

Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity
 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
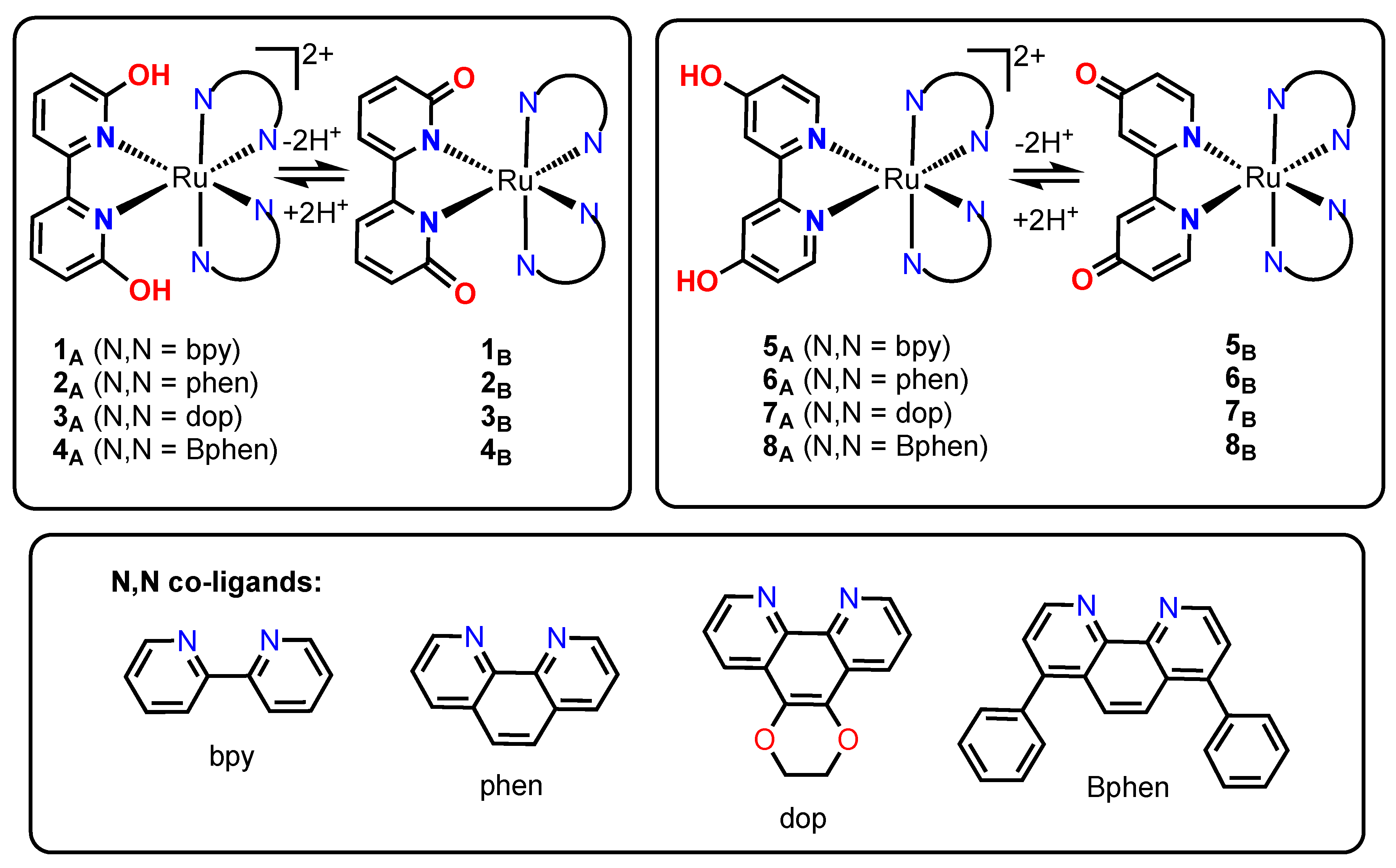
2.1. Synthesis
2.2. Characterization of the Compounds
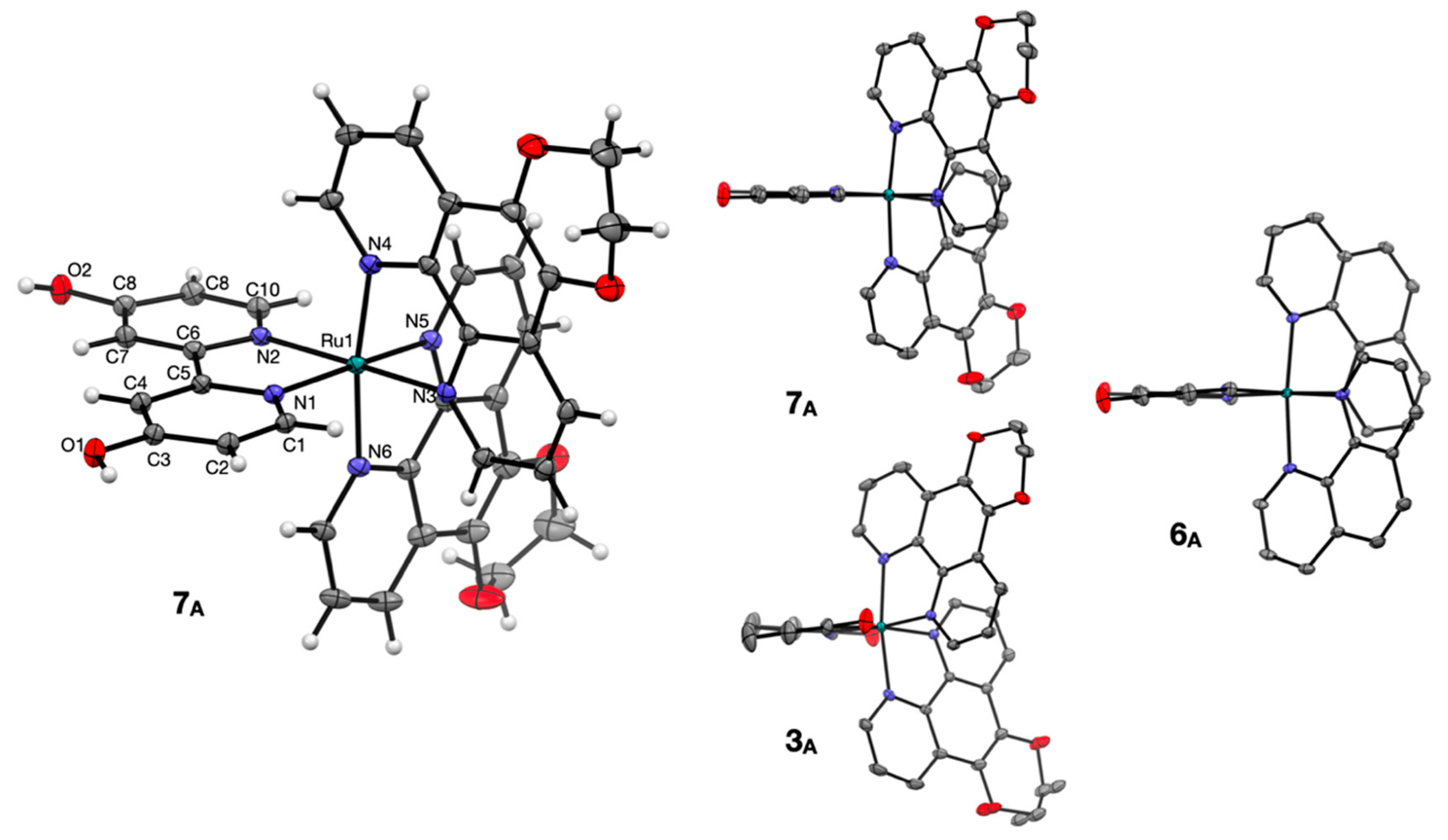
2.3. Crystallography
2.4. UV-Vis Spectroscopy
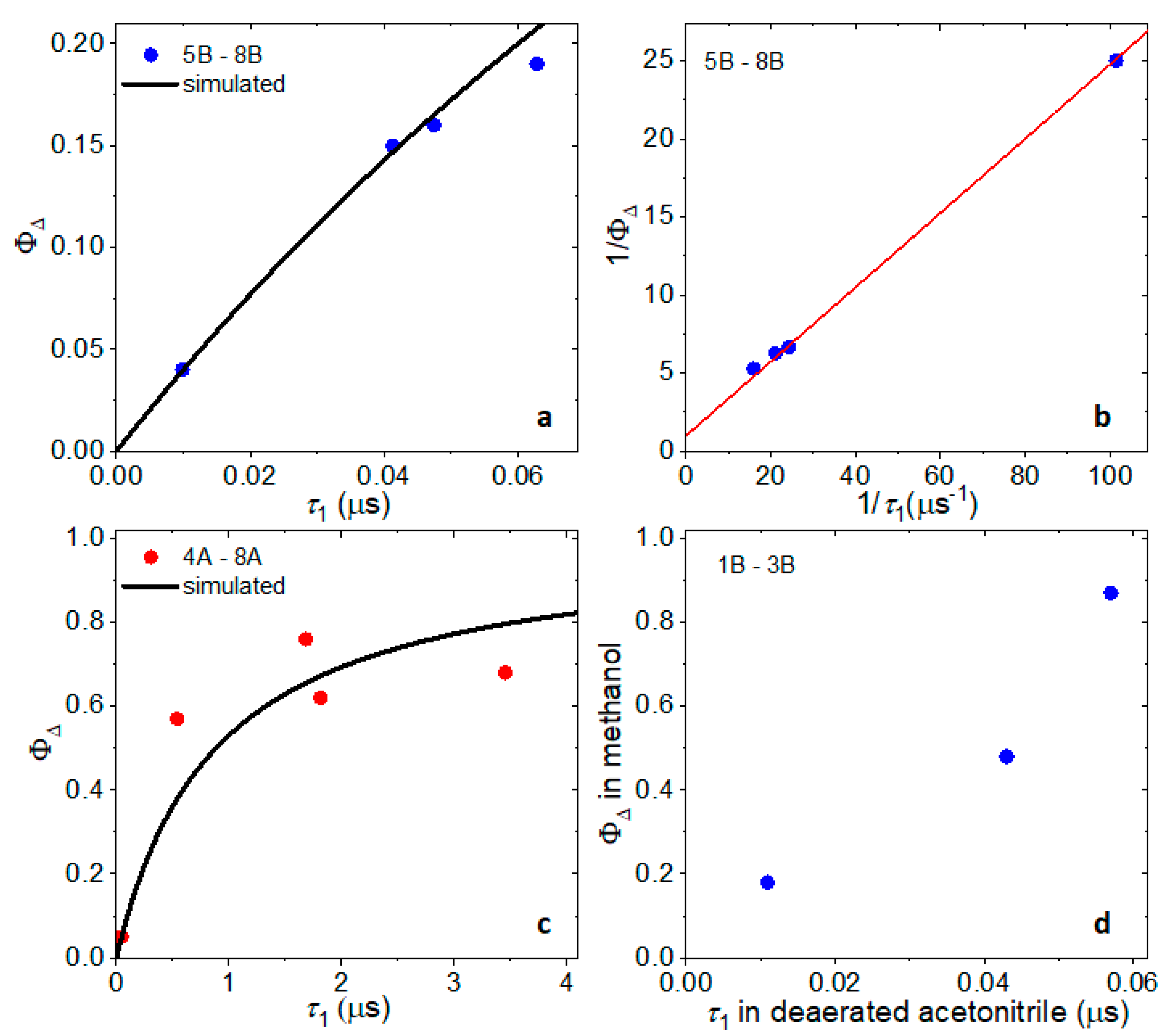
2.5. Singlet Oxygen Quantum Yields
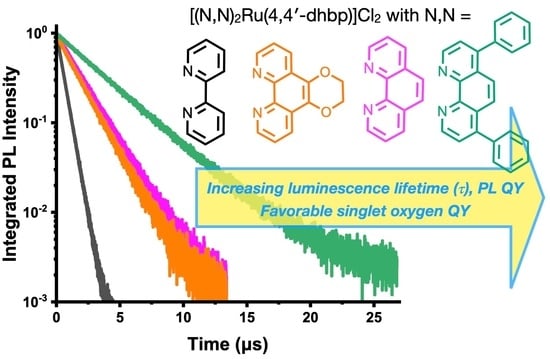
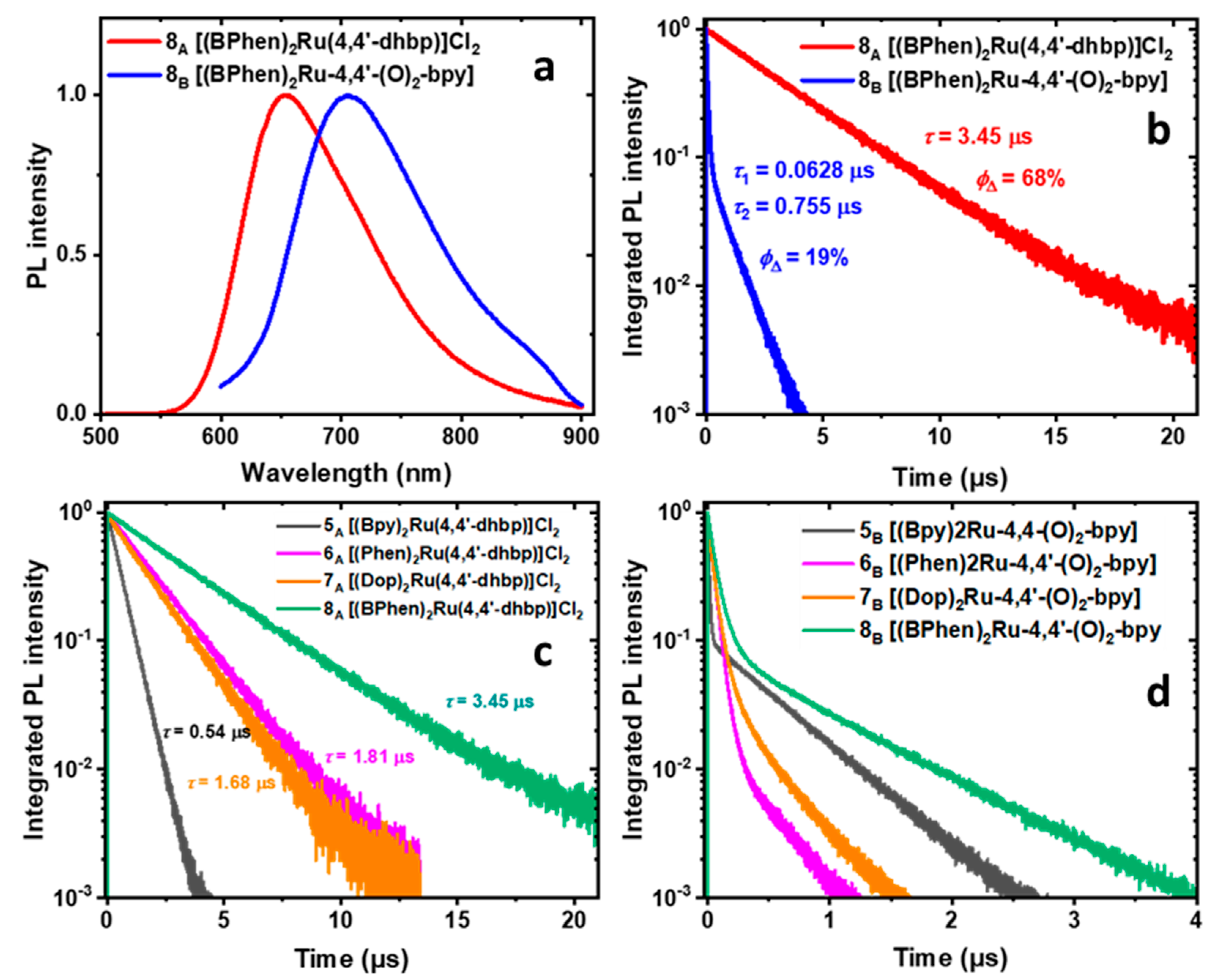
2.6. Photoluminescence, Photoluminescence Quantum Yield, and Luminescence Lifetime Studies
2.7. Cellular Cytotoxicity and Photocytotoxicity Data as Related to the Log(Do/w) Values and Photophysical Properties of These Compounds
3. Materials and Methods
3.1. Synthesis and Characterization Data
3.1.1. General
3.1.2. Synthesis and Characterization of 7A = [(dop)2Ru(4,4′-dhbp)]Cl2
3.1.3. Synthesis and Characterization of the Deprotonated Complexes 5B, 6B, and 7B
3.2. Single Crystal X-ray Diffraction
3.3. Spectroscopy
3.3.1. Photoluminescence Measurements
3.3.2. Photoluminescence Quantum Yield Measurement
3.3.3. Luminescence Lifetime Measurements
3.3.4. Singlet Oxygen Quantum Yields
3.4. Log(Do/w) Measurements
3.5. Cellular Viability Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fiedler, E.C.; Hemann, M.T. Aiding and Abetting: How the Tumor Microenvironment Protects Cancer from Chemotherapy. Annu. Rev. Cancer Biol. 2019, 3, 409–428. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef]
- Papish, E.T.; Oladipupo, O. Factors that Influence Singlet Oxygen Formation vs. Ligand Substitution for Light Activated Ruthenium Anticancer Compounds. Curr. Opin. Chem. Biol. 2022, 68, 102143. [Google Scholar] [CrossRef] [PubMed]
- Hachey, A.C.; Havrylyuk, D.; Glazer, E.C. Biological activities of polypyridyl-type ligands: Implications for bioinorganic chemistry and light-activated metal complexes. Curr. Opin. Chem. Biol. 2021, 61, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Roque, J., III; Havrylyuk, D.; Barrett, P.C.; Sainuddin, T.; McCain, J.; Colón, K.; Sparks, W.T.; Bradner, E.; Monro, S.; Heidary, D.; et al. Strained, Photoejecting Ru(II) Complexes that are Cytotoxic Under Hypoxic Conditions. Photochem. Photobiol. 2020, 96, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bai, L.; Zhang, P.; Zhao, H.; Zhou, Q. The Development of Ru(II)-Based Photoactivated Chemotherapy Agents. Molecules 2021, 26, 5679. [Google Scholar] [CrossRef] [PubMed]
- Lanquist, A.P.; Gupta, S.; Al-Afyouni, K.F.; Al-Afyouni, M.; Kodanko, J.J.; Turro, C. Trifluoromethyl substitution enhances photoinduced activity against breast cancer cells but reduces ligand exchange in Ru(II) complex. Chem. Sci. 2021, 12, 12056–12067. [Google Scholar] [CrossRef]
- White, J.K.; Schmehl, R.H.; Turro, C. An overview of photosubstitution reactions of Ru(II) imine complexes and their application in photobiology and photodynamic therapy. Inorg. Chim. Acta 2017, 454, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Qu, F.; Lamb, R.W.; Cameron, C.G.; Park, S.; Oladipupo, O.; Gray, J.L.; Xu, Y.; Cole, H.D.; Bonizzoni, M.; Kim, Y.; et al. Singlet Oxygen Formation vs Photodissociation for Light-Responsive Protic Ruthenium Anticancer Compounds: The Oxygenated Substituent Determines Which Pathway Dominates. Inorg. Chem. 2021, 60, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kar, P.; Das, A.; Ghosh, H.N. Photophysical properties of ligand localized excited state in ruthenium(ii) polypyridyl complexes: A combined effect of electron donor–acceptor ligand. Dalton Trans. 2011, 40, 9765–9773. [Google Scholar] [CrossRef] [PubMed]
- Toupin, N.P.; Nadella, S.; Steinke, S.J.; Turro, C.; Kodanko, J.J. Dual-Action Ru(II) Complexes with Bulky π-Expansive Ligands: Phototoxicity without DNA Intercalation. Inorg. Chem. 2020, 59, 3919–3933. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Monro, S.; Sobotta, F.H.; Colón, K.L.; Sainuddin, T.; Stephenson, M.; Sampson, E.; Roque, J.; Yin, H.; Brendel, J.C.; et al. Predictive Strength of Photophysical Measurements for in Vitro Photobiological Activity in a Series of Ru(II) Polypyridyl Complexes Derived from π-Extended Ligands. Inorg. Chem. 2019, 58, 3156–3166. [Google Scholar] [CrossRef]
- Plaetzer, K.; Krammer, B.; Berlanda, J.; Berr, F.; Kiesslich, T. Photophysics and photochemistry of photodynamic therapy: Fundamental aspects. Lasers Med. Sci. 2009, 24, 259–268. [Google Scholar] [CrossRef]
- Van Straten, D.; Mashayekhi, V.; De Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Soupart, A.; Alary, F.; Heully, J.-L.; Elliott, P.I.P.; Dixon, I.M. Recent progress in ligand photorelease reaction mechanisms: Theoretical insights focusing on Ru(II) 3MC states. Coord. Chem. Rev. 2020, 408, 213184. [Google Scholar] [CrossRef] [Green Version]
- Loftus, L.M.; Rack, J.J.; Turro, C. Photoinduced ligand dissociation follows reverse energy gap law: Nitrile photodissociation from low energy 3MLCT excited states. Chem. Commun. 2020, 56, 4070–4073. [Google Scholar] [CrossRef]
- Mukuta, T.; Tanaka, S.I.; Inagaki, A.; Koshihara, S.-Y.; Onda , K. Direct Observation of the Triplet Metal-Centered State in [Ru(bpy)3]2+ Using Time-Resolved Infrared Spectroscopy. ChemistrySelect 2016, 1, 2802–2807. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Stevens, K.; Parkin, S.; Glazer, E.C. Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features. Inorg. Chem. 2020, 59, 1006–1013. [Google Scholar] [CrossRef]
- Loftus, L.M.; Al-Afyouni, K.F.; Turro, C. New RuII Scaffold for Photoinduced Ligand Release with Red Light in the Photodynamic Therapy (PDT) Window. Chem.—A Eur. J. 2018, 24, 11550–11553. [Google Scholar] [CrossRef]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed. 2017, 56, 11549–11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; White, J.K.; Arora, K.; Herroon, M.K.; Martin, P.D.; Schlegel, H.B.; Podgorski, I.; Turro, C.; Kodanko, J.J. Selective Release of Aromatic Heterocycles from Ruthenium Tris(2-pyridylmethyl)amine with Visible Light. Inorg. Chem. 2016, 55, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Cuello-Garibo, J.-A.; Meijer, M.S.; Bonnet, S. To cage or to be caged? The cytotoxic species in ruthenium-based photoactivated chemotherapy is not always the metal. Chem. Commun. 2017, 53, 6768–6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufziger, K.T.; Thowfeik, F.S.; Charboneau, D.J.; Nieto, I.; Dougherty, W.G.; Kassel, W.S.; Dudley, T.J.; Merino, E.J.; Papish, E.T.; Paul, J.J. Ruthenium Dihydroxybipyridine Complexes are Tumor Activated Prodrugs due to Low pH and Blue Light Induced Ligand Release. J. Inorg. Biochem. 2014, 130, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oladipupo, O.; Brown, S.R.; Lamb, R.W.; Gray, J.L.; Cameron, C.G.; DeRegnaucourt, A.R.; Ward, N.A.; Hall, J.F.; Xu, Y.; Petersen, C.M.; et al. Light-responsive and Protic Ruthenium Compounds Bearing Bathophenanthroline and Dihydroxybipyridine Ligands Achieve Nanomolar Toxicity towards Breast Cancer Cells. Photochem. Photobiol. 2022, 98, 102–116. [Google Scholar] [CrossRef]
- Qu, F.; Martinez, K.; Arcidiacono, A.M.; Park, S.; Zeller, M.; Schmehl, R.H.; Paul, J.J.; Kim, Y.; Papish, E.T. Sterically demanding methoxy and methyl groups in ruthenium complexes lead to enhanced quantum yields for blue light triggered photodissociation. Dalton Trans. 2018, 47, 15685–15693. [Google Scholar] [CrossRef]
- Qu, F.; Park, S.; Martinez, K.; Gray, J.L.; Thowfeik, F.S.; Lundeen, J.A.; Kuhn, A.E.; Charboneau, D.J.; Gerlach, D.L.; Lockart, M.M.; et al. Ruthenium Complexes are pH-Activated Metallo Prodrugs (pHAMPs) with Light-Triggered Selective Toxicity Toward Cancer Cells. Inorg. Chem. 2017, 56, 7519–7532. [Google Scholar] [CrossRef]
- Park, S.; Gray, J.L.; Altman, S.D.; Hairston, A.R.; Beswick, B.T.; Kim, Y.; Papish, E.T. Cellular uptake of protic ruthenium complexes is influenced by pH dependent passive diffusion and energy dependent efflux. J. Inorg. Biochem. 2020, 203, 110922. [Google Scholar] [CrossRef]
- Reichardt, C.; Sainuddin, T.; Wächtler, M.; Monro, S.; Kupfer, S.; Guthmuller, J.; Gräfe, S.; McFarland, S.; Dietzek, B. Influence of Protonation State on the Excited State Dynamics of a Photobiologically Active Ru(II) Dyad. J. Phys. Chem. A. 2016, 120, 6379–6388. [Google Scholar] [CrossRef]
- Tardito, S.; Bassanetti, I.; Bignardi, C.; Elviri, L.; Tegoni, M.; Mucchino, C.; Bussolati, O.; Franchi-Gazzola, R.; Marchiò, L. Copper Binding Agents Acting as Copper Ionophores Lead to Caspase Inhibition and Paraptotic Cell Death in Human Cancer Cells. J. Am. Chem. Soc. 2011, 133, 6235–6242. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, Y.; Huang, H.; Wang, J.; Zhao, D.; Ji, L.; Chao, H. Cyclometalated Ruthenium(II) Anthraquinone Complexes Exhibit Strong Anticancer Activity in Hypoxic Tumor Cells. Chem. —A Eur. J. 2015, 21, 15308–15319. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, L.; Chiniforoshan, H. New Ru IIpincer complexes: Synthesis, characterization and biological evaluation for photodynamic therapy. Dalton Trans. 2016, 45, 18333–18345. [Google Scholar] [CrossRef]
- Mehanna, S.; Mansour, N.; Audi, H.; Bodman-Smith, K.; Mroueh, M.A.; Taleb, R.I.; Daher, C.F.; Khnayzer, R.S. Enhanced cellular uptake and photochemotherapeutic potential of a lipophilic strained Ru( ii) polypyridyl complex. RSC Adv. 2019, 9, 17254–17265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.L. Light-Activated Protic Ruthenium Anticancer Compounds: Structure Function Relationships and Determining Which Factors Influence Toxicity. Ph.D. Thesis, The University of Alabama, Tuscaloosa, AL, USA, 2020; p. 209. [Google Scholar]
- DePasquale, J.; Nieto, I.; Reuther, L.E.; Herbst-Gervasoni, C.J.; Paul, J.J.; Mochalin, V.; Zeller, M.; Thomas, C.M.; Addison, A.W.; Papish, E.T. Iridium Dihydroxybipyridine Complexes Show That Ligand Deprotonation Dramatically Speeds Rates of Catalytic Water Oxidation. Inorg. Chem. 2013, 52, 9175–9183. [Google Scholar] [CrossRef]
- Jeffrey, G.A. Hydrogen-Bonding: An update. Crystallogr. Rev. 2003, 9, 135–176. [Google Scholar] [CrossRef]
- Yao, W.; Das, S.; DeLucia, N.A.; Qu, F.; Boudreaux, C.M.; Vannucci, A.K.; Papish, E.T. Determining the Catalyst Properties That Lead to High Activity and Selectivity for Catalytic Hydrodeoxygenation with Ruthenium Pincer Complexes. Organometallics 2020, 39, 662–669. [Google Scholar] [CrossRef]
- Yao, W.; DeRegnaucourt, A.R.; Shrewsbury, E.D.; Loadholt, K.H.; Silprakob, W.; Brewster, T.P.; Papish, E.T. Reinvestigating Catalytic Alcohol Dehydrogenation with an Iridium Dihydroxybipyridine Catalyst. Organometallics 2020, 39, 3656–3662. [Google Scholar] [CrossRef]
- Brown, R.T.; Fletcher, N.C.; Danos, L.; Halcovitch, N.R. A Tripodal Ruthenium(II) Polypyridyl Complex with pH Controlled Emissive Quenching. Eur. J. Inorg. Chem. 2019, 2019, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsson, M.; Jäger, M.; Kumar, R.J.; Österman, T.; Persson, P.; Becker, H.-C.; Johansson, O.; Hammarström, L. Bistridentate Ruthenium(II)polypyridyl-Type Complexes with Microsecond 3MLCT State Lifetimes: Sensitizers for Rod-Like Molecular Arrays. J. Am. Chem. Soc. 2008, 130, 15533–15542. [Google Scholar] [CrossRef]
- Hidayatullah, A.N.; Wachter, E.; Heidary, D.K.; Parkin, S.; Glazer, E.C. Photoactive Ru(II) Complexes With Dioxinophenanthroline Ligands Are Potent Cytotoxic Agents. Inorg. Chem. 2014, 53, 10030–10032. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.R.; Concepcion, J.J.; Glasson, C.R.K.; Fang, Z.; Lapides, A.M.; Ashford, D.L.; Templeton, J.L.; Meyer, T.J. Synthesis of Phosphonic Acid Derivatized Bipyridine Ligands and Their Ruthenium Complexes. Inorg. Chem. 2013, 52, 12492–12501. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, A71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. [Google Scholar] [CrossRef]
- Ishida, H.; Tobita, S.; Hasegawa, Y.; Katoh, R.; Nozaki, K. Recent advances in instrumentation for absolute emission quantum yield measurements. Coord. Chem. Rev. 2010, 254, 2449–2458. [Google Scholar] [CrossRef]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233-234, 351–371. [Google Scholar] [CrossRef]
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Drug interaction and location in liposomes: Correlation with polar surface areas. Int. J. Pharm. 2005, 292, 179–185. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line a | EC50_light (μM) | EC50_dark (μM) | PI | pKa avg | log(Do/w) at pH 7.4 | ΦPS (%) d | ΦΔ (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1A | MCF7 | >500 b | >500 | ~1 | 6.3 | 1.4(1) | 0.58 | 4.1 g | [11,25], [28,29] |
| 1B | 0.12 | 18 g | |||||||
| 2A | MCF7 | 180 b | 490 | 2.8 | 6.0(1) | 1.6(1) | 0.20 | 4.8 g | [11,28,29] |
| 2B | 3.6 × 10−3 | 87 g | |||||||
| 3A | MCF7 | 4.1 b | 490 | 120 | 5.9(1) | 1.8(1) | 0.1 | 4.8 g | [11,28,29] |
| 3B | 2.2 × 10−2 | 48 g | |||||||
| 4A | MCF7 | 2.0 c | 18 | 8.9 | 5(1) | >3 | e | 5 h | [26] |
| 4B | <1 h | ||||||||
| 5A | HeLa | >100 b | >100 | ~1 | n.d. | −1.2(2) | f | 57 h | This work, [25] |
| 5B | 4 h | ||||||||
| 6A | MCF7 | 194 b | >300 | 1.5 | 6.01(8) | −0.1(2) | f | 62 h | This work, [28] |
| 6B | 16, 19 h,i | ||||||||
| 7A | MCF7 | >100 b | >100 | ~1 | n.d. | 0.8(2) | f | 76 h | This work |
| 7B | 15 h,j | ||||||||
| 8A | MCF7 | 0.5 c | >100 | >200 | 5.9(3) | >3 | f | 68 h | [26] |
| 8B | 19 h |
| Bond Lengths (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Ru1-N1 | 2.053(2) | N1-Ru1-N2 | 78.38(8) |
| Ru1-N2 | 2.060(2) | N1-Ru1-N3 | 96.24(8) |
| Ru1-N3 | 2.060(2) | N1-Ru1-N4 | 89.49(9) |
| Ru1-N4 | 2.061(2) | N1-Ru1-N5 | 172.40(9) |
| Ru1-N5 | 2.071(2) | N2-Ru1-N4 | 97.68(9) |
| Ru1-N6 | 2.051(2) | N2-Ru1-N5 | 95.68(9) |
| C3-O1 | 1.336(3) | N3-Ru1-N2 | 174.24(9) |
| C8-O2 | 1.341(3) | N3-Ru1-N4 | 80.09(9) |
| N3-Ru1-N5 | 89.86(9) | ||
| N4-Ru1-N5 | 96.06(9) | ||
| N6-Ru1-N1 | 95.05(9) | ||
| N6-Ru1-N2 | 89.96(9) | ||
| N6-Ru1-N3 | 92.58(9) | ||
| N6-Ru1-N4 | 171.78(9) | ||
| N6-Ru1-N5 | 80.09(9) | ||
| Compound | ΦPL | Measured Monoexponential Lifetime, τmeas (μs) | Calculated Radiative Lifetime, τrad (µs) |
|---|---|---|---|
| [(bpy)3Ru]Cl2 | 9.4% | 0.882 | 9.4 |
| 5A | 5.3% | 0.538 | 10.2 |
| 6A | 12.8% | 1.81 | 14.1 |
| 7A | 15.3% | 1.682 | 11.0 |
| 8A | 18.7% | 3.45 | 18.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oladipupo, O.E.; Prescott, M.C.; Blevins, E.R.; Gray, J.L.; Cameron, C.G.; Qu, F.; Ward, N.A.; Pierce, A.L.; Collinson, E.R.; Hall, J.F.; et al. Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity. Int. J. Mol. Sci. 2023, 24, 5980. https://doi.org/10.3390/ijms24065980
Oladipupo OE, Prescott MC, Blevins ER, Gray JL, Cameron CG, Qu F, Ward NA, Pierce AL, Collinson ER, Hall JF, et al. Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity. International Journal of Molecular Sciences. 2023; 24(6):5980. https://doi.org/10.3390/ijms24065980
Chicago/Turabian StyleOladipupo, Olaitan E., Meredith C. Prescott, Emily R. Blevins, Jessica L. Gray, Colin G. Cameron, Fengrui Qu, Nicholas A. Ward, Abigail L. Pierce, Elizabeth R. Collinson, James Fletcher Hall, and et al. 2023. "Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity" International Journal of Molecular Sciences 24, no. 6: 5980. https://doi.org/10.3390/ijms24065980
APA StyleOladipupo, O. E., Prescott, M. C., Blevins, E. R., Gray, J. L., Cameron, C. G., Qu, F., Ward, N. A., Pierce, A. L., Collinson, E. R., Hall, J. F., Park, S., Kim, Y., McFarland, S. A., Fedin, I., & Papish, E. T. (2023). Ruthenium Complexes with Protic Ligands: Influence of the Position of OH Groups and π Expansion on Luminescence and Photocytotoxicity. International Journal of Molecular Sciences, 24(6), 5980. https://doi.org/10.3390/ijms24065980