
In Silico Exploration of Metabolically Active Peptides as Potential Therapeutic Agents against Amyotrophic Lateral Sclerosis

 ,
, 
 and
and 
Abstract
1. Introduction
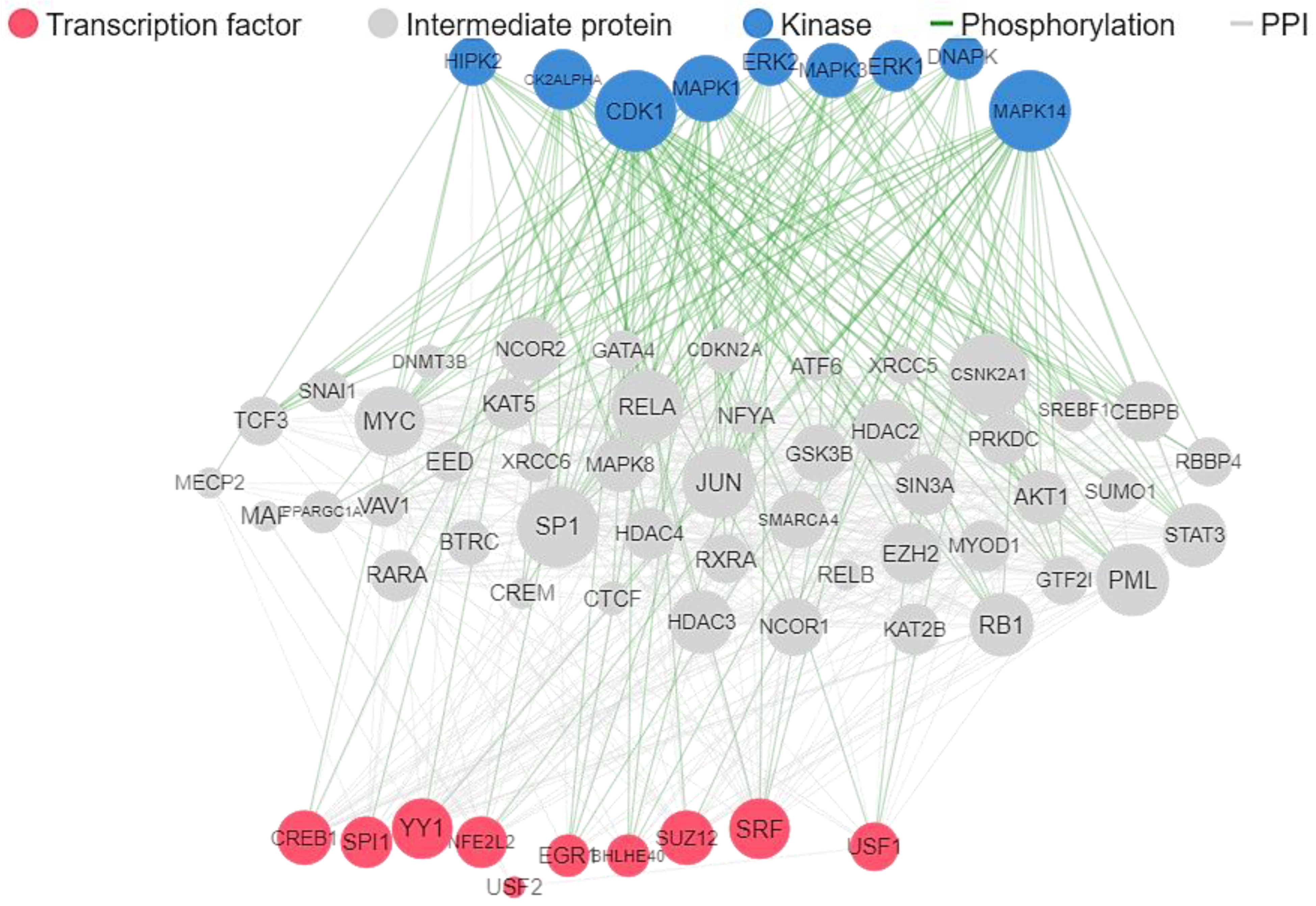
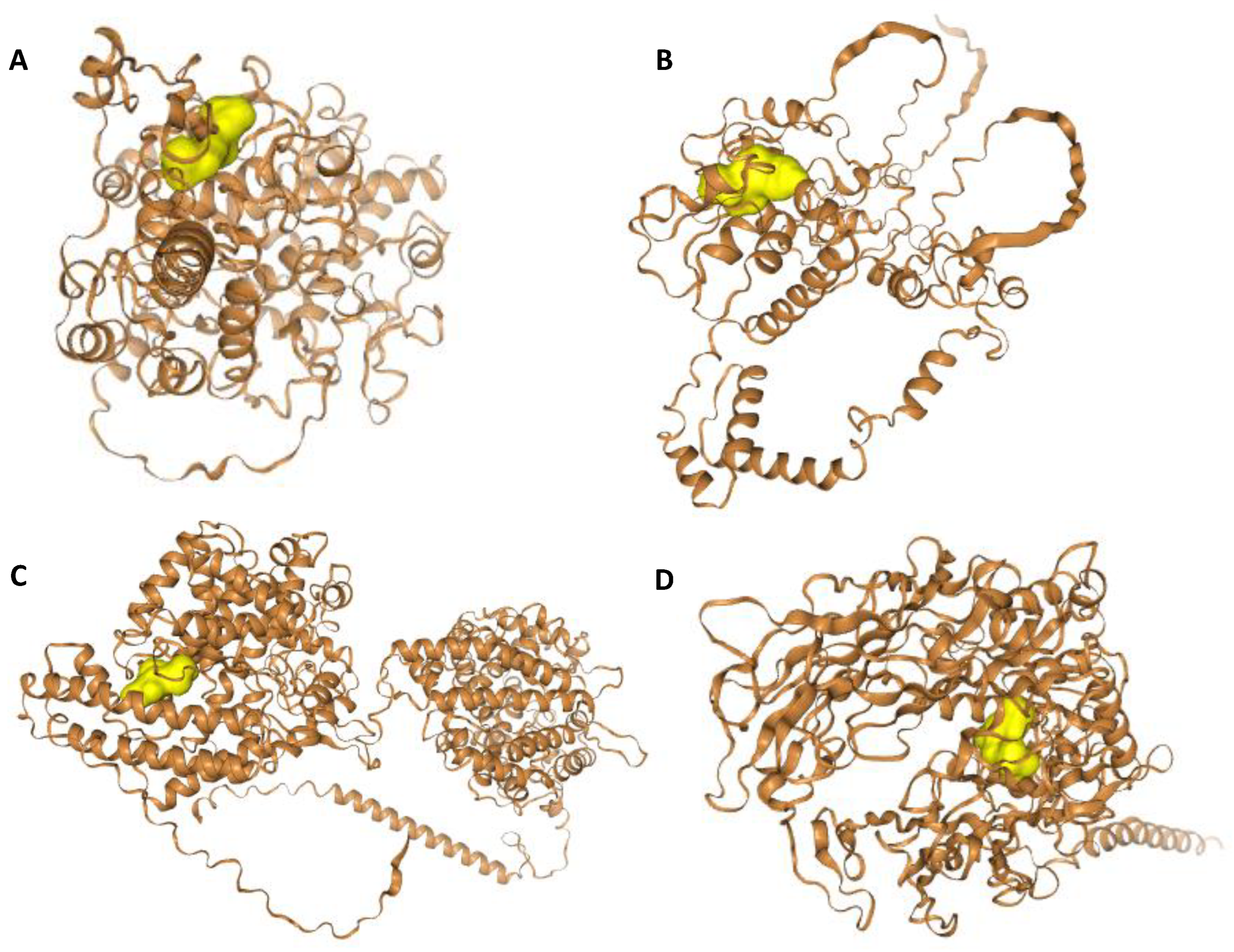
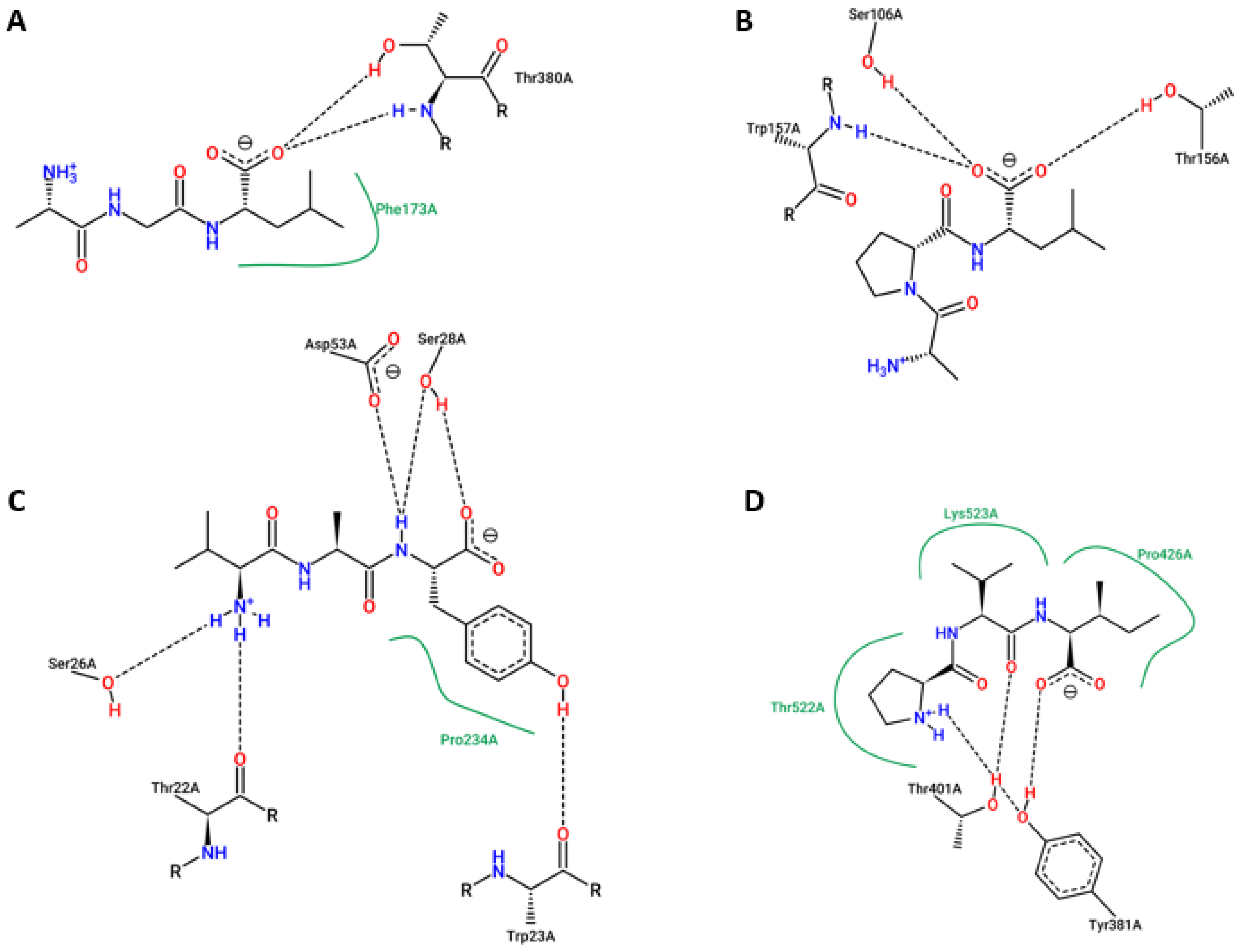
2. Results
3. Discussion
4. Materials and Methods
4.1. Preparation of ALS-Associated Genes
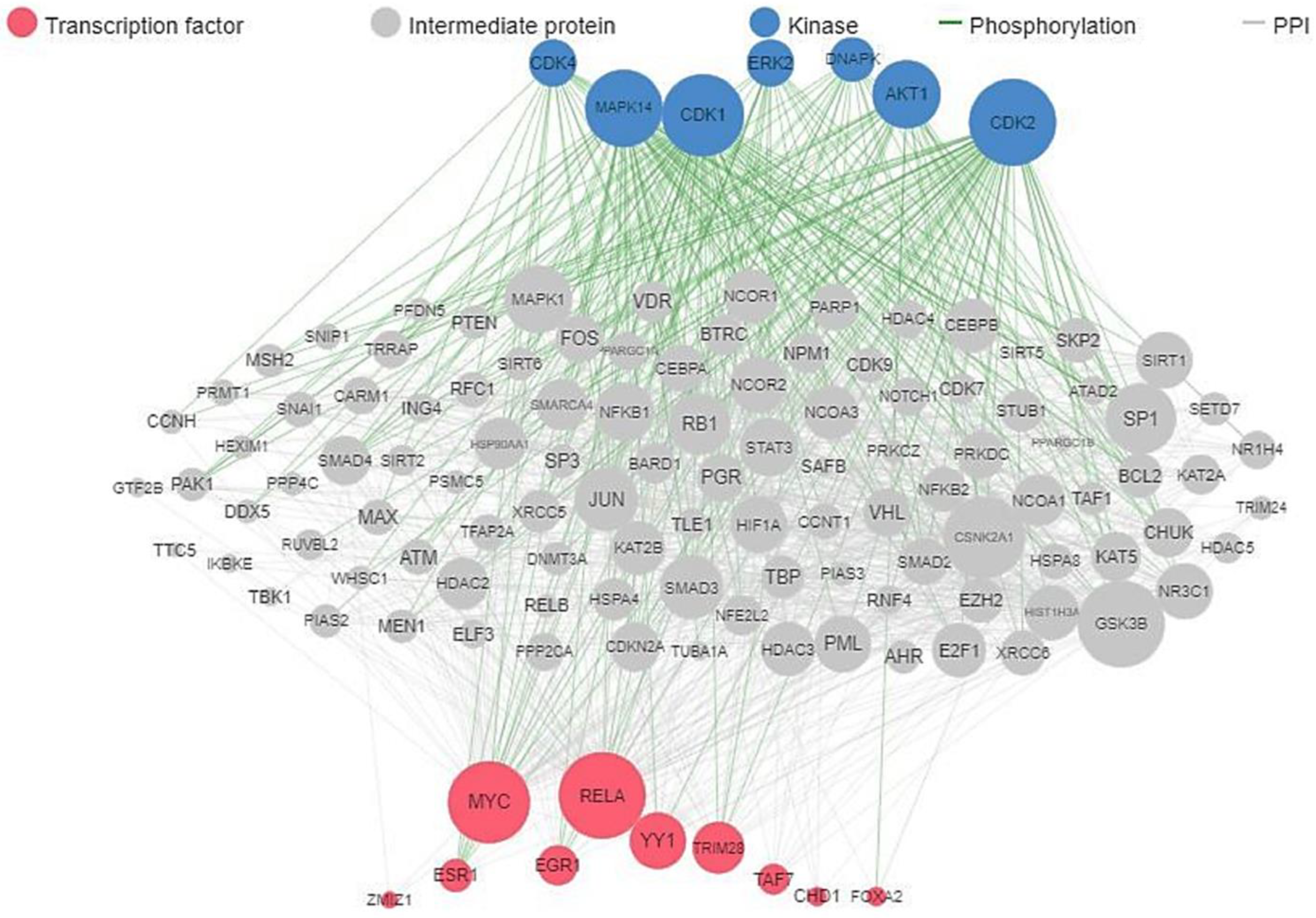
4.2. Target Genes Network Analysis
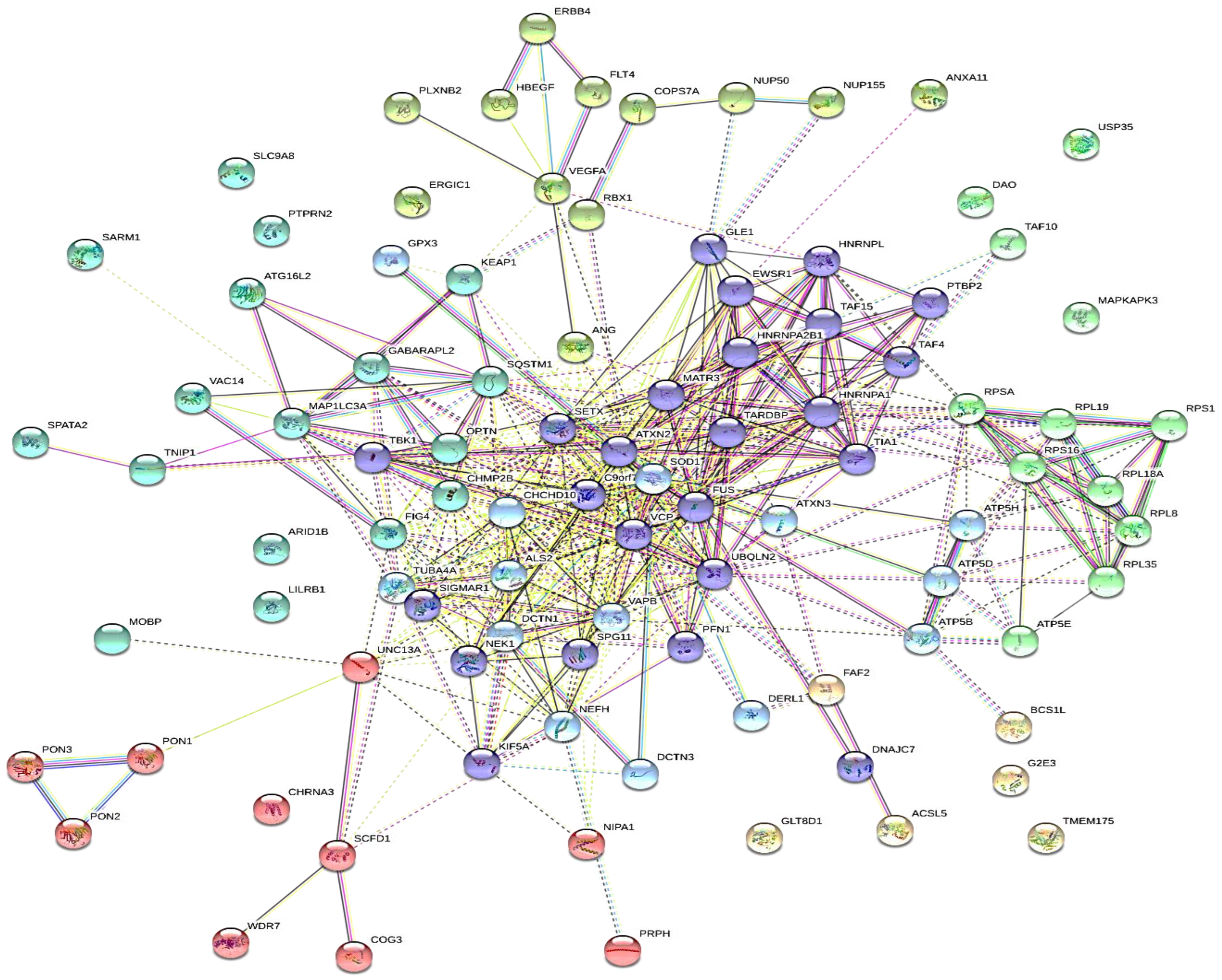
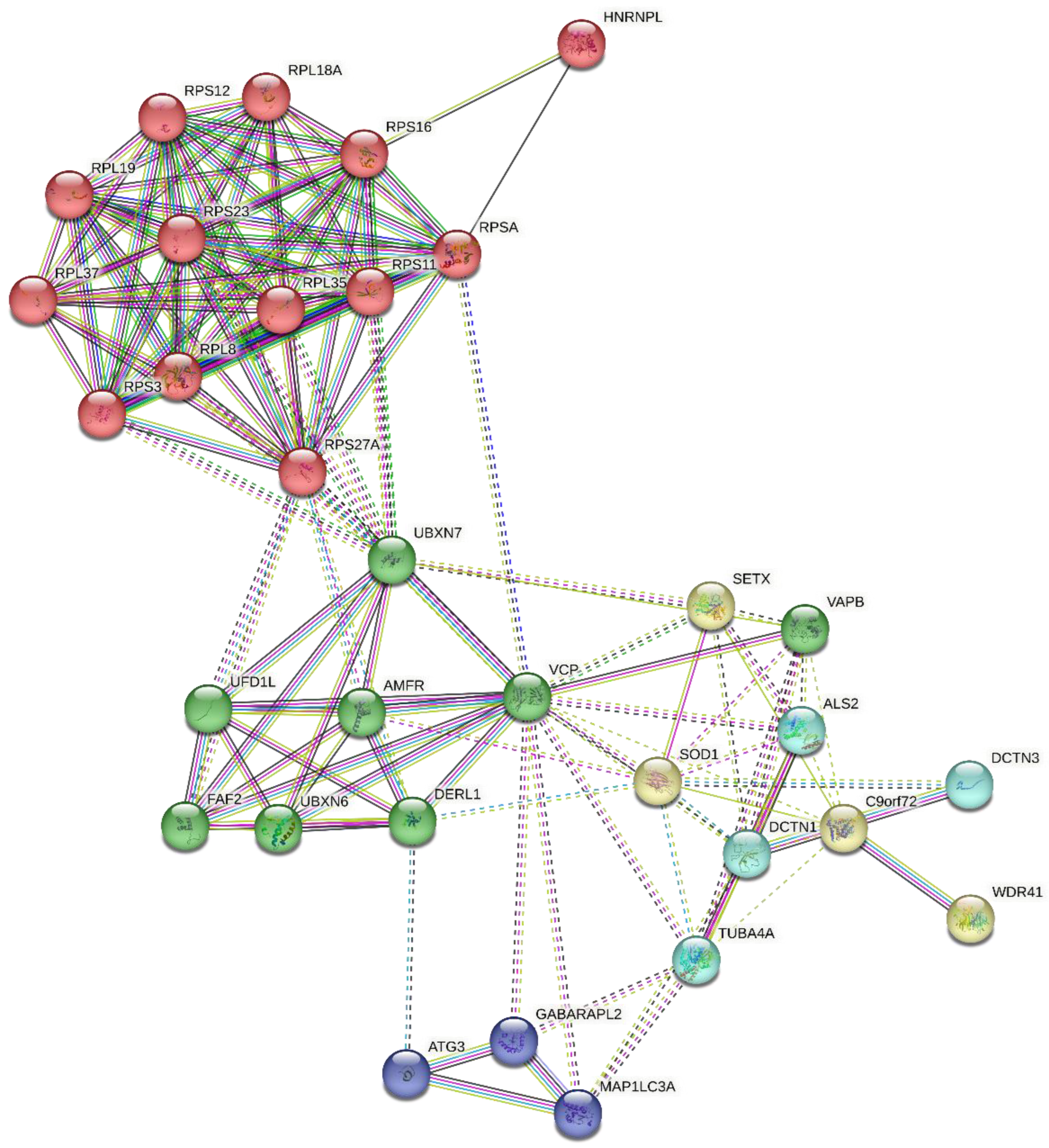
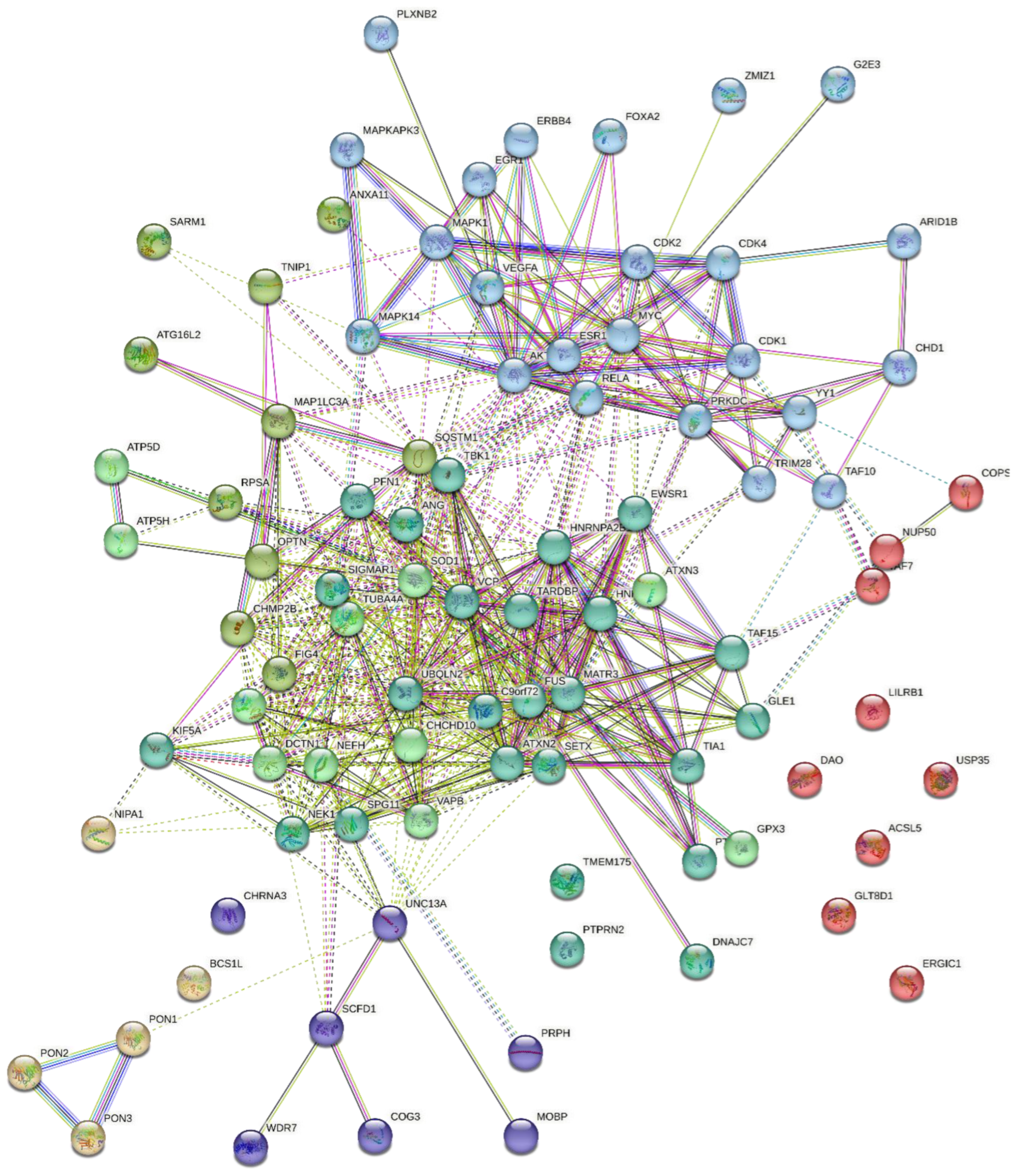
4.3. Protein-Protein Interaction
4.4. Protein Hydrolysis and Bioactivity Assessment
4.5. Target Prediction and Reconstruction of Gene Network
4.6. In Silico ADME and Allergenicity Prediction
4.7. Molecular Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saez-Atienzar, S.; Bandres-Ciga, S.; Langston, R.G.; Kim, J.J.; Choi, S.W.; Reynolds, R.H.; International ALS Genomics Consortium; ITALSGEN; Abramzon, Y.; Dewan, R.; et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv. 2021, 7, eabd9036. [Google Scholar] [CrossRef]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front. Mol. Neurosci. 2018, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Quansah, E.; Karikari, T.K. Motor Neuron Diseases in Sub-Saharan Africa: The Need for More Population-Based Studies. BioMed Res. Int. 2015, 2015, 298409. [Google Scholar] [CrossRef]
- Luna, J.; Diagana, M.; Aissa, L.A.; Tazir, M.; Pacha, L.A.; Kacem, I.; Gouider, R.; Henning, F.; Basse, A.; Cisse, O.; et al. Clinical features and prognosis of amyotrophic lateral sclerosis in Africa: The TROPALS study. J. Neurol. Neurosurg. Psychiatry 2018, 90, 20–29. [Google Scholar] [CrossRef]
- Fatoki, T.H.; Chukwuejim, S.; Ibraheem, O.; Oke, C.; Ejimadu, B.; Olaoye, I.; Oyegbenro, O.; Salami, T.; Basorun, R.; Oluwadare, O.; et al. Harmine and 7,8-dihydroxyflavone synergistically suitable for amyotrophic lateral sclerosis management: An insilico study. Res. Results Pharmacol. 2022, 8, 49–61. [Google Scholar] [CrossRef]
- Malik, R.; Wiedau, M. Therapeutic Approaches Targeting Protein Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 98. [Google Scholar] [CrossRef]
- Smukowski, S.N.; Maioli, H.; Latimer, C.S.; Bird, T.D.; Jayadev, S.; Valdmanis, P.N. Progress in Amyotrophic Lateral Sclerosis Gene Discovery. Neurol. Genet. 2022, 8, e669. [Google Scholar] [CrossRef]
- Pun, F.W.; Liu, B.H.M.; Long, X.; Leung, H.W.; Leung, G.H.D.; Mewborne, Q.T.; Gao, J.; Shneyderman, A.; Ozerov, I.V.; Wang, J.; et al. Identification of Therapeutic Targets for Amyotrophic Lateral Sclerosis Using PandaOmics–An AI-Enabled Biological Target Discovery Platform. Front. Aging Neurosci. 2022, 14, 914017. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Ruiz, M.D.V.P.; Perez, D.I.; Gil, C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2017, 26, 403–414. [Google Scholar] [CrossRef]
- Pioro, E.P.; Brooks, B.R.; Cummings, J.; Schiffer, R.; Thisted, R.A.; Wynn, D.; Hepner, A.; Kaye, R. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann. Neurol. 2010, 68, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Pape, J.A.; Grose, J.H. The effects of diet and sex in amyotrophic lateral sclerosis. Rev. Neurol. 2020, 176, 301–315. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- D’Amico, E.; Grosso, G.; Nieves, J.W.; Zanghì, A.; Factor-Litvak, P.; Mitsumoto, H. Metabolic Abnormalities, Dietary Risk Factors and Nutritional Management in Amyotrophic Lateral Sclerosis. Nutrients 2021, 13, 2273. [Google Scholar] [CrossRef] [PubMed]
- Muscaritoli, M.; Kushta, I.; Molfino, A.; Inghilleri, M.; Sabatelli, M.; Fanelli, F.R. Nutritional and metabolic support in patients with amyotrophic lateral sclerosis. Nutrition 2012, 28, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Oudart, H.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Dobak, S. Nutritional Care of the Patient with Amyotrophic Lateral Sclerosis. Pract. Gastroenterol. 2022, XLVI, 60–67. [Google Scholar]
- Rodríguez-Sánchez, S.; Valiente, N.; Seseña, S.; Cabrera-Pinto, M.; Rodriguez, A.; Aranda, A.; Palop, L.; Fernandez-Martos, C.M. Ozone modified hypothalamic signaling enhancing thermogenesis in the TDP-43A315T transgenic model of Amyotrophic Lateral. Sci. Rep. 2022, 12, 20814. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Layman, D.K.; Tome, D.; Schaafsma, G. Protein quality assessment: Impact of expanding understanding of protein and amino acid needs for optimal health. Am. J. Clin. Nutr. 2008, 87, 1576S–1581S. [Google Scholar] [CrossRef]
- Fatoki, T.H.; Aluko, R.E.; Udenigwe, C.C. In silico investigation of molecular targets, pharmacokinetics, and biological activities of chicken egg ovalbumin protein hydrolysates. J. Food Bioact. 2022, 17, 34–48. [Google Scholar] [CrossRef]
- Silver, N.; Hamill, M.; Samayoa, P.; Hou, J.; Hamm, L.; Berry, D. Nutritive Polypeptides, Formulations and Methods for Treating Disease and Improving Muscle Health and Maintenance. International Patient (WO2014134225A2, PCT/US2014/018807), 4 September 2014. [Google Scholar]
- Combaret, L.; Dardevet, D.; Bechet, D.; Taillandier, D.; Mosoni, L.; Attaix, D. Skeletal muscle proteolysis in aging. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 37–41. [Google Scholar] [CrossRef]
- Churchward-Venne, T.A.; Burd, N.A.; Mitchell, C.J.; West, D.W.; Philp, A.; Marcotte, G.R.; Baker, S.K.; Baar, K.; Phillips, S.M. Supplementation of a suboptimal protein dose with leucine or essential amino acids: Effects on myofibrillar protein synthesis at rest and following resistance exercise in men. J. Physiol. 2012, 590, 2751–2765. [Google Scholar] [CrossRef] [PubMed]
- Magne, H.; Savary-Auzeloux, I.; Migne, C.; Peyron, M.A.; Combaret, L.; Remond, D.; Dardevet, D. Unilateral hindlimb casting induced a delayed generalized muscle atrophy during rehabilitation that is prevented by a whey or a high protein diet but not a free leucine-enriched diet. PLoS ONE 2013, 8, e70130. [Google Scholar] [CrossRef]
- Davis, D.A.; Cox, P.A.; Banack, S.A.; Lecusay, P.D.; Garamszegi, S.P.; Hagan, M.J.; Powell, J.T.; Metcalf, J.S.; Palmour, R.M.; Beierschmitt, A.; et al. L-Serine Reduces Spinal Cord Pathology in a Vervet Model of Preclinical ALS/MND. J. Neuropathol. Exp. Neurol. 2020, 79, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Kiaei, M.; Damiano, M.; Hiller, A.; Wille, E.; Manfredi, G.; Calingasan, N.Y.; Szeto, H.H.; Beal, M.F. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2006, 98, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Post, J.; Kogel, V.; Schaffrath, A.; Lohmann, P.; Shah, N.J.; Langen, K.-J.; Willbold, D.; Willuweit, A.; Kutzsche, J. A Novel Anti-Inflammatory D-Peptide Inhibits Disease Phenotype Progression in an ALS Mouse Model. Molecules 2021, 26, 1590. [Google Scholar] [CrossRef]
- Layman, D.K.; Anthony, T.G.; Rasmussen, B.B.; Adams, S.H.; Lynch, C.J.; Brinkworth, G.D.; Davis, T.A. Defining meal requirements for protein to optimize metabolic roles of amino acids. Am. J. Clin. Nutri. 2015, 101, 1330S–1338S. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Kwak, S.G.; Park, J.S.; Park, D. Relationship between statins and the risk of amyotrophic lateral sclerosis: A PRISMA-compliant meta-analysis. Medicine 2021, 100, e26751. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Crippa, V.; Cicardi, M.E.; Tedesco, B.; Ferrari, V.; Chierichetti, M.; Casarotto, E.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. A Crucial Role for the Protein Quality Control System in Motor Neuron Diseases. Front. Aging Neurosci. 2020, 12, 191. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Megat, S.; Mora, N.; Sanogo, J.; Catanese, A.; Alami, N.O.; Freischmidt, A.; Mingaj, X.; de Calbiac, H.; Muratet, F.; Dirrig-Grosch, S.; et al. Loss of nucleoporin Nup50 is a risk factor for amyotrophic lateral sclerosis. medRxiv 2021. [Google Scholar] [CrossRef]
- Al-Khleifat, A.; Iacoangeli, A.; van Vugt, J.; Bowles, H.; Moisse, M.; Zwamborn, R.A.J.; van der Spek, R.A.A.; Shatunov, A.; Cooper-Knock, J.; Topp, S.; et al. Structural variation analysis of 6,500 whole genome sequences in amyotrophic lateral sclerosis. NPJ Genom. Med. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.L.; Dittlau, K.S.; Bosch, L.V.D. Axonal Transport Defects and Neurodegeneration: Molecular Mechanisms and Therapeutic Implications. Semin. Cell Dev. Biol. 2019, 99, 133–150. [Google Scholar] [CrossRef]
- Morfini, G.A.; Bosco, D.A.; Brown, H.; Gatto, R.G.; Kaminska, A.; Song, Y.; Molla, L.; Baker, L.; Marangoni, M.N.; Berth, S.; et al. Inhibition of Fast Axonal Transport by Pathogenic SOD1 Involves Activation of p38 MAP Kinase. PLoS ONE 2013, 8, e65235. [Google Scholar] [CrossRef]
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 2018, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef]
- Zhang, F.; Ström, A.L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 2007, 282, 16691–16699. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef]
- Raghav, Y.; Dilliot, A.A.; Petrozziello, T.; Kim, S.E.; Berry, J.D.; Cudkowicz, M.E.; Vakili, K.; NYGC ALS Consortium; Fraenkel, E.; Farhan, S.M.; et al. Identification of gene fusions associated with amyotrophic lateral sclerosis. medRxiv 2022. [Google Scholar] [CrossRef]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef]
- Chiba, K.; Ori-McKenney, K.M.; Niwa, S.; McKenney, R.J. Reconstitution of a Synergistic Kinesin-1 Activation Mechanism. Cell Rep. 2022, 39, 110900. [Google Scholar] [CrossRef] [PubMed]
- Pant, C.D.; Parameswarn, J.; Rao, L.; Loss, I.; Chilukuri, G.; Parlato, R.; Shi, L.; Glass, J.D.; Bassell, G.J.; Koch, P.; et al. ALS-linked KIF5A ΔExon27 mutant causes neuronal toxicity through gain-of-function. EMBO Rep. 2022, 23, e54234. [Google Scholar] [CrossRef] [PubMed]
- Nakano, J.; Chiba, K.; Niwa, S. An ALS-associated KIF5A mutant forms oligomers and aggregates and induces neuronal toxicity. Genes Cells 2022, 27, 421–435. [Google Scholar] [CrossRef]
- Srinivasan, E.; Rajasekaran, R. Rational design of linear tripeptides against the aggregation of human mutant SOD1 protein causing amyotrophic lateral sclerosis. J. Neurol. Sci. 2019, 405, 116425. [Google Scholar] [CrossRef]
- Raposo, G.; Campagne, C.; Delevoye, C. Skin Whitening Peptide Agents. International Patent (WO2016034541A1), 10 March 2016. [Google Scholar]
- Pampanin, D.M.; Larssen, E.; Provan, F.; Sivertsvik, M.; Ruoff, P.; Sydnes, M.O. Detection of small bioactive peptides from Atlantic herring (Clupea harengus L.). Peptides 2012, 34, 423–426. [Google Scholar] [CrossRef]
- Pampanin, D.M.; Haarr, M.B.; Sydnes, M.O. Natural peptides with antioxidant activity from Atlantic cod and Atlantic salmon residual material. Int. J. Appl. Res. Nat. Prod. 2016, 9, 1–8. [Google Scholar]
- Petersen, M.C.; Nielsen, M.S.; Jacobsen, C.; Tauris, J.; Jacobsen, L.; Gliemann, J.; Moestrup, S.K.; Madsen, P.S. Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. EMBO J. 1999, 18, 595–604. [Google Scholar] [CrossRef]
- Mazella, J.; Petrault, O.; Lucas, G.; Deval, E.; Beraud-Dufour, S.; Gandin, C.; El-Yacoubi, M.; Widmann, C.; Guyon, A.; Chevet, E.; et al. Spadin, a Sortilin Derived Peptide, Targeting Rodent TREK-1 Channels: A New Concept in the Antidepressant Drug Design. PLoS Biol. 2010, 8, e1000355. [Google Scholar] [CrossRef]
- Heurteaux, C.; Mazella, J.; Borsotto, M. Spadin, a Sortilin-derived peptide: A new concept in the antidepressant drug design. OCL 2011, 18, 202–207. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Thome, J.; Sakai, N.; Shin, K.; Steffen, C.; Zhang, Y.J.; Impey, S.; Storm, D.; Duman, R.S. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J. Neurosci. 2000, 20, 4030–4036. [Google Scholar] [CrossRef] [PubMed]
- Ciesler, J.; Sari, Y. Neurotrophic Peptides: Potential Drugs for Treatment of Amyotrophic Lateral Sclerosis and Alzheimer’s disease. Open J. Neurosci. 2013, 3, 1–11. [Google Scholar]
- Li, J.; Bollati, C.; Bartolomei, M.; Mazzolari, A.; Arnoldi, A.; Vistoli, G.; Lammi, C. Hempseed (Cannabis sativa) Peptide H3 (IGFLIIWV) Exerts Cholesterol-Lowering Effects in Human Hepatic Cell Line. Nutrients 2022, 14, 1804. [Google Scholar] [CrossRef]
- Cruz-Chamorro, I.; Santos-Sánchez, G.; Bollati, C.; Bartolomei, M.; Li, J.; Arnoldi, A.; Lammi, C. Hempseed (Cannabis sativa) Peptides WVSPLAGRT and IGFLIIWV Exert Anti-inflammatory Activity in the LPS-Stimulated Human Hepatic Cell Line. J. Agric. Food Chem. 2022, 70, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, J.; Scicli, A.G.; Carretero, O.A.; Slaughter, C.; Moomaw, C.; Hersh, L.B. The Hydrolysis of Endothelins by Neutral Endopeptidase 24.11 (Enkephalinase). J. Biol. Chem. 1990, 265, 14150–14155. [Google Scholar] [CrossRef]
- Jones, C.R.; Hiley, C.R.; Pelton, J.T.; Miller, R.C. Endothelin receptor heterogeneity; structure activity, autoradiographic and functional studies. J. Recept. Res. 1991, 11, 299–310. [Google Scholar] [CrossRef]
- Ranno, E.; D’Antoni, S.; Spatuzza, M.; Berretta, A.; Laureanti, F.; Bonaccorso, C.M.; Pellitteri, R.; Longone, P.; Spalloni, A.; Iyer, A.M.; et al. Endothelin-1 is over-expressed in amyotrophic lateral sclerosis and induces motor neuron cell death. Neurobiol. Dis. 2014, 65, 160–171. [Google Scholar] [CrossRef]
- D’Antoni, S.; Ranno, E.; Spatuzza, M.; Cavallaro, S.; Catania, M.V. Endothelin-1 induces degeneration of cultured motor neurons through a mechanism mediated by nitric oxide and PI3K/Akt pathway. Neurotox/Res. 2017, 32, 58–70. [Google Scholar] [CrossRef]
- Okeke, E.B.; Abioye, R.O.; Ventura-Santana, E.; Sun, X.; Udenigwe, C.C. TNKPVI, a Putative Bioaccessible Pharmacophore of Anti-Inflammatory Potato Patatin-Derived Decapeptide DIKTNKPVIF. Molecules 2022, 27, 3869. [Google Scholar] [CrossRef]
- Pałasz, A.; Rojczyk, E.; Bogus, K.; Worthington, J.J.; Wiaderkiewicz, R. The novel neuropeptide phoenixin is highly co-expressed with nesfatin-1 in the rat hypothalamus, an immunohistochemical study. Neurosci. Lett. 2015, 592, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Schalla, M.A.; Scharner, S.; Kühne, S.G.; Goebel-Stengel, M.; Kobelt, P.; Rose, M.; Stengel, A. Intracerebroventricular injection of phoenixin alters feeding behavior and activates nesfatin-1 immunoreactive neurons in rats. Brain Res. 2019, 1715, 188–195. [Google Scholar] [CrossRef]
- Wei, P.; Keller, C.; Li, L. Neuropeptides in gut-brain axis and their influence on host immunity and stress. Comput. Struct. Biotechnol. J. 2020, 18, 843–885. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Rothstein, J.D. Inhibition of cyclooxygenase-2 protects motor neurons in an organotypic model of amyotrophic lateral sclerosis. Ann. Neurol. 2000, 48, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Frank, K.; Dykes-Hoberg, M.; Teismann, P.; Almer, G.; Przedborski, S.; Rothstein, J.D. Cyclooxygenase 2 Inhibition Protects Motor Neurons and Prolongs Survival in a Transgenic Mouse Model of ALS. Ann. Neurol. 2002, 52, 771–778. [Google Scholar] [CrossRef]
- Almer, G.; Guegan, C.; Teismann, P.; Naini, A.; Rosoklija, G.; Hays, A.P.; Chen, C.; Przedborski, S. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann. Neurol. 2001, 49, 176–185. [Google Scholar] [CrossRef]
- Yasojima, K.; Tourtellotte, W.W.; McGeer, E.G.; McGeer, P.L. Marked increase in cyclooxygenase-2 in ALS spinal cord Implications for therapy. Neurology 2001, 57, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Li, M.W.; Ona, V.O.; Guegan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.; Olszewski, A.; Stieg, P.; Lee, J.; Przedborski, S.; et al. Functional roles of caspase-1 and caspase-2 in an ALS transgenic mouse model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef]
- Samad, T.A.; Moore, K.A.; Sapirstein, A.; Billet, S.; Allchorne, A.; Poole, S.; Bonventre, J.V.; Woolf, C.J. Interleukin-1 beta mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001, 410, 471–475. [Google Scholar] [CrossRef]
- Nogawa, S.; Zhang, F.Y.; Ross, M.E.; Iadecola, C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J. Neurosci. 1997, 17, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Uchimura, K.; Zhu, R.L.; Nagayama, T.; Rose, M.E.; Stetler, R.A.; Isakson, P.C.; Chen, J.; Graham, S.H. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 10954–10959. [Google Scholar] [CrossRef]
- Okuno, T.; Nakatsuji, Y.; Kumanogoh, A.; Koguchi, K.; Moriya, M.; Fujimura, H.; Kikutani, H.; Sakoda, S. Induction of cyclooxygenase-2 in reactive glial cells by the CD40 pathway: Relevance to amyotrophic lateral sclerosis. J. Neurochem. 2004, 91, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Kaila, K. Carbonic anhydrases and Brain pH in the control of neuronal excitability. Subcell. Biochem. 2014, 75, 271–290. [Google Scholar] [CrossRef]
- Mumby, S.; Chung, K.F.; Adcock, I.M. Transcriptional Effects of Ozone and Impact on Airway Inflammation. Front. Immunol. 2019, 10, 1610. [Google Scholar] [CrossRef]
- Cenci, A.; Macchia, I.; La Sorsa, V.; Sbarigia, C.; Di Donna, V.; Pietraforte, D. Mechanisms of Action of Ozone Therapy in Emerging Viral Diseases: Immunomodulatory Effects and Therapeutic Advantages With Reference to SARS-CoV-2. Front. Microbiol. 2022, 13, 871645. [Google Scholar] [CrossRef]
- Zou, Y.-H.; Guan, P.-P.; Zhang, S.-Q.; Guo, Y.-S.; Wang, P. Rofecoxib Attenuates the Pathogenesis of Amyotrophic Lateral Sclerosis by Alleviating Cyclooxygenase-2-Mediated Mechanisms. Front. Neurosci. 2020, 14, 817. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Petri, S.; Choi, D.K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Integrative role of cPLA with COX-2 and the effect of non-steriodal anti-inflammatory drugs in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2005, 93, 403–411. [Google Scholar] [CrossRef]
- Lin, F.-C.; Tsai, C.-P.; Kuang-Wu, L.J.; Wu, M.-T.; Tzu-Chi, L.C. Angiotensin-converting enzyme inhibitors and amyotrophic lateral sclerosis risk: A total population-based case-control study. JAMA Neurol. 2015, 72, 40–48. [Google Scholar] [CrossRef]
- Hazato, T.; Kase, R. Isolation of angiotensin-converting enzyme inhibitor from porcine plasma. Biochem. Biophys. Res. Commun. 1986, 139, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Aluko, R.E.; Nakai, S. Structural requirements of angiotensin I-converting enzyme inhibitory peptides: Quantitative structure−activity relationship study of di- and tripeptides. J. Agric. Food Chem. 2006, 54, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.J.; Jung, W.K.; Lee, S.H.; Byun, H.G.; Kim, S.K. Antihypertensive effect of an angiotensin I-converting enzyme inhibitory peptide from bullfrog (Rana catesbeiana Shaw) muscle protein in spontaneously hypertensive rats. Process Biochem. 2007, 42, 1443–1448. [Google Scholar] [CrossRef]
- Xia, K.; Zhang, L.; Tang, L.; Huang, T.; Fan, D. Assessing the role of blood pressure in amyotrophic lateral sclerosis: A Mendelian randomization study. Orphanet J. Rare Dis. 2022, 17, 56. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ichikawa, Y.; Igarash, O.; Ikeda, K.; Kinoshita, M. Influence of temocapril on cultured ventral spinal cord neurons. Neurochem. Res. 2003, 28, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Holdom, C.J.; Steyn, F.J.; Henderson, R.D.; McCombe, P.A.; Rogers, M.-L.; Ngo, S.T. Biofluid Biomarkers of Amyotrophic Lateral Sclerosis. In Neurodegenerative Diseases Biomarkers: Towards Translating Research to Clinical Practice; Philip, V., Peplow, B.M., Gennarelli, T.A., Eds.; Springer: Berlin/Heidelberg, Germany. [CrossRef]
- Holst, J.J. On the physiology of GIP and GLP-1. Horm. Metab. Res. 2004, 36, 747–754. [Google Scholar] [CrossRef]
- Zhang, J.; Xue, C.; Zhu, T.; Vivekanandan, A.; Pennathur, S.; Ma, Z.A.; Chen, Y.E. A tripeptide diapin effectively lowers blood glucose levels in male type 2 diabetes mice by increasing blood levels of insulin and GLP-1. PLoS ONE 2013, 8, e83509. [Google Scholar] [CrossRef]
- Ahren, B.; Landin-Olsson, M.; Jansson, P.A.; Svensson, M.; Holmes, D.; Schweizer, A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 2078–2084. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.S.; Deacon, C.F.; Holst, J.J. Do we know the true mechanism of action of the DPP-4 inhibitors? Diabetes Obes. Metab. 2018, 20, 34–41. [Google Scholar] [CrossRef]
- Darsalia, V.; Ortsäter, H.; Olverling, A.; Darlöf, E.; Wolbert, P.; Nyström, T.; Klein, T.; Sjöholm, Å.; Patrone, C. The DPP-4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: A comparison with glimepiride. Diabetes 2013, 62, 1289–1296. [Google Scholar] [CrossRef]
- Lin, C.L.; Huang, C.N. The neuroprotective effects of the anti-diabetic drug linagliptin against Aβ-induced neurotoxicity. Neural. Regen. Res. 2016, 11, 236–237. [Google Scholar]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.; Arsdale, T.; Zhou, Q.; Srinivasula, S.; Alnemri, E.; Salvesen, S.; Reed, J. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Leo, E.; Stennicke, H.; Welsh, K.; Salvesen, G.S.; Reed, J. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999, 18, 5242–5251. [Google Scholar] [CrossRef]
- Wootz, H.; Hansson, I.; Korhonen, L.; Lindholm, D. XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice. Exp. Cell Res. 2006, 312, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Guegan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophiclateral sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef]
- Ishigaki, S.; Liang, Y.; Yamamoto, M.; Niwa, J.; Ando, Y.; Yoshihara, T.; Takeuchi, H.; Doyu, M.; Sobue, G. X-Linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J. Neurochem. 2002, 82, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Mufti, A.R.; Burstein, E.; Duckett, C.S. XIAP: Cell death regulation meets copper homeostasis. Arch. Biochem. Biophys. 2007, 463, 168–174. [Google Scholar] [CrossRef]
- Burstein, E.; Ganesh, L.; Dick, R.D.; van De Sluis, B.; Wilkinson, J.C.; Klomp, L.W.J.; Wijmenga, C.; Brewer, G.J.; Nabel, G.J.; Duckett, C.S. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004, 23, 244–254. [Google Scholar] [CrossRef]
- Brady, G.F.; Galban, S.; Liu, X.; Basrur, V.; Gitlin, J.D.; Elenitoba-Johnson, K.S.J.; Wilson, T.E.; Duckett, C.S. Regulation of the Copper Chaperone CCS by XIAP-Mediated Ubiquitination. Mol. Cell. Biol. 2010, 30, 1923–1936. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanno, T.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Ikeda, J.E. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 232, 41–52. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanno, T.; Yanagisawa, Y.; Yasutake, K.; Inoue, S.; Hirayama, N.; Ikeda, J.E. A novel acylaminoimidazole derivative, WN1316, alleviates disease progression via suppression of glial inflammation in ALS mouse model. PLoS ONE 2014, 9, e87728. [Google Scholar] [CrossRef]
- Kano, O.; Tanaka, K.; Kanno, T.; Iwasaki, Y.; Ikeda, J.E. Neuronal apoptosis inhibitory protein is implicated in amyotrophic lateral sclerosis symptoms. Sci. Rep. 2018, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, R.; Chan, C.; Curley, P.; Sharp, J.; Chiong, J.; Rannard, S.; Siccardi, M.; Owen, A. Inhibitory effects of commonly used excipients on P-glycoprotein in vitro. Mol. Pharm. 2018, 15, 4835–4842. [Google Scholar] [CrossRef]
- Fatoki, T.H.; Awofisayo, O.A.; Ibraheem, O.; Oyedele, A.S.; Akinlolu, O.S. In silico Investigation of First-Pass Effect on Selected Small Molecule Excipients and Structural Dynamics of P-glycoprotein. Bioinform. Biol. Insight 2020, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS barrier impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Markandaiah, S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Blood-Brain Barrier Driven Pharmacoresistance in Amyotrophic Lateral Sclerosis and Challenges for Effective Drug Therapies. AAPS J. 2017, 19, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Lichter, J.; Sarlo, M.; Markandaiah, S.S.; McAvoy, K.; Richard, J.P.; Jablonski, M.R.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-glycoprotein) in endothelial cells of the blood-brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia 2016, 64, 1298–1313. [Google Scholar] [CrossRef]
- Rentzsch, R.; Renard, B.Y. Docking small peptides remains a great challenge: An assessment using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stark, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.; De Klein, N.; Westra, H.J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in Amyotrophic Lateral Sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. medRxiv 2021. [Google Scholar] [CrossRef]
- Clarke, D.J.B.; Kuleshov, M.V.; Schilder, B.M.; Torre, D.; Duffy, M.E.; Keenan, A.B.; Lachmann, A.; Feldmann, A.S.; Gundersen, G.W.; Silverstein, M.C.; et al. eXpression2Kinases (X2K) Web: Linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018, 46, 171–179. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Lammi, C.; Fassi, E.M.A.; Li, J.; Bartolomei, M.; Benigno, G.; Roda, G.; Arnoldi, A.; Grazioso, G. Computational Design and Biological Evaluation of Analogs of Lupin Peptide P5 Endowed with Dual PCSK9/HMG-CoAR Inhibiting Activity. Pharmaceutics 2022, 14, 665. [Google Scholar] [CrossRef] [PubMed]
- Yacila, G.; Sari, Y. Potential Therapeutic Drugs and Methods for the Treatment of Amyotrophic Lateral Sclerosis. Curr. Med. Chem. 2014, 21, 3583–3593. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mori, Y.; Asakura, S.; Yamamoto, A.; Odagiri, S.; Yamada, D.; Sekiguchi, M.; Wada, K.; Sato, M.; Kurabayashi, A.; Suzuki, H.; et al. Characterization of soy-deprestatin, a novel orally active decapeptide that exerts antidepressant-like effects via gut-brain communication. FASEB J. 2018, 32, 568–575. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Scientific Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2-a server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Hetenyi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discovery Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic. Acids Res. 2018, 6, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 8. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Active Hydrolysate Peptide | % Probability of Predicted Targets | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SN | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | |
| 1 | AGL | 65 | 20 | 20 | 20 | 20 | ||||||||||||||||||
| 2 | AIF | 25 | 25 | 20 | 55 | 35 | 25 | 20 | 20 | |||||||||||||||
| 3 | APL | 80 | 20 | 75 | 25 | 50 | 20 | |||||||||||||||||
| 4 | APVSIPQ | 35 | 30 | 20 | ||||||||||||||||||||
| 5 | AVK | 45 | 25 | 20 | 20 | 20 | 20 | 20 | ||||||||||||||||
| 6 | AVY | 35 | 20 | 40 | 50 | 50 | 30 | 25 | ||||||||||||||||
| 7 | IGF | 30 | 20 | 30 | ||||||||||||||||||||
| 8 | IIW | 45 | 40 | 25 | 50 | 30 | 20 | |||||||||||||||||
| 9 | PVI | 20 | 50 | 20 | 80 | 80 | ||||||||||||||||||
| 10 | SDPF | 20 | 20 | 45 | 30 | 25 | 25 | |||||||||||||||||
| 11 | SGGVVK | 20 | 20 | 45 | 20 | |||||||||||||||||||
| 12 | VAY | 20 | 40 | 20 | 35 | 45 | 45 | 20 | 30 | 25 | ||||||||||||||
| SN | Hydrolysate Peptides (Ligands) | Predicted ADME Parameter | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | MR | TPSA (Å2) | Log P | ESOL Log S | ESOL Class | GIA | BBB | P-gp | CYPs Inhibitor | Log Kp (cm/s) | BS | SA | Allergenicity | ||
| 1 | AGL | 259.3 | 65.47 | 121.52 | −0.76 | 0.99 | Highly soluble | High | No | No | None | −9.95 | 0.55 | 2.69 | Non-allergen |
| 2 | AIF | 349.42 | 94.77 | 121.52 | 0.56 | −0.4 | Very soluble | High | No | No | None | −9.62 | 0.55 | 3.49 | Non-allergen |
| 3 | APL | 299.37 | 81.79 | 112.73 | −0.37 | 0.65 | Highly soluble | High | No | Yes | None | −10.17 | 0.55 | 3.21 | Non-allergen |
| 4 | APVSIPQ | 710.82 | 186.29 | 283.66 | −1.48 | −0.24 | Very soluble | Low | No | No | None | −13.36 | 0.17 | 6.34 | Non-allergen |
| 5 | AVK | 316.4 | 82.6 | 147.54 | −0.83 | 1.74 | Highly soluble | Low | No | No | None | −11.33 | 0.55 | 3.41 | Non-allergen |
| 6 | AVY | 351.4 | 91.98 | 141.75 | −0.18 | 0.33 | Highly soluble | Low | No | No | None | −10.55 | 0.55 | 3.27 | Allergen |
| 7 | IGF | 335.4 | 89.96 | 121.52 | 0.51 | −0.41 | Very soluble | High | No | No | None | −9.43 | 0.55 | 3.19 | Non-allergen |
| 8 | IIW | 430.54 | 121.05 | 137.31 | 1.82 | −1.98 | Very soluble | High | No | Yes | CYP3A4 | −8.8 | 0.55 | 4.32 | Non-allergen |
| 9 | PVI | 327.42 | 91.4 | 107.53 | 0.35 | −0.04 | Very soluble | High | No | Yes | None | −9.61 | 0.55 | 3.58 | Non-allergen |
| 10 | SDPF | 464.47 | 117.02 | 199.36 | −1.72 | 1.4 | Highly soluble | Low | No | No | None | −12.89 | 0.11 | 4.14 | Allergen |
| 11 | SGGVVK | 545.63 | 136.03 | 255.07 | −1.88 | 1.09 | Highly soluble | Low | No | Yes | None | −12.77 | 0.17 | 4.89 | Non-allergen |
| 12 | VAY | 351.4 | 91.98 | 141.75 | −0.1 | 0.21 | Highly soluble | Low | No | No | None | −10.41 | 0.55 | 3.27 | Allergen |
| Selected Active Hydrolysate Peptides | HPEPDOCK Docking Score | |||||||
| A | B | C | D | F | G | H | Q | |
| AGL | −91.468 | |||||||
| AIF | −116.310 | |||||||
| APL | −100.047 | −112.268 | −91.378 | |||||
| AVK | −99.983 | |||||||
| IIW | −172.819 | −188.427 | −147.996 | |||||
| PVI | −128.131 | −118.817 | −119.050 | |||||
| VAY | −137.310 | −144.350 | −114.143 | |||||
| Selected Active Hydrolysate Peptides | AutoDock Vina Binding Affinity (kcal.mol−1) | |||||||
| A | B | C | D | F | G | H | Q | |
| AGL | −5.462 | |||||||
| AIF | −6.723 | |||||||
| APL | −6.909 | −5.380 | −5.734 | |||||
| AVK | −5.679 | |||||||
| IIW | −7.912 | −6.660 | −6.897 | |||||
| PVI | −5.083 | −5.832 | −5.194 | |||||
| VAY | −7.420 | −4.452 | −5.988 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatoki, T.H.; Chukwuejim, S.; Udenigwe, C.C.; Aluko, R.E. In Silico Exploration of Metabolically Active Peptides as Potential Therapeutic Agents against Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 5828. https://doi.org/10.3390/ijms24065828
Fatoki TH, Chukwuejim S, Udenigwe CC, Aluko RE. In Silico Exploration of Metabolically Active Peptides as Potential Therapeutic Agents against Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2023; 24(6):5828. https://doi.org/10.3390/ijms24065828
Chicago/Turabian StyleFatoki, Toluwase Hezekiah, Stanley Chukwuejim, Chibuike C. Udenigwe, and Rotimi E. Aluko. 2023. "In Silico Exploration of Metabolically Active Peptides as Potential Therapeutic Agents against Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 24, no. 6: 5828. https://doi.org/10.3390/ijms24065828
APA StyleFatoki, T. H., Chukwuejim, S., Udenigwe, C. C., & Aluko, R. E. (2023). In Silico Exploration of Metabolically Active Peptides as Potential Therapeutic Agents against Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 24(6), 5828. https://doi.org/10.3390/ijms24065828