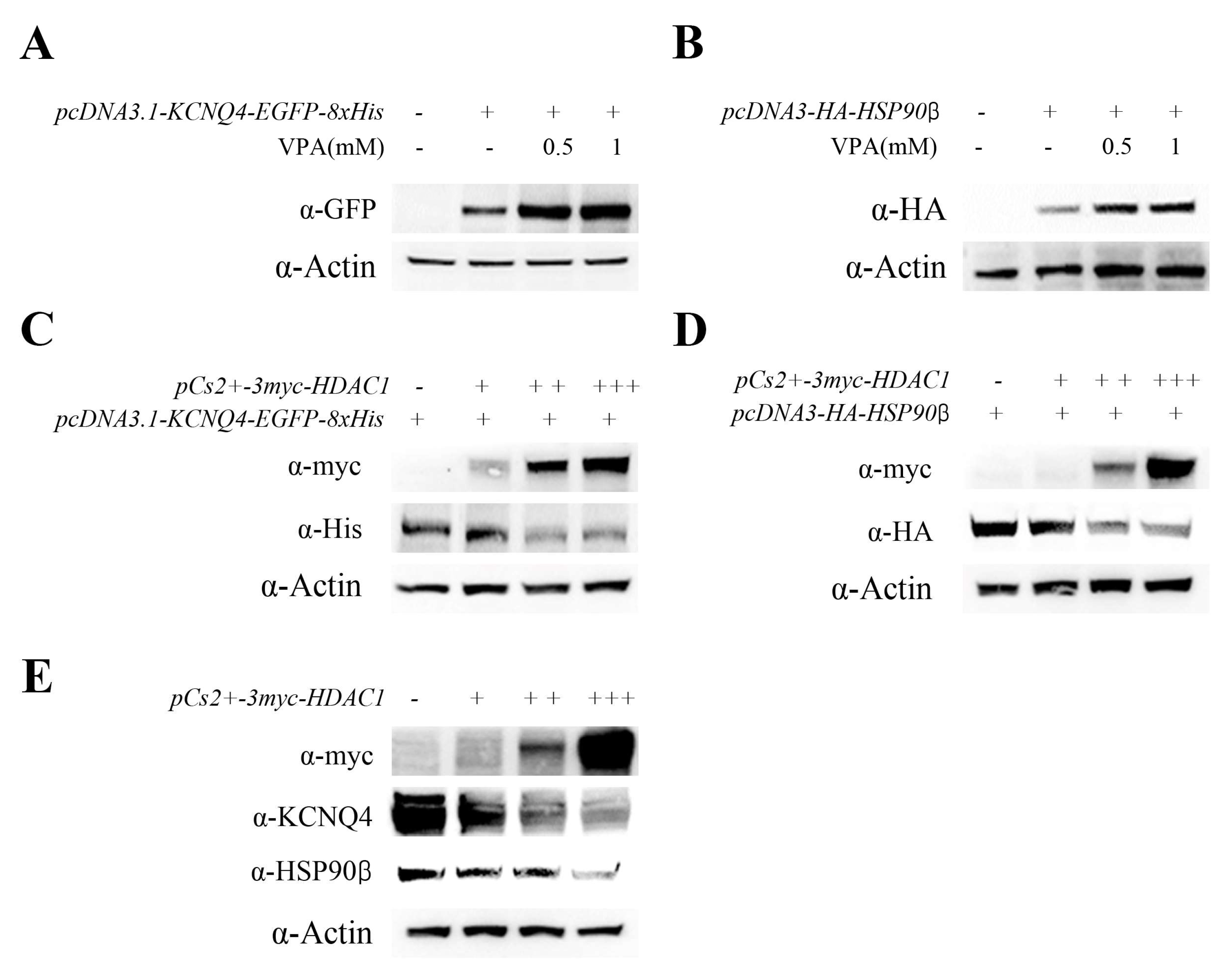
2.4. VPA Upregulated KCNQ4 Expression by Inhibiting HDAC1 Activity
2.5. VPA-Induced HSP90β Expression and HSP90β–KCNQ4 Interaction
3. Discussion
4. Materials and Methods
4.1. VPA Treatment and Generation of Hearing Loss Animals
4.2. Auditory Brainstem Response for an Animal Hearing Evaluation
4.3. Expression Constructs
4.4. Immunohistochemistry for OHCs
4.5. Counting of Hair Cells
4.6. Antibodies
4.7. Protein Preparation
4.8. Co-Immunoprecipitation
4.9. In Vitro HEK293T Culture and Transfection
4.10. RNA Isolation and Real-Time Polymerase Chain Reaction
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Marazita, M.L.; Ploughman, L.M.; Rawlings, B.; Remington, E.; Arnos, K.S.; Nance, W.E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 1993, 46, 486–491. [Google Scholar] [CrossRef]
- Sharma, N.; Kumari, D.; Panigrahi, I.; Khetarpal, P. A systematic review of the monogenic causes of Non-Syndromic Hearing Loss (NSHL) and discussion of Current Diagnosis and Treatment options. Clin. Genet. 2023, 103, 16–34. [Google Scholar] [CrossRef]
- Sogaard, R.; Ljungstrom, T.; Pedersen, K.A.; Olesen, S.P.; Jensen, B.S. KCNQ4 channels expressed in mammalian cells: Functional characteristics and pharmacology. Am. J. Physiol. Cell Physiol. 2001, 280, C859–C866. [Google Scholar] [CrossRef] [Green Version]
- Kharkovets, T.; Hardelin, J.P.; Safieddine, S.; Schweizer, M.; El-Amraoui, A.; Petit, C.; Jentsch, T.J. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4333–4338. [Google Scholar] [CrossRef] [Green Version]
- Boettger, T.; Hubner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef]
- Kharkovets, T.; Dedek, K.; Maier, H.; Schweizer, M.; Khimich, D.; Nouvian, R.; Vardanyan, V.; Leuwer, R.; Moser, T.; Jentsch, T.J. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006, 25, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Choi, H.B.; Koh, Y.I.; Rim, J.H.; Choi, H.J.; Kim, S.H.; Lee, J.H.; An, J.; Kim, A.; Lee, J.S.; et al. Whole-exome sequencing identifies two novel mutations in KCNQ4 in individuals with nonsyndromic hearing loss. Sci. Rep. 2018, 8, 16659. [Google Scholar] [CrossRef]
- Coucke, P.J.; Van Hauwe, P.; Kelley, P.M.; Kunst, H.; Schatteman, I.; Van Velzen, D.; Meyers, J.; Ensink, R.J.; Verstreken, M.; Declau, F.; et al. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Mol. Genet. 1999, 8, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y. Pathophysiology of age-related hearing loss (peripheral and central). Korean J. Audiol. 2013, 17, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Chen, D.; Xu, M.; Wu, B.; Chen, L. Histone deacetylases in hearing loss: Current perspectives for therapy. J. Otol. 2017, 12, 47–54. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Chen, X.; Zhang, P.Z.; Shi, Z.T.; Wen, L.T.; Qiu, J.H.; Chen, F.Q. Histone deacetylase inhibitor sodium butyrate attenuates gentamicin-induced hearing loss in vivo. Am. J. Otolaryngol. 2015, 36, 242–248. [Google Scholar] [CrossRef]
- Gurvich, N.; Tsygankova, O.M.; Meinkoth, J.L.; Klein, P.S. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004, 64, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, H.Y.; Greene, D.L.; Kang, S.; Kosenko, A.; Hoshi, N. M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Investig. 2015, 125, 3904–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, W.J.; Hinton, A.S.; Herby, J.T.J.; Salt, A.N.; Hartsock, J.J.; Wilson, S.; Lucchino, D.L.; Lenarz, T.; Warnecke, A.; Prenzler, N.; et al. Improved Speech Intelligibility in Subjects With Stable Sensorineural Hearing Loss Following Intratympanic Dosing of FX-322 in a Phase 1b Study. Otol. Neurotol. 2021, 42, e849–e857. [Google Scholar] [CrossRef]
- Avila, A.M.; Burnett, B.G.; Taye, A.A.; Gabanella, F.; Knight, M.A.; Hartenstein, P.; Cizman, Z.; Di Prospero, N.A.; Pellizzoni, L.; Fischbeck, K.H.; et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2007, 117, 659–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernochan, L.E.; Russo, M.L.; Woodling, N.S.; Huynh, T.N.; Avila, A.M.; Fischbeck, K.H.; Sumner, C.J. The role of histone acetylation in SMN gene expression. Hum. Mol. Genet. 2005, 14, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Gao, Y.; Yechikov, S.; Vazquez, A.E.; Chen, D.; Nie, L. Distinct roles of molecular chaperones HSP90alpha and HSP90beta in the biogenesis of KCNQ4 channels. PLoS ONE 2013, 8, e57282. [Google Scholar]
- Monti, B.; Polazzi, E.; Contestabile, A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2009, 2, 95–109. [Google Scholar] [CrossRef]
- Li, R.F.; Cao, S.S.; Fang, W.J.; Song, Y.; Luo, X.T.; Wang, H.Y.; Wang, J.G. Roles of HDAC2 and HDAC8 in Cardiac Remodeling in Renovascular Hypertensive Rats and the Effects of Valproic Acid Sodium. Pharmacology 2017, 99, 27–39. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G. Valproic Acid Improves Glucose Homeostasis by Increasing Beta-Cell Proliferation, Function, and Reducing its Apoptosis through HDAC Inhibition in Juvenile Diabetic Rat. J. Biochem. Mol. Toxicol. 2016, 30, 438–446. [Google Scholar] [CrossRef]
- Sun, X.Y.; Qin, H.J.; Zhang, Z.; Xu, Y.; Yang, X.C.; Zhao, D.M.; Li, X.N.; Sun, L.K. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2016, 13, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Gupta, S.; Verma, I.; Morsy, M.A.; Nair, A.B.; Ahmed, A.F. Hidden pharmacological activities of valproic acid: A new insight. Biomed. Pharmacother. 2021, 142, 112021. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.Q.; Schacht, J.; Sha, S.H. Aminoglycoside-induced histone deacetylation and hair cell death in the mouse cochlea. J. Neurochem. 2009, 108, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.T.; Wang, J.; Wang, Y.; Chen, F.Q. Association between histone deacetylases and the loss of cochlear hair cells: Role of the former in noise-induced hearing loss. Int. J. Mol. Med. 2015, 36, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drottar, M.; Liberman, M.C.; Ratan, R.R.; Roberson, D.W. The histone deacetylase inhibitor sodium butyrate protects against cisplatin-induced hearing loss in guinea pigs. Laryngoscope 2006, 116, 292–296. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Mazziotta, C.; Lanzillotti, C.; Gafa, R.; Touze, A.; Durand, M.A.; Martini, F.; Rotondo, J.C. The Role of Histone Post-Translational Modifications in Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 832047. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- Kramer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Piao, J.; Li, N.; Yang, Y.; Kim, K.Y.; Lin, Z. Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. FEBS J. 2020, 287, 2118–2133. [Google Scholar] [CrossRef] [PubMed]
- Forthun, R.B.; Sengupta, T.; Skjeldam, H.K.; Lindvall, J.M.; McCormack, E.; Gjertsen, B.T.; Nilsen, H. Cross-species functional genomic analysis identifies resistance genes of the histone deacetylase inhibitor valproic acid. PLoS ONE 2012, 7, e48992. [Google Scholar] [CrossRef] [Green Version]
- Bulatov, E.; Khaiboullina, S.; dos Reis, H.J.; Palotas, A.; Venkataraman, K.; Vijayalakshmi, M.; Rizvanov, A. Ubiquitin-Proteasome System: Promising Therapeutic Targets in Autoimmune and Neurodegenerative Diseases. Bionanoscience 2016, 6, 341–344. [Google Scholar] [CrossRef]
- Wakizono, T.; Nakashima, H.; Yasui, T.; Noda, T.; Aoyagi, K.; Okada, K.; Yamada, Y.; Nakagawa, T.; Nakashima, K. Growth factors with valproic acid restore injury-impaired hearing by promoting neuronal regeneration. JCI Insight 2021, 6, e139171. [Google Scholar] [CrossRef]
- Armon, C.; Brown, E.; Carwile, S.; Miller, P.; Shin, C. Sensorineural hearing loss: A reversible effect of valproic acid. Neurology 1990, 40, 1896–1898. [Google Scholar] [CrossRef] [PubMed]
- Hori, A.; Kataoka, S.; Sakai, K.; Hirose, G.; Iwasaki, N.; Horiguchi, A.; Takashima, M.; Tomoda, K. Valproic acid-induced hearing loss and tinnitus. Intern. Med. 2003, 42, 1153–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yechikov, S.; Vazquez, A.E.; Chen, D.; Nie, L. Impaired surface expression and conductance of the KCNQ4 channel lead to sensorineural hearing loss. J. Cell. Mol. Med. 2013, 17, 889–900. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- Noh, B.; Rim, J.H.; Gopalappa, R.; Lin, H.; Kim, K.M.; Kang, M.J.; Gee, H.Y.; Choi, J.Y.; Kim, H.H.; Jung, J. In vivo outer hair cell gene editing ameliorates progressive hearing loss in dominant-negative Kcnq4 murine model. Theranostics 2022, 12, 2465–2482. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.L.; Brigande, J.V. Fetal gene therapy and pharmacotherapy to treat congenital hearing loss and vestibular dysfunction. Hear. Res. 2020, 394, 107931. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, Y.S.; Choi, Y.M.; Lee, S.; Cho, H.-H. Valproic Acid Inhibits Progressive Hereditary Hearing Loss in a KCNQ4 Variant Model through HDAC1 Suppression. Int. J. Mol. Sci. 2023, 24, 5695. https://doi.org/10.3390/ijms24065695
Nam YS, Choi YM, Lee S, Cho H-H. Valproic Acid Inhibits Progressive Hereditary Hearing Loss in a KCNQ4 Variant Model through HDAC1 Suppression. International Journal of Molecular Sciences. 2023; 24(6):5695. https://doi.org/10.3390/ijms24065695
Chicago/Turabian StyleNam, Yoon Seok, Young Mi Choi, Sungsu Lee, and Hyong-Ho Cho. 2023. "Valproic Acid Inhibits Progressive Hereditary Hearing Loss in a KCNQ4 Variant Model through HDAC1 Suppression" International Journal of Molecular Sciences 24, no. 6: 5695. https://doi.org/10.3390/ijms24065695
APA StyleNam, Y. S., Choi, Y. M., Lee, S., & Cho, H.-H. (2023). Valproic Acid Inhibits Progressive Hereditary Hearing Loss in a KCNQ4 Variant Model through HDAC1 Suppression. International Journal of Molecular Sciences, 24(6), 5695. https://doi.org/10.3390/ijms24065695