1. Introduction
Aldehyde dehydrogenases (ALDHs) comprise a large group of NAD(P)
+-dependent proteins responsible for the oxidation of aldehydes to their carboxylic acids. Due to their ability to detoxify a wide spectrum of endogenous and exogenous aldehydes, they are considered crucial for cellular antioxidant defense mechanisms [
1]. Apart from possessing mere metabolic roles, many ALDH members have been characterized as multifunctional proteins as they are involved in various cellular processes, such as embryogenesis, development, and differentiation [
2].
Aldehyde dehydrogenase 3A1 (ALDH3A1) is a homodimeric, cytoplasmic protein, and is a member of the ALDH superfamily. It is constitutively expressed in tissues with high antioxidant needs, such as the stomach, urinary bladder, skin, cornea, and lung, and its expression is inducible in the liver as a response to xenobiotic exposure [
3,
4,
5]. Due to its high expression in the cornea of most mammals (up to 50% of the water-soluble proteins), it has been characterized as a corneal crystallin and it has been attributed with certain structural properties [
6,
7,
8]. ALDH3A1 oxidizes medium-chain aldehydes, such as octanal, hexanal, 4-hydroxy-nonenal, and benzaldehyde [
9].
Considering its specificity for toxic aldehydes that are by-products of lipid peroxidation, ALDH3A1 is a crucial component of the antioxidant cellular defense system. It is well established that ALDH3A1 protects corneal epithelial cells from 4-HNE and UV-induced protein oxidation and apoptosis [
7,
10]. Nevertheless, novel experimental data support that ALDH3A1 exerts its cytoprotective role through additional, non-catalytic functions. For instance, it has been proposed that ALDH3A1 exhibits chaperone-like activity thus protecting corneal epithelial cells from protein aggregation, and consequently preventing the formation of corneal opacities. Quite interestingly,
Aldh3a1 knockout mice presented with a phenotype of corneal haze or clouding, analogous to lens cataracts [
11]. Additionally, ALDH3A1 appears to be implicated in cellular functions, such as cell cycle progression, and cellular proliferation [
8,
12].
The implication of ALDH3A1 in key homeostatic processes has been demonstrated by previous research findings. Specifically, transfected corneal epithelial cells expressing human ALDH3A1 exhibited lower plating efficiency, decreased DNA synthesis, elongated cell cycle, and altered expression/activation of cell cycle regulators [
13,
14]. Similarly, ALDH3A1 has been associated with the protection of corneal cells against 4-HNE-, UVC-, hydrogen peroxide-, etoposide-, and mitomycin-induced apoptosis [
13]. Finally, the expression of ALDH3A1 was downregulated in replicating explant primary cultures of the human corneal epithelium, suggesting an inverse association of ALDH3A1 with proliferation [
14].
Previously we established, via stable transfection, an isogenic cell line pair of HCE-2 (Human Corneal Epithelial) cells differing in the expression of ALDH3A1. We demonstrated that HCE-2 cells overexpressing ALDH3A1 exhibited higher viability after hydrogen peroxide and tert-butyl peroxide treatments compared to the mock-transfected HCE-2 cells [
12]. Similarly, ALDH3A1 expression resulted in higher resistance against hydrogen peroxide- and tert-butyl peroxide-induced genotoxic and oxidative effects in HCE-2 cells. Finally, the isogenic cell lines exhibited differential expression of a panel of genes related to DNA damage response, apoptosis, cell cycle regulation, DNA damage repair, and ATM/ATR signaling, as illustrated by a pathway-focused RT2 profiler™ PCR array [
5]. Based on these observations, in the present study, we sought to further investigate the modes of action underlying the cytoprotective role(s) of ALDH3A1 regarding its association with (i) cell cycle progression and proliferation, (ii) DDR, and (iii) apoptosis in human corneal epithelial cells in an approach to better understand the molecular mechanisms underlying corneal epithelial physiology.
3. Discussion
ALDH3A1 is a multifaceted enzyme, expressed in tissues with increased antioxidant needs. In the cornea, it exhibits, apart from its metabolic role, other functions associated with the cell growth and optical properties of the tissue. In a previous report, our group demonstrated the protective properties of ALDH3A1 under stress conditions and its effect on the regulation of genes involved in the DNA damage response (e.g., ATM/ATR signaling, and DNA damage repair) [
5]. In this study, we focused on the unclarified cytoprotective properties of ALDH3A1 in human corneal epithelium. For this reason, we utilized a previously established isogenic HCE-2 (human corneal epithelial) cell line pair differing only in the expression of ALDH3A1 [
5,
12]. Our findings demonstrated that ALDH3A1 expression was associated with a slower proliferation rate, higher mobility, morphological alterations, and differential expression of
E-cadherin,
ZEB1,
CDK3, and
p57. Additionally, flow cytometry analysis of the cell cycle revealed an accumulation of ALDH3A1/HCE-2 cells in the G2/M phase. Treatments of the isogenic HCE-2 cells revealed that the ALDH3A1 expression protected HCE-2 cells from H
2O
2- and etoposide-induced apoptosis. The anti-apoptotic and cell-cycle-altering effects of ALDH3A1 were accompanied by the altered expression/regulation of γ-H2AΧ and p53. Finally, ALDH3A1 was found to be localized in the nucleus, and treatment with oxidative agents (H
2O
2 or etoposide) did not alter its cellular compartmentalization. The potential NLS signal of ALDH3A1 that was previously reported (a bipartite NLS signal) [
14] was further validated in this study and found not to be responsible for the nuclear translocation of the protein.
Our experimental data showed that the ALDH3A1-expressing HCE-2 cells exhibited certain differentiations in their morphological characteristics, including a small change in their size and a less-dense cellular culture. This phenotype was accompanied by a significant downregulation of
E-cadherin. E-Cadherin is a calcium-dependent protein involved in the formation of adherent junctions among neighboring cells [
17]. Apart from its role in cell–cell adhesion, it is considered a crucial factor for the maintenance of the epithelial phenotype of cells as well as for contact inhibition [
18]. Consequently, downregulation of E-cadherin is associated with higher cellular mobility and metastasis in cancer [
19]. The downregulation of
E-cadherin as a result of ALDH3A1 expression is in line with our optical observations demonstrating the formation of denser cultures with more adhered cells in the case of the non-expressing, compared to the ALDH3A1-expressing, HCE-2 cells. To the extent of our knowledge, this is the first time that the cell morphology and the expression of
E-cadherin in relation to ALDH3A1 are being studied in corneal epithelial cells. However, the association of ALDH3A1 with E-cadherin expression has been previously studied in cancer cells. Specifically, Terzuoli et al. (2019) demonstrated that ALDH3A1
high non-small-cell lung carcinoma (NSCLC) and melanoma cells exhibited downregulation of E-cadherin compared to ALDH3A1
low cells [
20]. Similarly, Wu et al. (2016) reported that ALDH
bright SGC-7901 (gastric cancer cell lines) cells had lower mRNA levels of E-cadherin compared to ALDH
low SGC-7901 cells [
21].
Next, we examined the wound-healing potential of ALDH3A1 and showed that the ALDH3A1/HCE-2 cells have higher mobility/wound-healing capacity compared to the mock-transfected HCE-2 cells. We also examined, through QPCR, the expression rates of
slug,
vimentin,
ZEB1, and
ZEB2, and showed that
ZEB1 is significantly upregulated in the ALDH3A1-expressing HCE-2 cells. ZEB1 (Zinc finger E-box binding homeobox 1) is a transcription factor, known for its role in the Epithelial–Mesenchymal Transition (EMT) in cancer. Specifically, overexpression of ZEB1 is associated with EMT, tumor invasion, and metastasis [
22]. In non-cancerous epithelial cells, ZEB1 attenuates cell adhesion, partly by downregulating E-cadherin and other cell-adhesion-related proteins [
23,
24]. Consequently, it promotes mobility and de-differentiation of epithelial cells [
24]. Feng et. al. (2004) examined the expression and enzymatic activity of ALDH3A1 after an alkali burn in mice cornea. They demonstrated that ALDH3A1 is downregulated as a result of alkali burn, however, its expression is gradually restored during healing [
25]. Additionally, Terzuoli et. al. (2019) showed that ALDH3A1 expression correlated with an increased expression of ZEB1 in HCC4006 (metastatic NSCLC) cells [
20]. To this end, our observations suggest that ALDH3A1 expression induces the transition of HCE-2 cells from a phenotype with tight cell–cell interactions towards a state with low cell–cell adhesion with high regenerative properties.
Subsequently, we demonstrated that ALDH3A1 had a negative effect on the cellular proliferation of HCE-2 and induced the downregulation of
CDK3, and
p57. Cyclin-dependent kinases (CDKs) comprise a subfamily of serine/threonine kinase proteins involved in the control of cell cycle progression and proliferation. CDKs activity is positively regulated through the formation of heterodimeric complexes with specific cyclins [
26]. This complex is responsible for modulating the progression of cell cycle phases. Protein p57, encoded by gene
CDKN1C, is a cyclin/CDK inhibitor that causes the arrest of cells at the G1 phase [
27]. In addition, p57 is involved in the differentiation of certain cell types both through CDKs inhibition and thus cell cycle exit, as well as through mechanisms that are not related to CDKs [
28]. The differential expression of these cell cycle regulators does not come as a surprise considering the effect of ALDH3A1 on cell growth. Our data are in line with a previous study by Pappa et al. (2005) showing that ALDH3A1 exhibits suppressive functions on cell proliferation accompanied by reduced CDK activity and altered protein levels of cyclins A, B, and E, E2F1 and p21 in HCE (human corneal epithelial) cells [
14]. Additionally, Qu et al. (2020) demonstrated that the stable transfection of SCC15 and SCC25 (oral squamous carcinoma cell lines) cells to express ALDH3A1 resulted in slower proliferation rates. However, in contrast to our study, they showed that the ALDH3A1-expressing SCC15 and SCC25 cells also exhibited weaker migration and invasion potential [
29].
Our results on the effect of ALDH3A1 on cell proliferation prompted us to study the progression of cell cycle on the isogenic cell line pair. Our flow cytometry analysis revealed a “delay” in ALDH3A1/HCE-2 cells at the G2/M phase. To the extent of our knowledge, this is the first time that the effect of ALDH3A1 on cell cycle progression is being studied in human corneal cells. Qu et al. (2020) performed a similar study, however in oral squamous carcinoma cells, and reported that, in agreement with our results, the ALDH3A1-expressing SCC15 and SCC25 cells also exhibited a delay at the G2/M phase [
29].
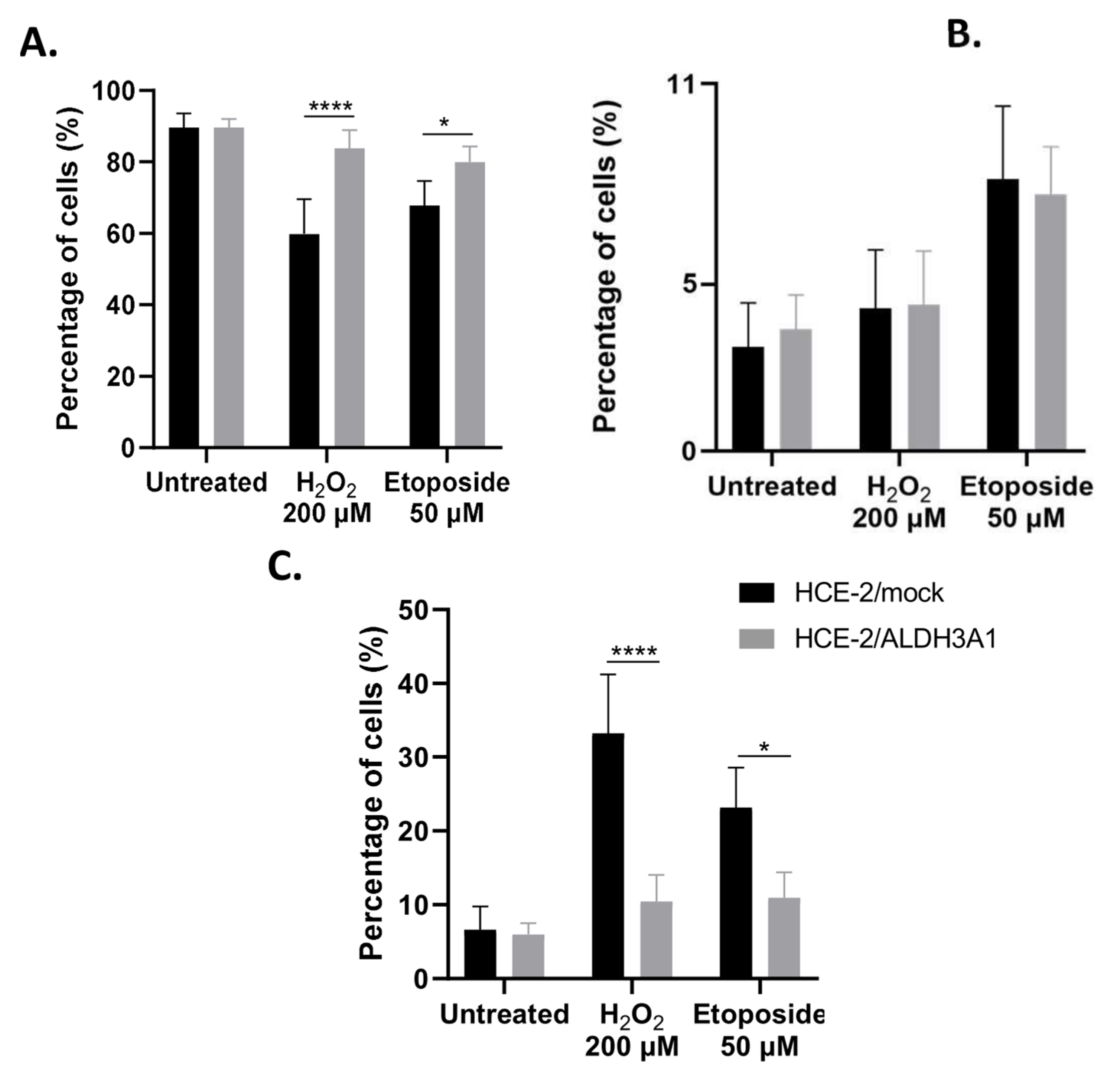
Next, we treated HCE-2 cells with either H
2O
2 or etoposide for 16 h and investigated their apoptotic status by flow cytometry. We chose these specific stressors due to their different mode of action. H
2O
2 is a common oxidative agent, while etoposide acts mainly through inducing double-strand DNA break formation by inhibiting topoisomerase II [
30,
31]. Our data revealed that ALDH3A1 expression exerted an anti-apoptotic role in HCE-2 cells under both conditions tested. Our findings agree with the findings reported in previous studies. Specifically, Saiki et al. (2018) reported that a higher percentage (2.2 fold) of salivary stem/progenitor cells (SSPCs) isolated from irradiated
Aldh3a1(-/-) murine submandibular glands (SMGs) were early or late apoptotic compared to the respective SSPCs isolated from wild-type SMGs [
32]. Similarly, the anti-apoptotic effect of ALDH3A1 was demonstrated in studies reporting that ALDH3A1 expression protected HCE cells from mitomycin C, VP-16, and H
2O
2-induced DNA fragmentation [
7,
33]. Along these lines, our group has previously shown that, after a 16 h treatment with either H
2O
2 or tert-butyl hydroperoxide, the ALDH3A1-expressing HCE-2 cells managed to retain their viability more effectively than the mock-transfected cells [
12].
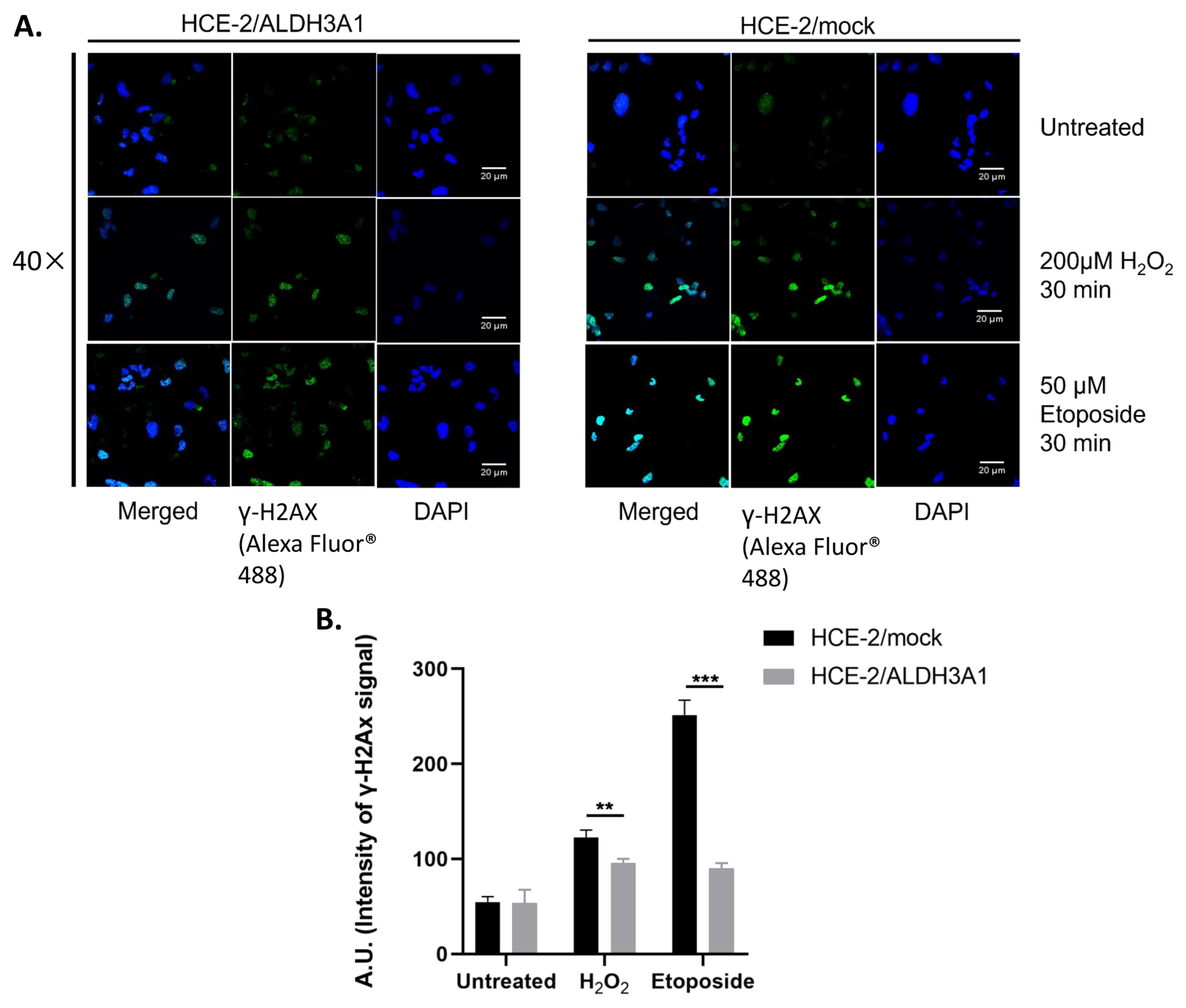
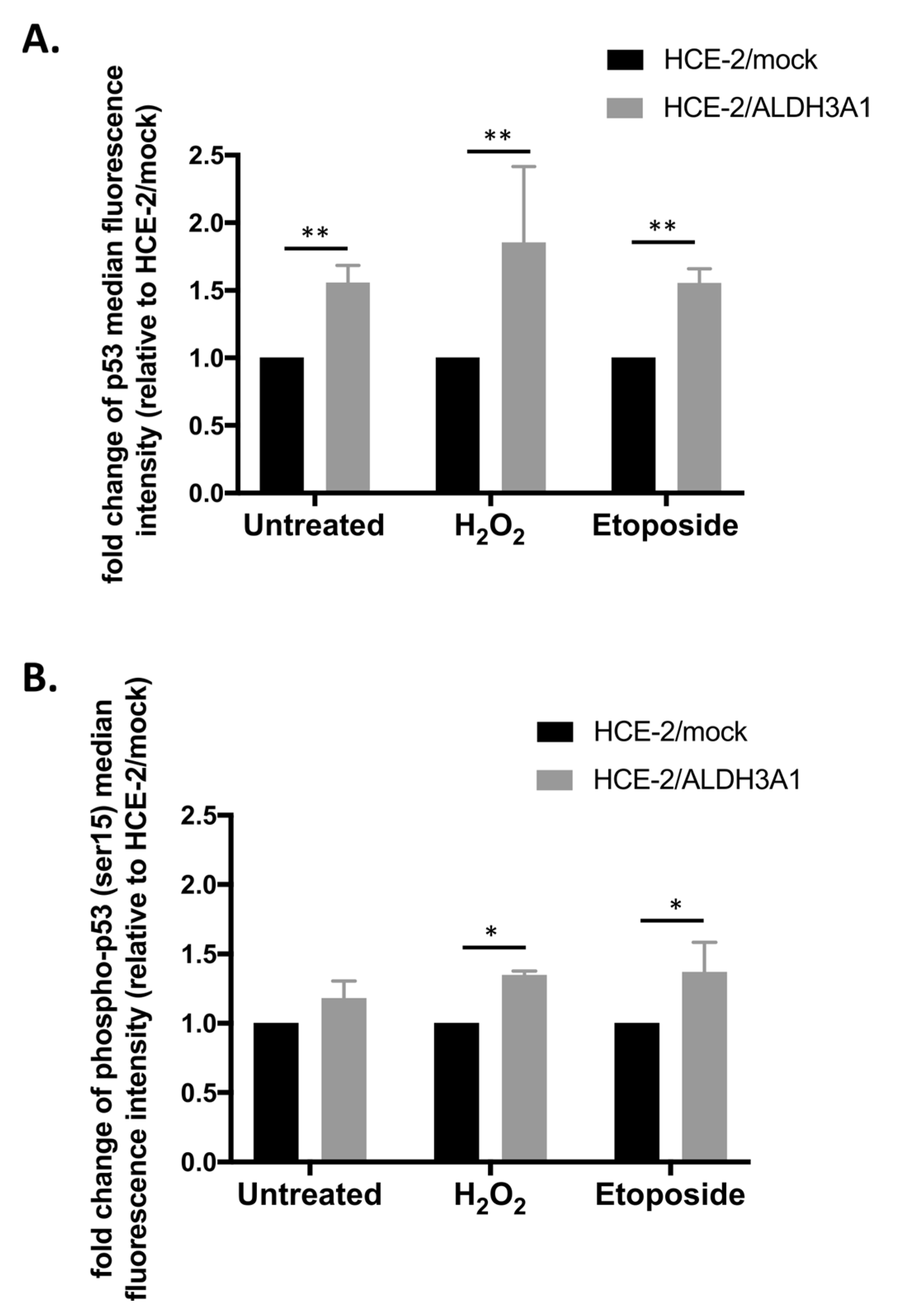
Next, we observed, through either immunofluorescence or flow cytometry, that the expression of ALDH3A1 is associated with lower activation/foci formation of γH2AX as well as higher protein levels of p53 and phospho-p53(ser15) after treatments with H
2O
2 or etoposide. H2AX is being phosphorylated at serine 139 (γ-H2AX) and thus being activated as a response to DNA damage and specifically double-strand breaks [
34]. The activation of γ-H2AX subsequently induces its accumulation at the DSBs sites leading to the formation of foci, which are visible through immunofluorescence microscopy. γH2AX is involved in the DNA damage response pathway by recruiting certain DNA repair proteins and it is considered an important, early DNA damage marker [
35]. Our findings are in accordance with previously published data, demonstrating lower H
2O
2−, tert-butyl peroxide-, and etoposide-induced DNA damage levels in ALDH3A1/HCE-2 cells compared to mock-transfected HCE-2 cells [
5]. Similarly, Jang et al. (2013) reported that ALDH3A1-overexpressing human bronchial epithelial cells exhibited lower cigarette-smoking-induced H2AX Ser-319 phosphorylation levels [
36].
The tumor protein p53 plays a crucial role in the response of cells under different stress conditions, by regulating cell-cycle progression and thus inducing growth arrest or apoptosis [
37]. p53 is involved in both antioxidant and DNA damage responses, while it promotes DNA damage repair [
38]. Specifically, DNA damage induces the stabilization/activation of p53 through phosphorylation at various sites, with Ser15 being a common phosphorylation target under this specific condition [
37]. Subsequently, the activation of p53 results in the induction of a panel of cellular signaling pathways related to DNA damage repair, cell death as well as cell cycle arrest, primarily at the G1/S, but also at the G2/M checkpoint [
39]. Indeed, the upregulation of p53 in the ALDH3A1-expressing untreated HCE-2 cells could be associated with the accumulation of the ALDH3A1/HCE-2 cells at the G2/M phase. The increased levels of p53 even under non-stress conditions could be beneficial by maintaining cells in a state of “alertness”. This would enable them to react faster in case of DNA damage, and thus, more effectively induce the DNA damage response. The enhancement of DDR would eventually lead to quicker and more efficient DNA damage repair and increased cell viability. Our data, demonstrating higher levels of total and phospho-p53 along with lower DNA damage and apoptosis levels in the ALDH3A1/HCE-2 cells, are in agreement with this hypothesis. In accordance with our findings, Fan et al. (2021) reported that ALDH3A1 was positively associated with the total p53 in tissues of lung adenocarcinoma and that a knockdown of ALDH3A1 in the lung adenocarcinoma cell line A549 resulted in lower protein levels of total p53 [
40]. Similarly, Koppaka et al. (2016), demonstrated that expression of ALDH3A1 in the corneal epithelium hTCEpi cell line was associated with increased cytosolic and nuclear p53 levels. Furthermore, even though knockout of
aldh3a1 did not have any effect on p53, dual knockout of both
aldh3a1 and
aldh1a1 in mice resulted in the total loss of total p53 and phospho-p53 in the corneal tissue [
41].
Finally, we studied the sub-cellular localization of ALDH3A1 and found that it is located mainly in the cytoplasmic compartment, but also, to a lesser extent, in the nucleus of the HCE-2 cells under non-stress conditions, and that treatments with either oxidative or genotoxic agents did not further enhance its nuclear localization. The nuclear localization of ALDH3A1 in human corneal epithelial cells has been reported in other studies [
14,
42]. Interestingly, Pappa et al. (2005) reported a potential bipartite (BP) NLS at the ALDH3A1 amino acid sequence [
14]. BP NLS consists of two basic clusters comprised of 2–3 amino acids and a 9–10 amino acid spacer that links these two positively charged regions [
43]. In the case of ALDH3A1, this putative NLS is comprised of two lysine regions linked by a 10-residue spacer (265KKSLKEFYGEDAKKSRD281). Therefore, to deactivate this putative NLS, we substituted its basic lysine residues with the non-polar amino acid alanine. Our experiments demonstrated that this substitution did not inhibit the translocation of ALDH3A1 at the nucleus of HCE-2, thus the mechanism underlying ALDH3A1 nuclear localization is not related to this specific sequence. The detection of the protein in the nucleus of corneal cells, implies novel, yet-uncharacterized ALDH3A1 functions, which warrants further investigation. Consequently, the existence of ALDH3A1 in the nucleus of cells could allow its direct interaction with certain regulatory proteins. This mode of action should be unsurprising in relation to the corneal epithelium. For instance, ferritin, an iron-sequestering molecule, mainly known as a metabolic, cytoplasmic protein, was also found to be nuclear in the avian corneal epithelium and the mechanistic studies for its nuclear localization identified a tissue-specific nuclear transporter termed ferritoid. In the nucleus, ferritin was found to directly interact both with DNA and gene regulatory proteins and to exert a protective role against UV-induced oxidative damage [
44,
45].
4. Materials and Methods
4.1. Materials
The human corneal epithelium cell line HCE-2 was obtained from ATCC (Manassas, VA, USA). Culture medium, antibiotics, fetal bovine serum (FBS), trypsin, and medium additives were either from Biosera (East Sussex, UK), Gibco (Life Technologies, Carlsbad, CA, USA) or Sigma-Aldrich Co. (St. Louis, MO, USA). Lipofectamine 2000 was purchased from Invitrogen (ThermoFischer Scientific, Waltham, MA, USA) and hygromycin was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Etoposide and hydrogen peroxide was purchased by Sigma-Aldrich (St. Louis, MO, USA). The Pierce™ BCA Protein assay was from Thermo Scientific (Rockford, IL, USA). Chemiluminescence reagents and polyvinylidene difluoride (PVDF) membranes were purchased from Millipore (Bedford, MA, USA). The primary antibodies anti-γH2AX, anti-p53, anti-phospho-p53 (ser15), and anti-H3 were from Cell Signaling Technology (Boston, MA, USA), while anti-β tubulin was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) and anti-ALDH3A1 from Abnova (Taipei City, Taiwan). Goat anti-mouse and anti-rabbit IgG horseradish-peroxidase-conjugated antibodies were purchased from Millipore (Bedford, MA, USA), goat anti-rabbit Alexa Fluor® 488-conjugated secondary antibodies for immunofluorescence were obtained from Jackson Immunoresearch (Cambridge House, St. Thomas Place, Ely CB7 4EX, UK), while goat anti-mouse and anti-rabbit Alexa Fluor® 647-conjugated secondary antibodies for flow cytometry were from Cell Signaling Technology (Boston, MA, USA). DAPI was obtained from Biotium (Landing Parkway Fremont, CA, USA), while NucleoZOL was from Macherey-Nagel (Düren, Germany). dNTPs, primers, random hexamers, and PrimeScript reverse transcriptase were from Invitrogen (ThermoFischer Scientific, Waltham, MA, USA). Finally, the KAPA SYBR Fast Master Mix was obtained from Kapa Biosystems (Hoffmann-La Roche, Basel, Switzerland).
4.2. Cell Cultures
The HCE-2 cell line was obtained from American Type Culture Collection (ATCC, Middlesex, UK) and was cultured in a mixture (1:1) of Ham’s F-12 nutrient mixture and Dulbecco’s modified Eagle’s medium (DMEM/F12) supplemented with 15% FBS, 0.1 μg/mL cholera toxin, 0.5% DMSO, 5 μg/mL insulin, 40 μg/mL gentamycin, 10 ng/mL epidermal growth factor and 100 units/mL penicillin/streptomycin solution. HCE-2/ALDH3A1 and HCE-2/mock stable transfected cells [
5,
12] were cultured in the same medium in the presence of 0.2 mg/mL hygromycin B. The cells were maintained at 37 °C with 5% CO
2 in a humidified incubator.
4.3. Analysis of Cellular Morphology
The cells were collected through trypsinization, resuspended in growth medium, washed with PBS, and finally analyzed with respect to their forward (FSC) and side scatter (SSC) profile in an Attune NxT flow cytometer. To quantify the morphological changes of HCE-2 cells concerning granularity, cellular size, and cell surface topography as a result of ALDH3A1 expression, the median fluorescence intensity values of FSC and SSC were determined with Flowjo software (BD Biosciences, Franklin Lakes, NJ, USA) [
15]. The cells were also photographed by a ZEISS Primovert light microscope (Zeiss, Göttingen, Germany) equipped with a digital camera (Axiocam ERc 5 s by Zeiss, Oberkochen, Germany) under the 20× and 40× lenses.
4.4. Real-Time PCR
Real-time PCR was performed as previously described with minor modifications [
5]. NucleoZOL reagent was used for isolating total RNA from cells according to the manufacturer’s instructions. Primers (
Table 1) were designed with the Primer Express 3.0 software (Applied Biosystems, Waltham, MA, USA).
Total RNA (4.5 μg) was used for cDNA synthesis with the SuperScript™ First-Strand Synthesis System according to the manufacturer’s instructions. For Real-time PCR, KAPA SYBR Fast Master Mix was used according to the manufacturer’s instructions. The reactions were performed with an Applied Biosystems Step One instrument. The reactions were performed in triplicate, while data derived from three independent experiments were used for the final quantification of the gene expression ratio.
β-Actin was used for normalizing gene expression according to the 2
−ΔΔCT method [
46].
4.5. Scratch Assay
For the scratch assay, 7.5 × 105 cells were seeded on 6-well plates a day prior to the experiment. Then, plates were washed twice with PBS and a straight scratch was introduced by drawing a sterilized pipette tip on the cellular confluent monolayer. The scratch closure was subsequently monitored every 12 h by a ZEISS Primovert light microscope (Zeiss, Göttingen, Germany) equipped with a digital camera (Axiocam ERc 5 s by Zeiss, Oberkochen, Germany). The acquired images were analyzed by ImageJ software (National Institute of Health, Bethesda, MD, USA) to estimate the average % percentage of gap closure determined as the change in gap width compared to the initial scratch.
4.6. Cell Counting and Growth Rate Determination
To investigate the growth rates of the isogenic cell line pair, 4 × 10
5 HCE-2/mock or HCE-2/ALDH3A1 cells were plated on 10 cm culture dishes. The number of cells was determined at various time points for up to 84 h after staining with trypan blue using a Neubauer hemocytometer. The growth rate (the number of doublings occurring/day) and the doubling time were determined by the online calculator
https://doubling-time.com/compute.php (accesed on 1 August 2022).
4.7. Cell Cycle Analysis
Approximately 1.5 × 106 cells were seeded in 10 cm culture dishes and were placed in a humidified incubator (37 °C 5% CO2). Seeding was considered as t0 and then the cells were further cultured for different time points (16, 24, 36, 48, 72 h). At each time point, the cells were washed with PBS, fixed with ice-cold 75% ethanol, and incubated for at least 24 h at −20 °C. The cells were then washed with PBS, counted, suspended in RNase A (1 mg/mL), stained with propidium iodide (50 μg/mL), and incubated in the dark for 40 min. Finally, the % percentage of cells in each cell cycle phase was determined through flow cytometry analysis.
4.8. Apoptosis Analysis
Approximately 1.5 × 106 of HCE-2/mock or HCE-2/ALDH3A1 cells were seeded on 10 cm culture dishes and placed in a humidified incubator (37 °C 5% CO2) a day prior to the experiment. Then, the cells were treated with either 200 μM H2O2 or 50 μM etoposide for 16 h. Subsequently, the early and late apoptotic percentage of cells was determined using an Annexin-FITC/PI apoptosis detection kit (BD Bioscience, San Jose, CA, USA) according to the manufacturer’s instructions. Briefly, the cells were collected with trypsin and washed twice with PBS. The cell pellet was then resuspended in Annexin V binding buffer to achieve a final concentration of 1 × 106 cells per 1 mL of buffer. Subsequently, 100 μL of the cell suspension (1 × 105 cells) was dyed with 5 μL of Annexin V and incubated for 13 min in the dark. Finally, the samples were stained with 5 μL of propidium iodide (PI) and analyzed by flow cytometry.
4.9. Immunofluorescence
Immunofluorescence was performed as previously described with few modifications [
16]. Specifically, 1 × 10
5 cells were grown in a monolayer on the surface of coverslips a day before the experiment. The cells were treated with either 200 μM H
2O
2 or 50 μM etoposide for 30 min and then washed with PBS and fixed with 4% formaldehyde in PBS for 20 min. Subsequently, the cells were washed with PBS (5 min, ×3) and paraformaldehyde was neutralized with the addition of 1 M of glycine (pH 8.5). The cells were permeabilized with 0.1% Triton Χ-100 in PBS for 5 min and blocking was performed by 5% (
w/v) BSA in PBS for 5 min. Primary anti-γH2AX was used at a dilution of 1:500 (1 h, RT). The samples were then washed with PBS (×3) before incubation with Alexa Fluor
® 488-conjugated secondary antibody in PBS at a dilution of 1:400 (30 min, RT). Finally, the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (1 μg/mL), washed, and finally mounted with MOWIOL (Calbiochem, Bad Soden, Germany). The samples were imaged with a 40×/NA 1.45 objective and an Andor Ixon + 885 digital camera on a customized Andor Revolution Spinning Disk Confocal System built around an IX81; Olympus stand (BioImaging-GR Facility, DUTH), while Andor IQ 2.7.1 software (Andor Technology, Belfast, UK) was used for image acquisition. Visualization of different samples was performed with the same exposure time and settings. To comparatively analyze the formation of γH2AX foci at the nucleus of cells, quantification methods were used, as previously described, with several modifications [
47,
48]. Specifically, each cell was scored as class 0 to 4 based on the intensity of Alexa Fluor-488 emission. Class 0 was representative of no signal, while class 4 was representative of a super bright signal (
Figure 9).
For each sample, 100 cells were classified, while each cell was assigned a value based on its class. The overall score of each sample (total values of 100 cells) consequently ranged from 0 (100% in class 0) up to 400 (100% of cells in class 4). By this method, the intensity of γH2AX staining was expressed in arbitrary units.
4.10. Flow Cytometric Analysis of Total P53 and Phospho-p53 (Ser15)
To analyze the levels of total p53 and phospho-p53 (Ser15), cells were treated with either 200 μM H2O2 or 50 μM etoposide for 24 h. Then, the cells were collected through trypsinization, washed with PBS, and fixed with 4% formaldehyde at 25 °C for 15 min. Subsequently, they were washed once more with PBS and permeabilized with 100% ice-cold methanol for 10 min on ice. After permeabilization, the cells were washed (PBS) and immunostained with primary rabbit anti-p53 (1:800) or mouse anti-phospho-p53 (Ser15) (1:1600) antibodies for 1 h at room temperature. After an additional step of washing with PBS, the cells were stained with secondary goat anti-mouse or anti-rabbit AlexaFluor-647-conjugated (1:1000) antibody for 30 min at room temperature under dark conditions. Finally, the samples were washed and analyzed on the Attune NxT flow cytometer.
4.11. Western Immunoblotting
Western immunoblotting was performed as described previously with minor modifications [
16]. To separate the nuclear and cytosolic parts, the cell pellets were lysed using a fractionation lysis buffer containing 10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 1.5 mM MgCl
2, 0.2% NP-40 and supplemented with proteinase inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 2.5 μg/mL aprotinin, 2.5 μg/mL pepstatin A, and 2.5 μg/mL leupeptin), and a phosphatase inhibitor cocktail. Cell lysates were incubated for 30 min at 4 °C, vortexed every 10 min, and centrifuged at 1000×
g for 10 min to isolate the cytosolic (supernatant) and nuclear (pellet) fractions. Protein quantification was performed with a Pierce™ BCA Protein Assay kit, according to the manufacturer’s instructions.
Approximately 40 μg of cytosolic and 15 μg of nuclear samples were prepared and subjected to SDS-PAGE on Tris-Glycine gels. The resultant separated proteins were transferred to PVDF membranes while non-specific sites were blocked with 5% non-fat dry milk in TBST (150 mM NaCl, 100 mM Tris pH 7.5, and 0.1% v/v Tween-20), at room temperature for 2 h. The membranes were hybridized overnight at 4 °C with primary antibodies against ALDH3A1 (1:1000), H3 (1:1000), and β-tubulin (1:5000). Then, the membranes were incubated with secondary horseradish-peroxidase-conjugated anti-rabbit and anti-mouse IgG antibodies (1:5000) for 1 h at RT and immunoblot bands were developed through Chemidoc MD Imaging System (Biorad, Hercules, CA, USA). When necessary, blots were stripped with a stripping buffer (62.5 mM Tris-HCl pH 6.7, 2% SDS, 6 μL/mL 2-mercaptoethanol) and re-hybridized with an alternate antibody. Protein expression levels were quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA), and the relative quantity of the protein with respect to β-tubulin or H3 protein was estimated.
4.12. Analysis of the Enzymatic Activity of ALDH3A1
The enzymatic activity of ALDH3A1 was determined in the cytosolic and nuclear extracts of the untreated HCE-2/ALDH3A1 cells as previously described [
49]. A molar extinction coefficient of 6.22 mM
−1/cm
−1 was applied for NADPH, while the activity is expressed as the number of nmoles of NADPH produced per min and per mg of (total) protein when using benzaldehyde as a substrate.
4.13. Site-Directed Mutagenesis
Site-directed mutagenesis was utilized to introduce eight specific base pair substitutions altering the codons for lysine 265, lysine 266, lysine 277 and lysine 278 to alanines to “deactivate” the potential bipartite Nuclear Localization Signal (NLS) (265KKSLKEFYGEDAKKSRD281) [
14] on the hALDH3A1 plasmid construct [
16]. Site-directed mutagenesis was performed as previously described [
12] via the Transformer TM kit by Clontech (TaKaRa Bio Inc., Kusatsu, Japa) according to the manufacturer’s instructions. Specifically, three oligonucleotides were synthesized. One acted as a selection primer, inducing the deactivation of the unique restriction site for NruI (underlined sequence) in the hALDH3A1 plasmid construct and two mutagenic primers were designed to introduce the desired base pair substitutions in the coding region of ALDH3A1 cDNA (underlined sequence): Selection primer: 5′-C AGC CTC GCG
GCG CG
C ACG CCA GCA AG-3′, Mutagenic primer A: 5′-CAA ATT GTG GAG AAG CTC
GCG
GCG TCA CTG AAA GAG TTC-3′ and Mutagenic primer B: 5′-C GGG GAA GAT GCT
GCG
GCA TCC CGG GAC TAT GGA AG-3′. Both the selection primer and the two mutagenic primers were simultaneously annealed to one strand of the denaturated hALDH3A1 plasmid DNA. Then, the mutated strand was further synthesized and ligated using T4 DNA polymerase and T4 DNA ligase. To ensure the selection of the constructs with the mutated NruI restriction site, digestion with NruI was performed. Subsequently, mutS
E. coli bacteria (defective mismatch repair mechanism) were transformed with the digested plasmids.
E. coli cells were then amplified and their plasmid DNA was isolated. The mixed plasmid pool was once more digested with NruI and re-transformed in mutS
E. coli cells. Finally, single mutS
E. coli colonies were amplified and their plasmid DNA was isolated and sequenced to confirm the Lys 265, Lys 266, Lys 277, and Lys 278 alterations (hALDH3A1_NLS_mut).
4.14. Transient Transfection
HCE-2 cells were transiently transfected with both hALDH3A1 (wild-type) and hALDH3A1_NLS_mut (mutated at the potent NLS region) constructs. For transient transfection, 2 × 105 HCE-2 cells were plated on 10 cm culture plates a day prior to the experiment. Then, 16 μg of DNA and 40 μL of Lipofectamine reagent 3000 were mixed and added in 3 mL of Opti-MEM reduced serum medium and applied to the culture plates. After a 6 h incubation (37 °C, 5% CO2), Opti-MEM medium was replaced with fully supplemented DMEM-F12. The cells were then washed and the expression and cellular localization of ALDH3A1 were monitored by Western immunoblotting analysis.
4.15. Statistical Analysis
At least three independent experiments were performed for each sample per condition tested. All values are expressed as the mean ± S.D. GraphPad Prism software (version 8.3.0, GraphPad, Boston, MA, USA) was used for statistical analyses and graph construction. A comparison of the results between the two groups was performed by the Student’s t-test. An analysis of two variables among multiple groups was performed with a two-way ANOVA, followed by Tukey’s multiple comparison tests. A value of p < 0.05 was considered significant.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}