

TNFα Activates the Liver X Receptor Signaling Pathway and Promotes Cholesterol Efflux from Human Brain Pericytes Independently of ABCA1


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
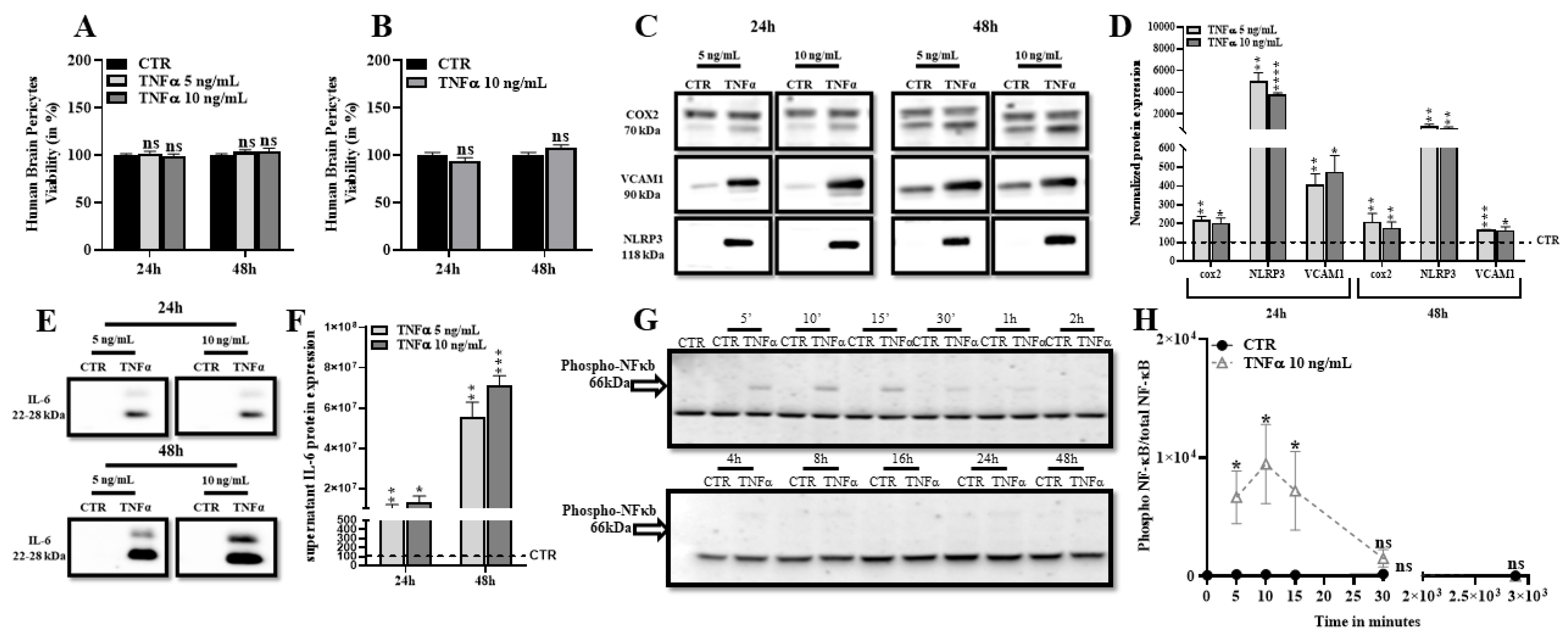
2.1. TNFα Promotes Expression of Inflammatory Markers and Does Not Affect Human Brain Pericytes Survival
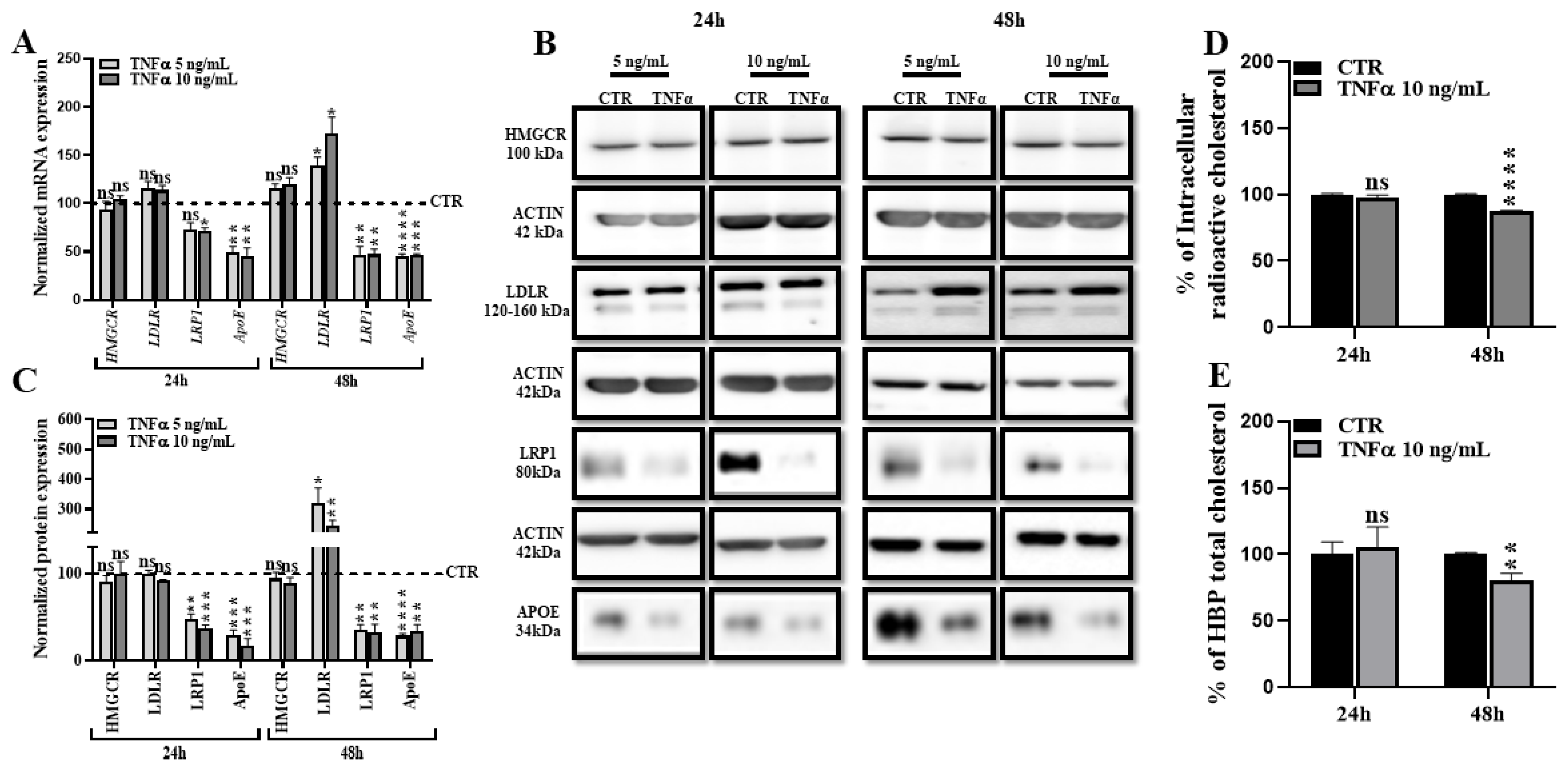
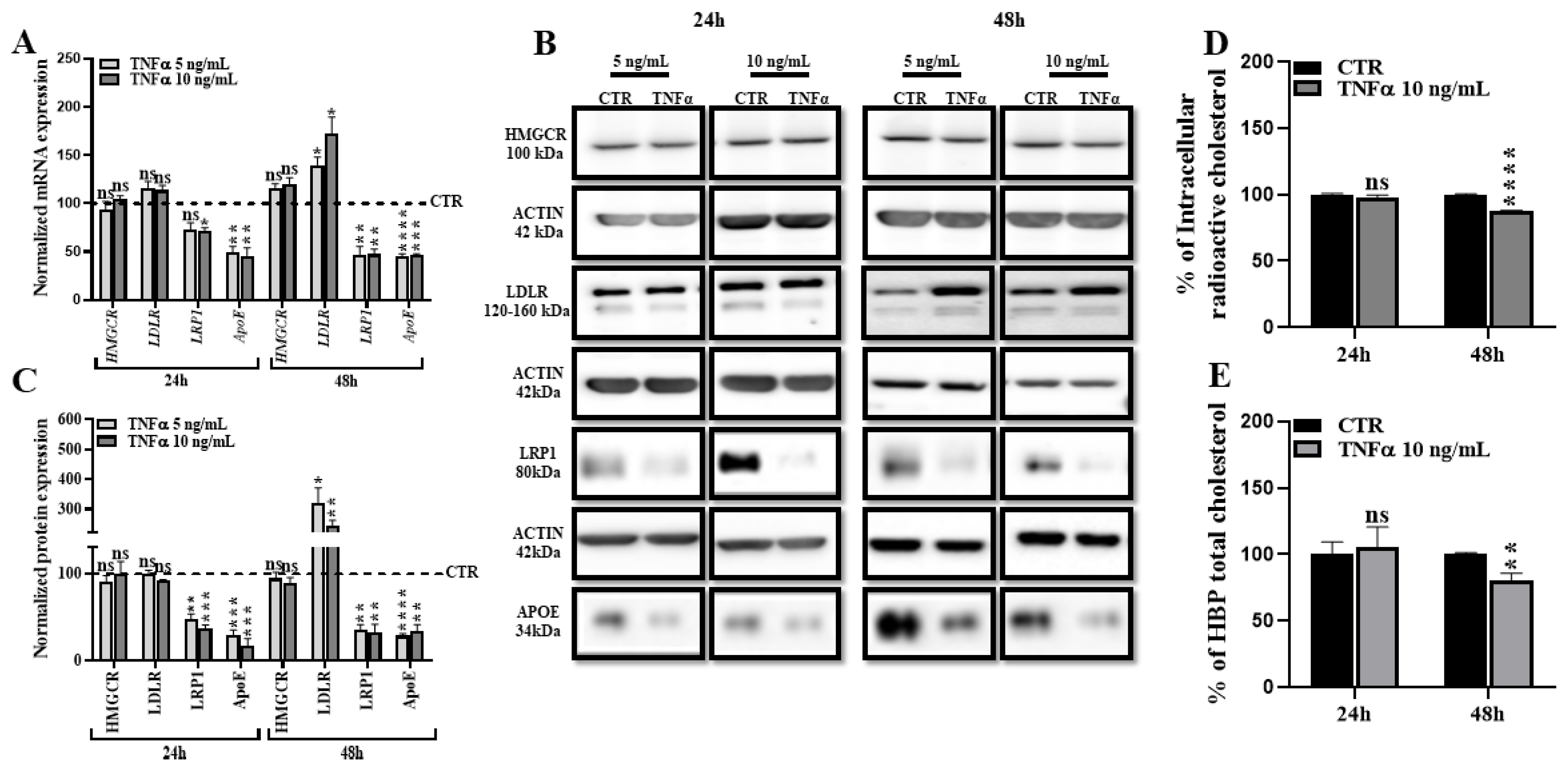
2.2. TNFα Alters HBP Cholesterol Metabolism
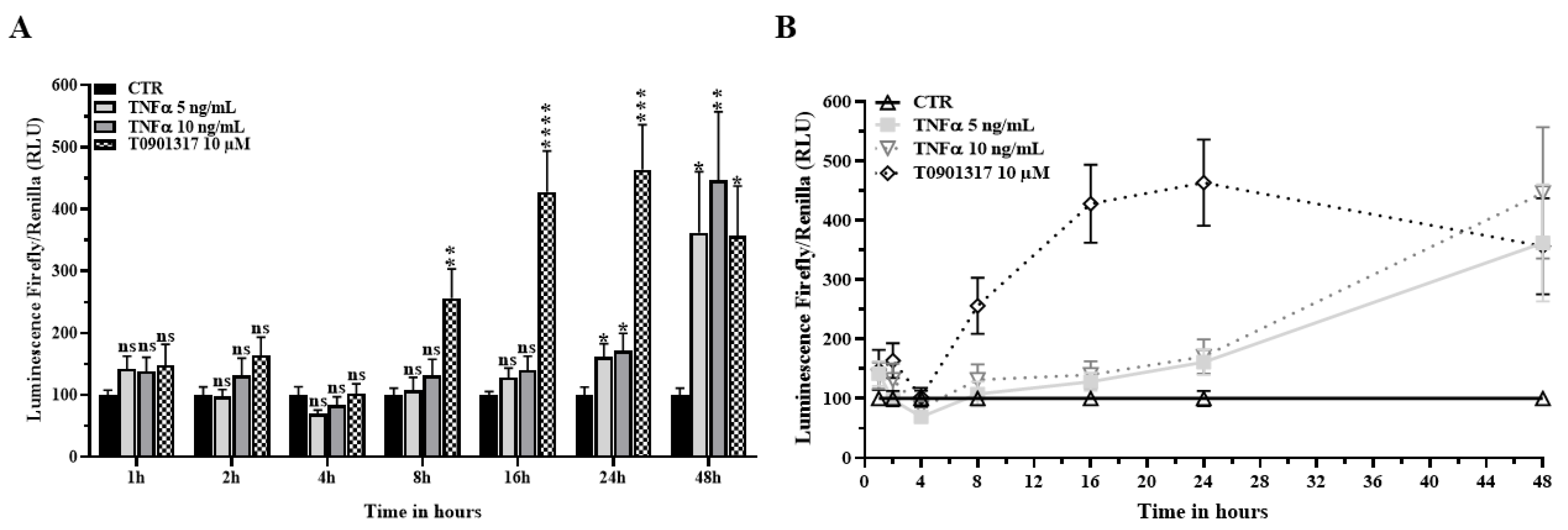
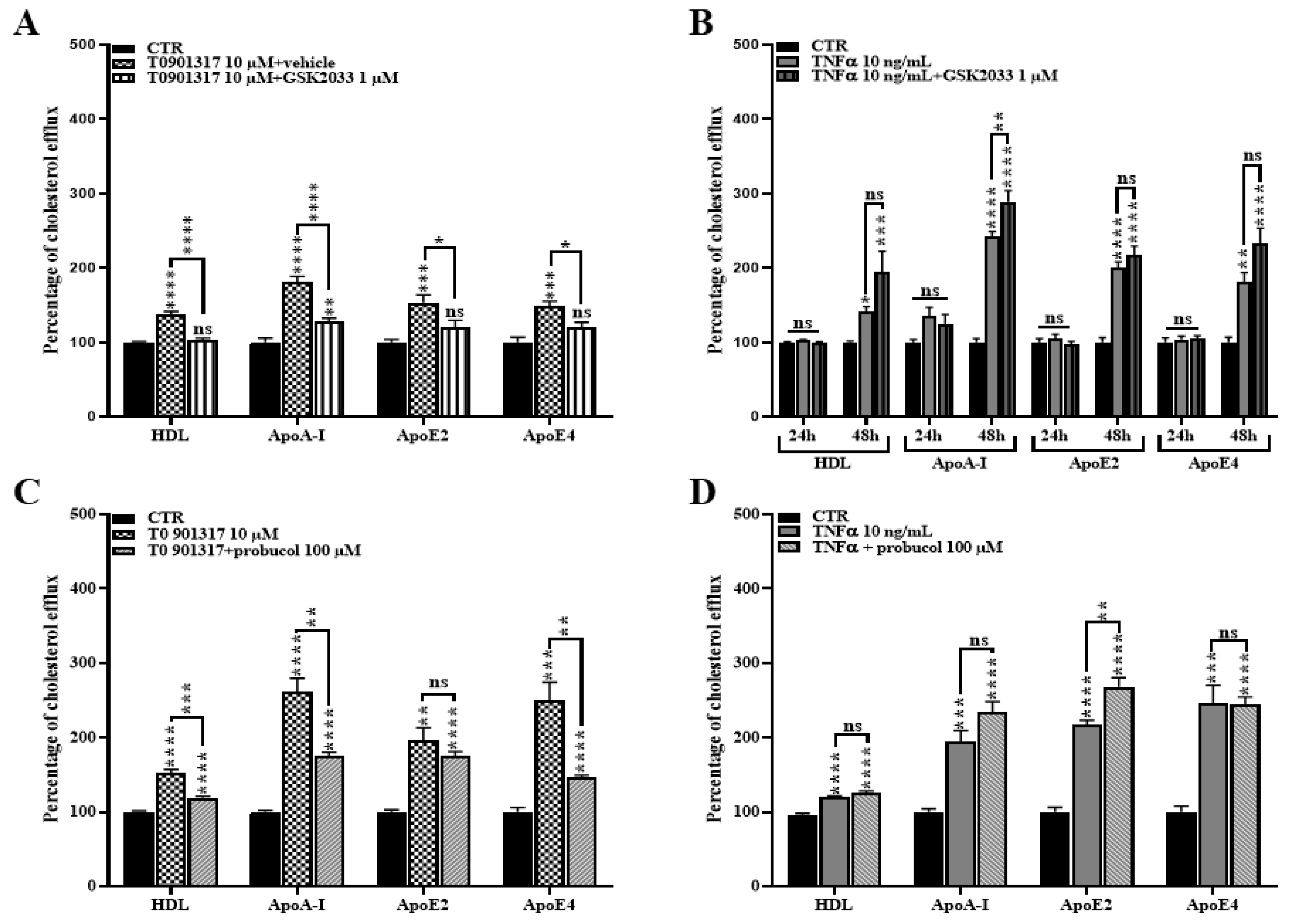
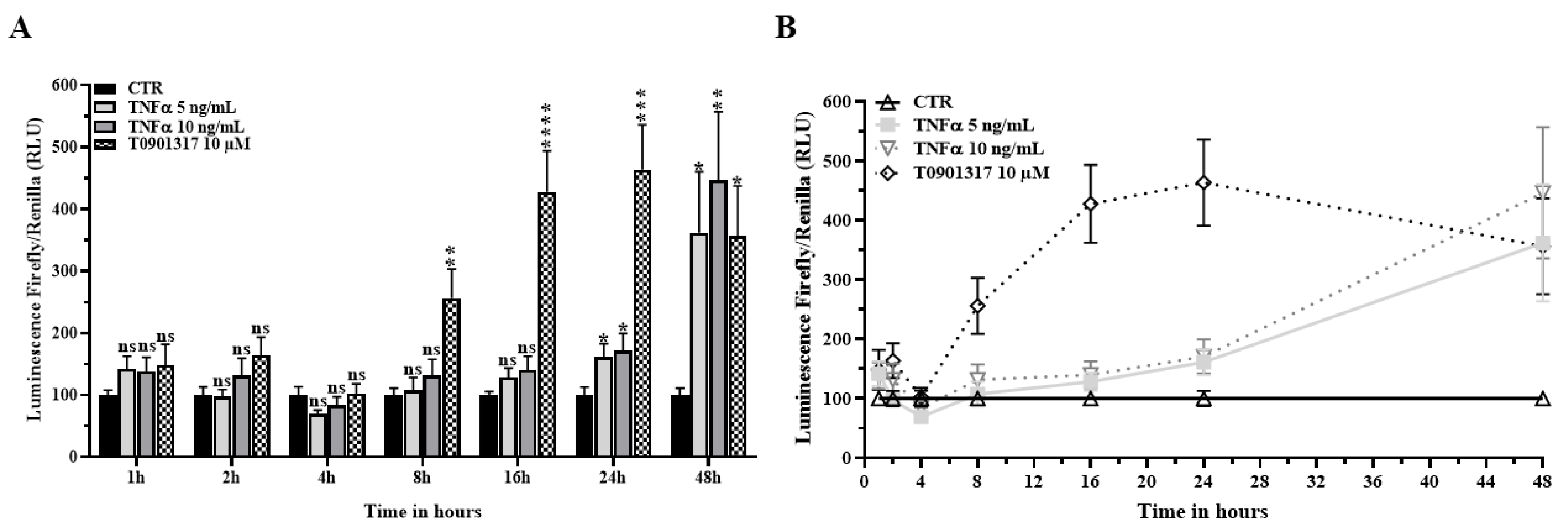
2.3. TNFα Activates the LXR Signaling Pathway
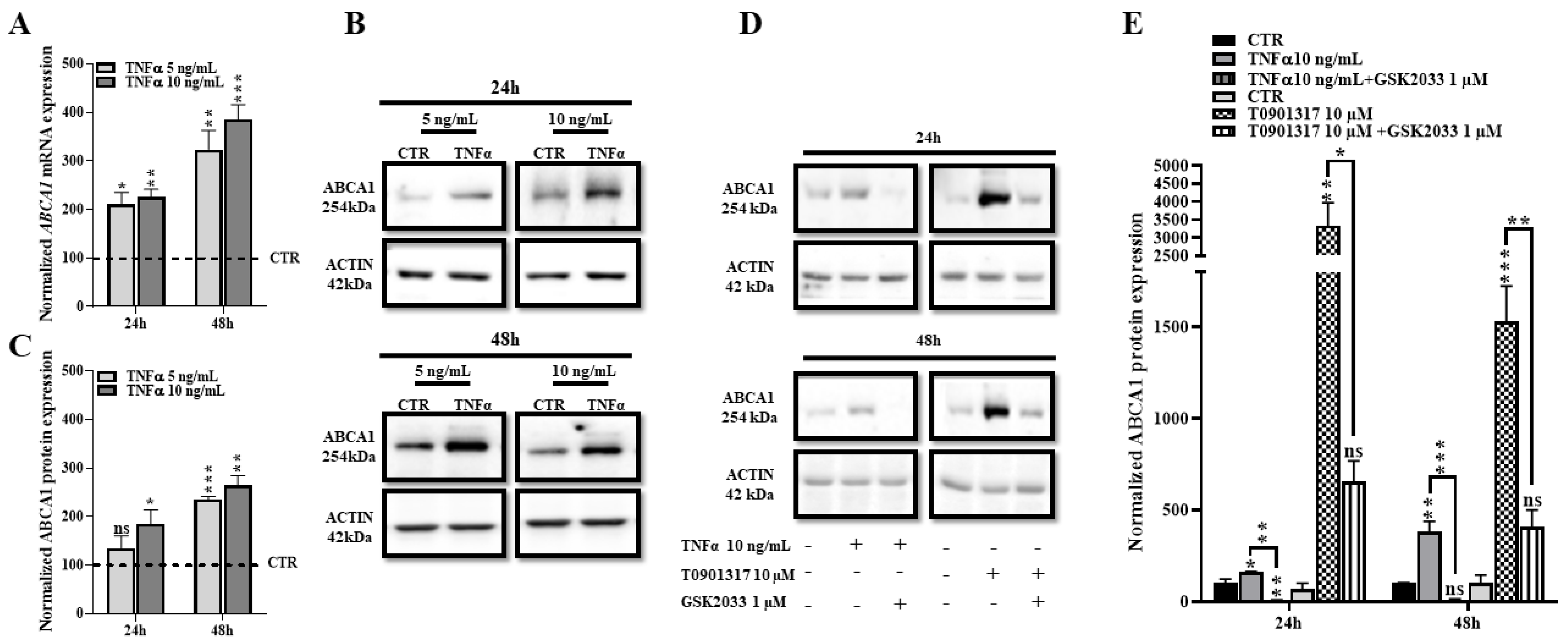
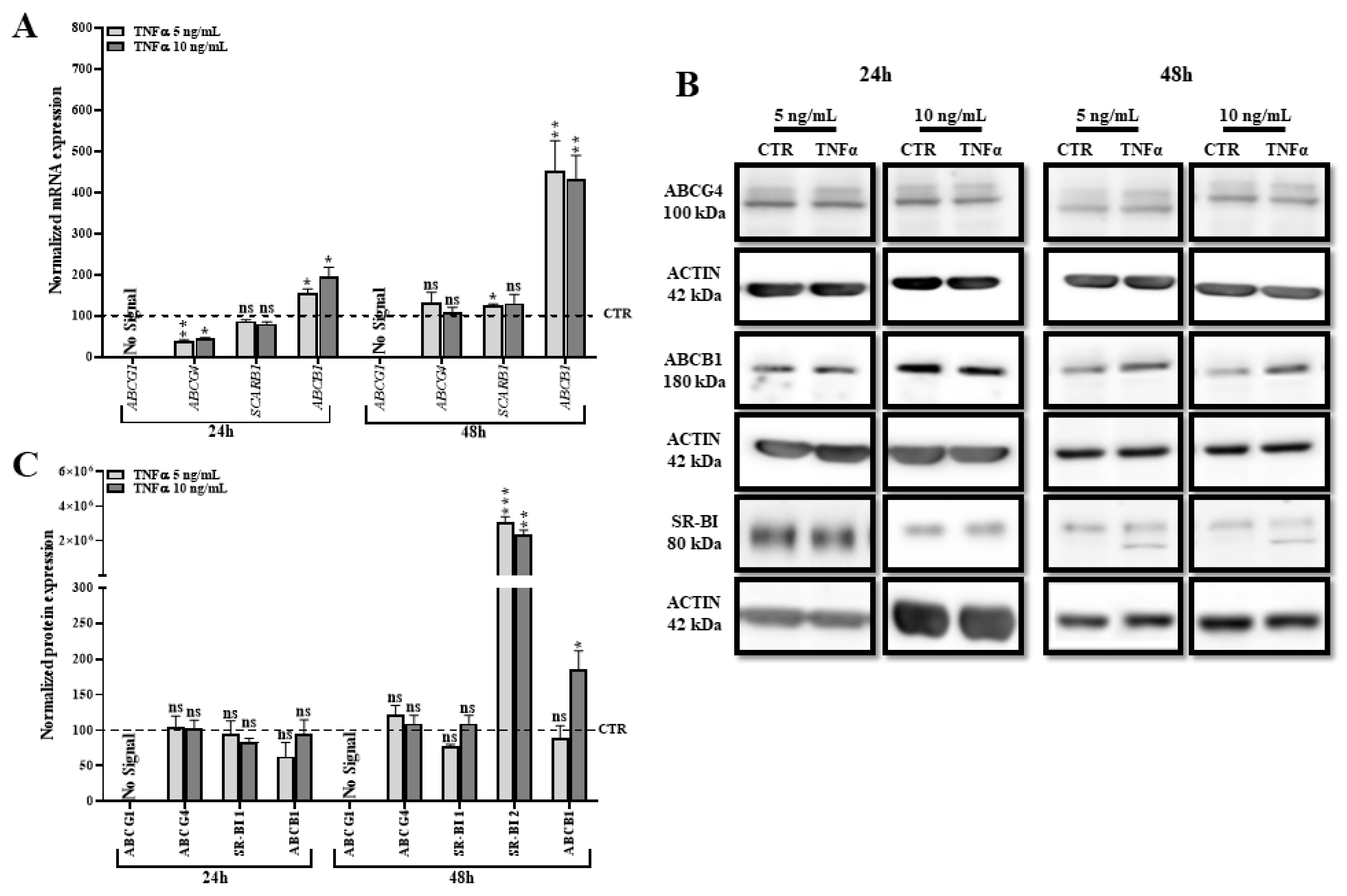
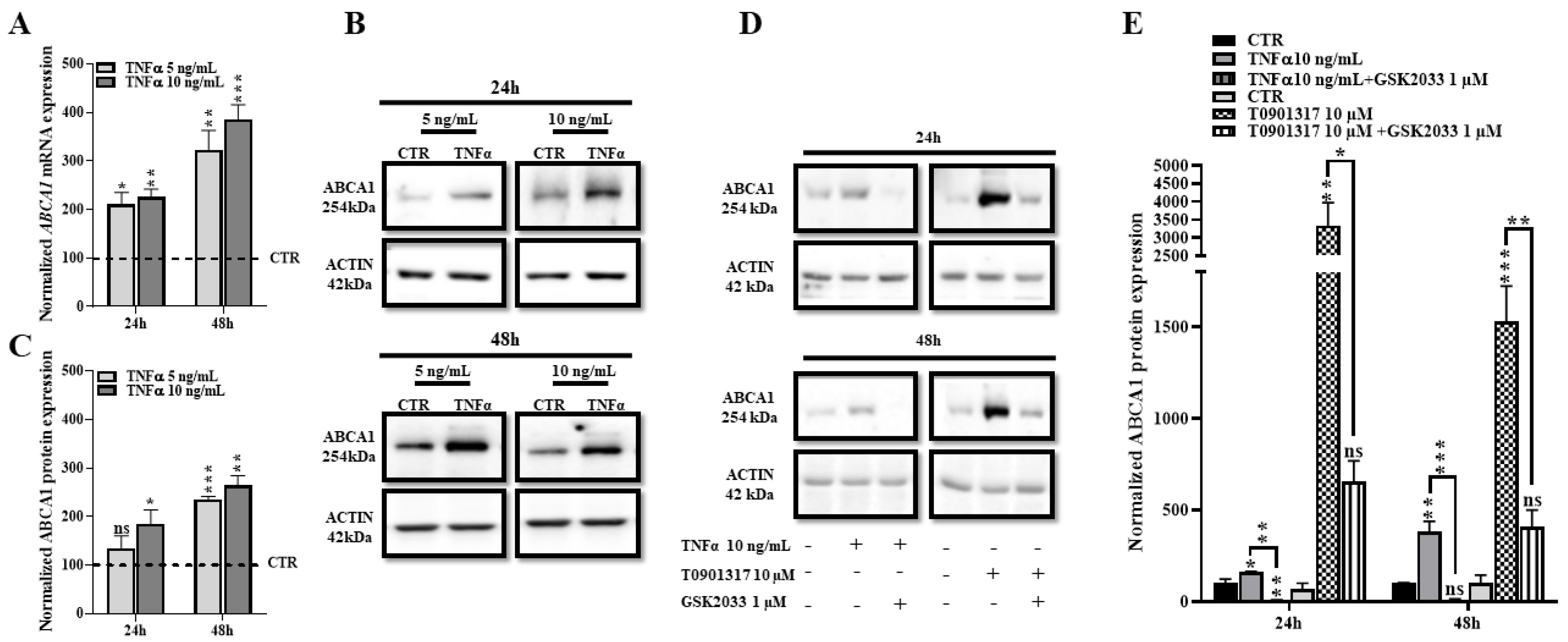
2.4. TNFα Increases Expression of ABCA1 at the Transcriptional and Protein Levels
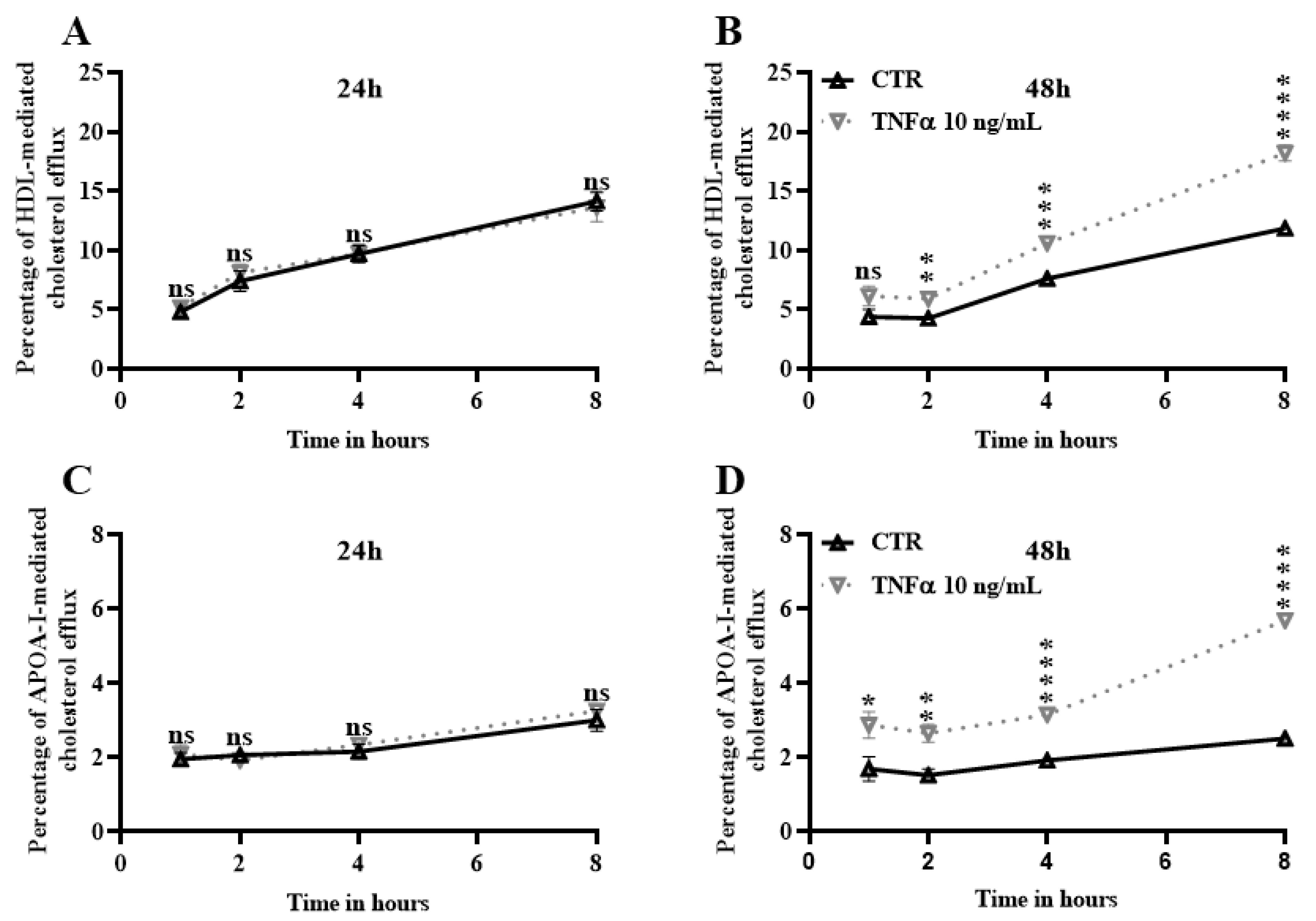
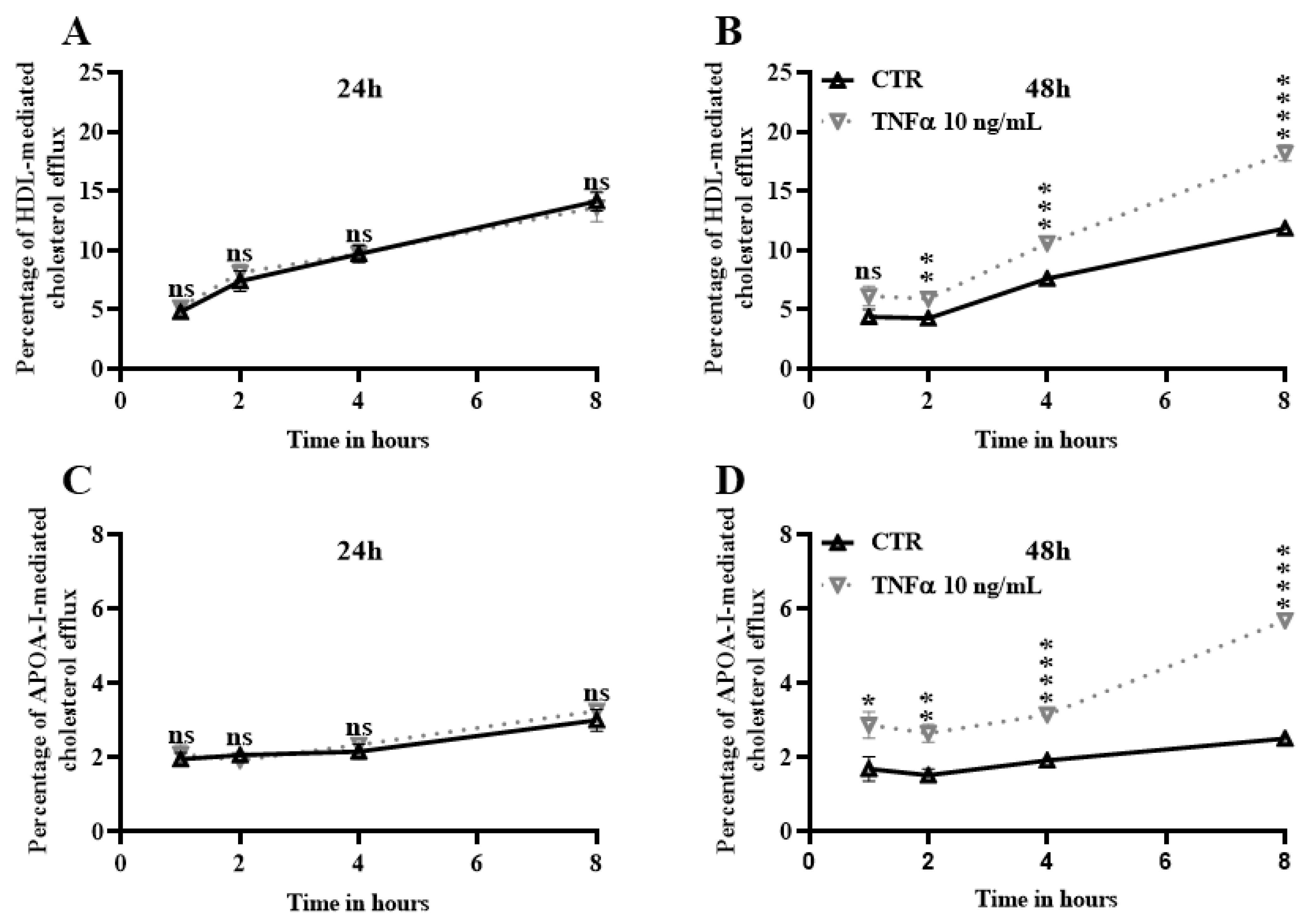
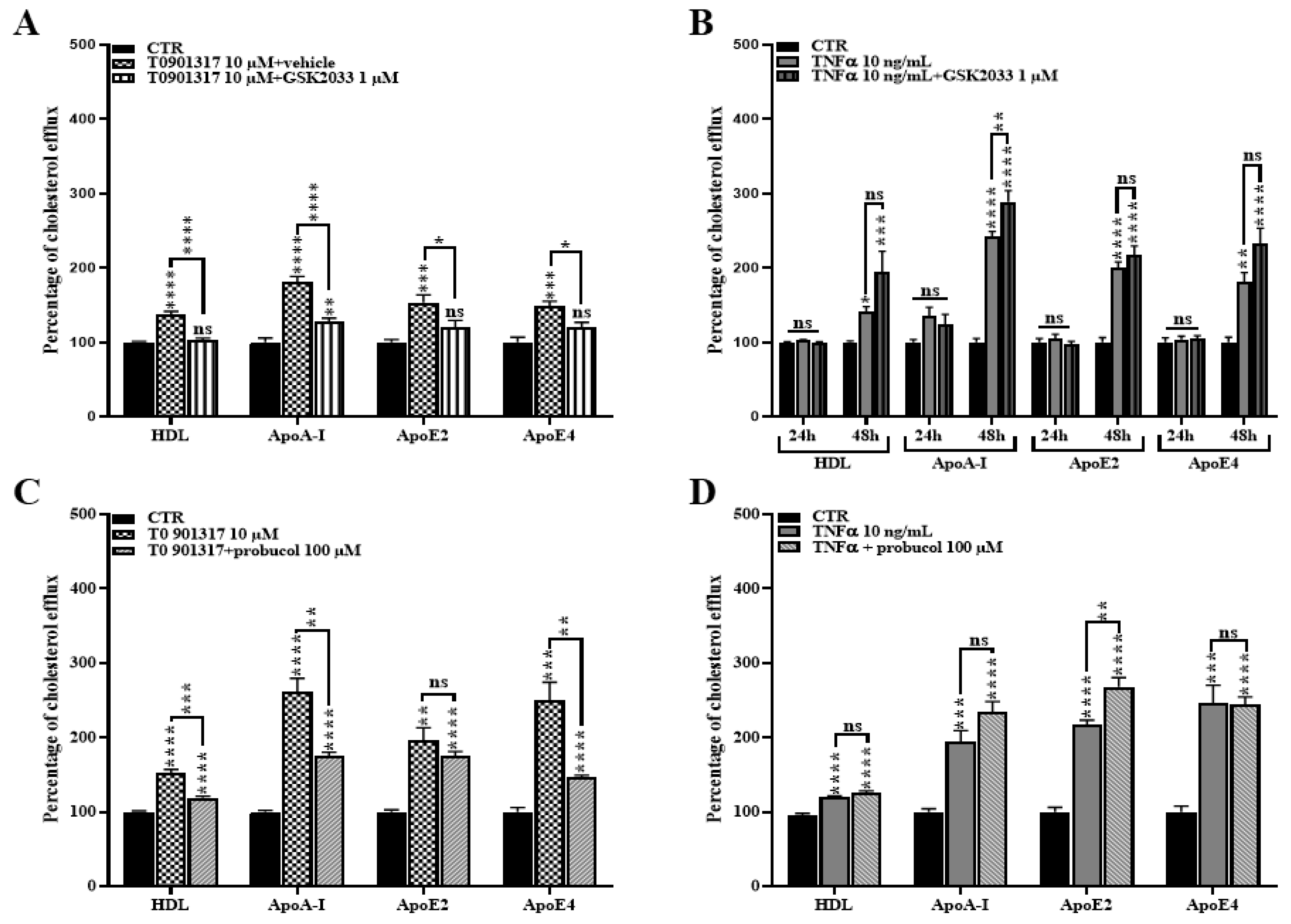
2.5. TNFα Increases Cholesterol Efflux
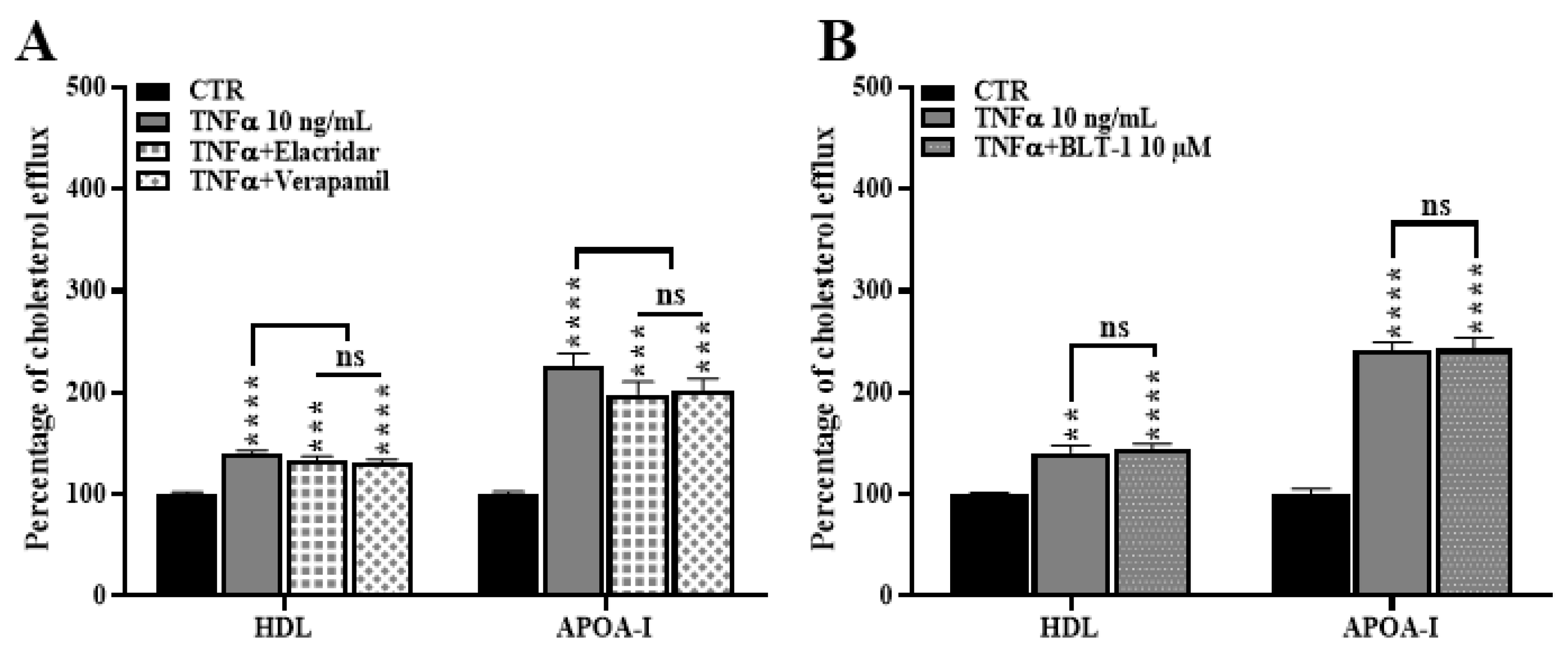
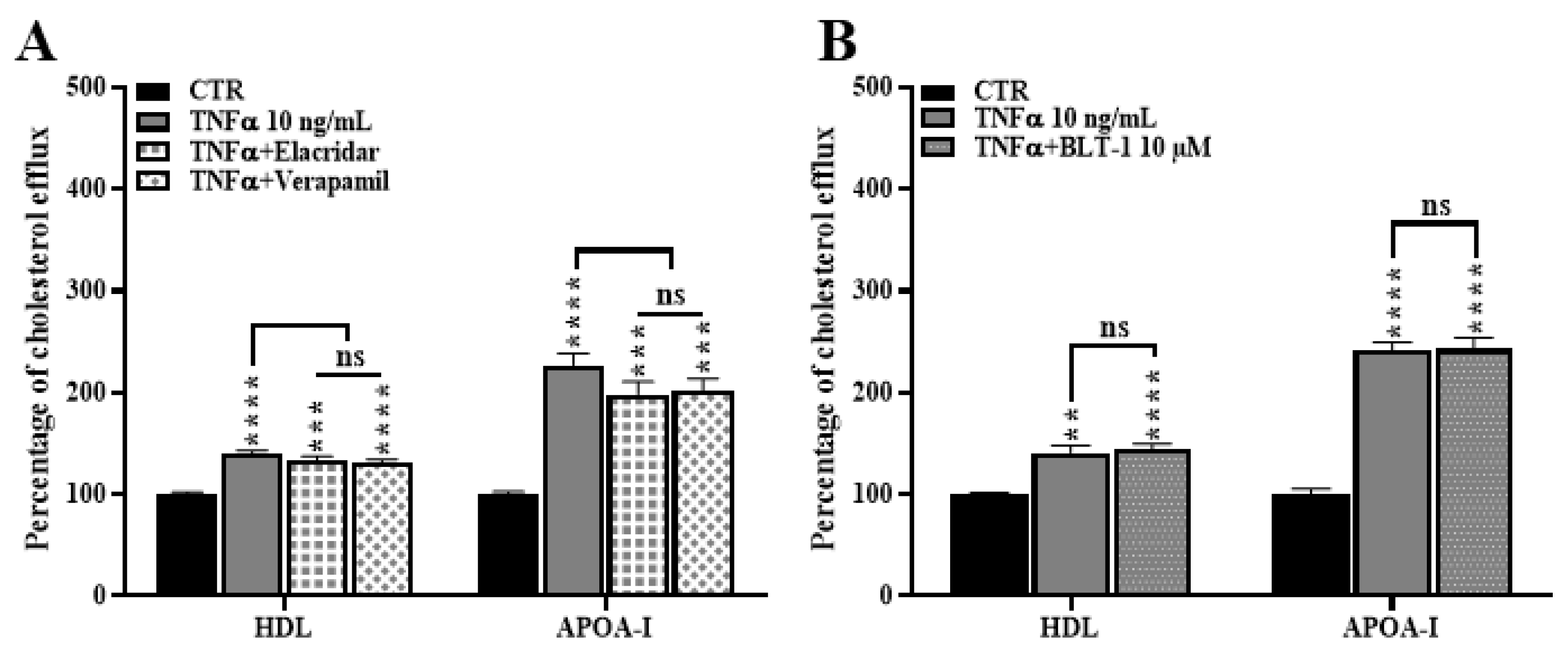
2.6. ABCA1 Inhibition Does Not Rescue the TNFα Induced Cholesterol Efflux
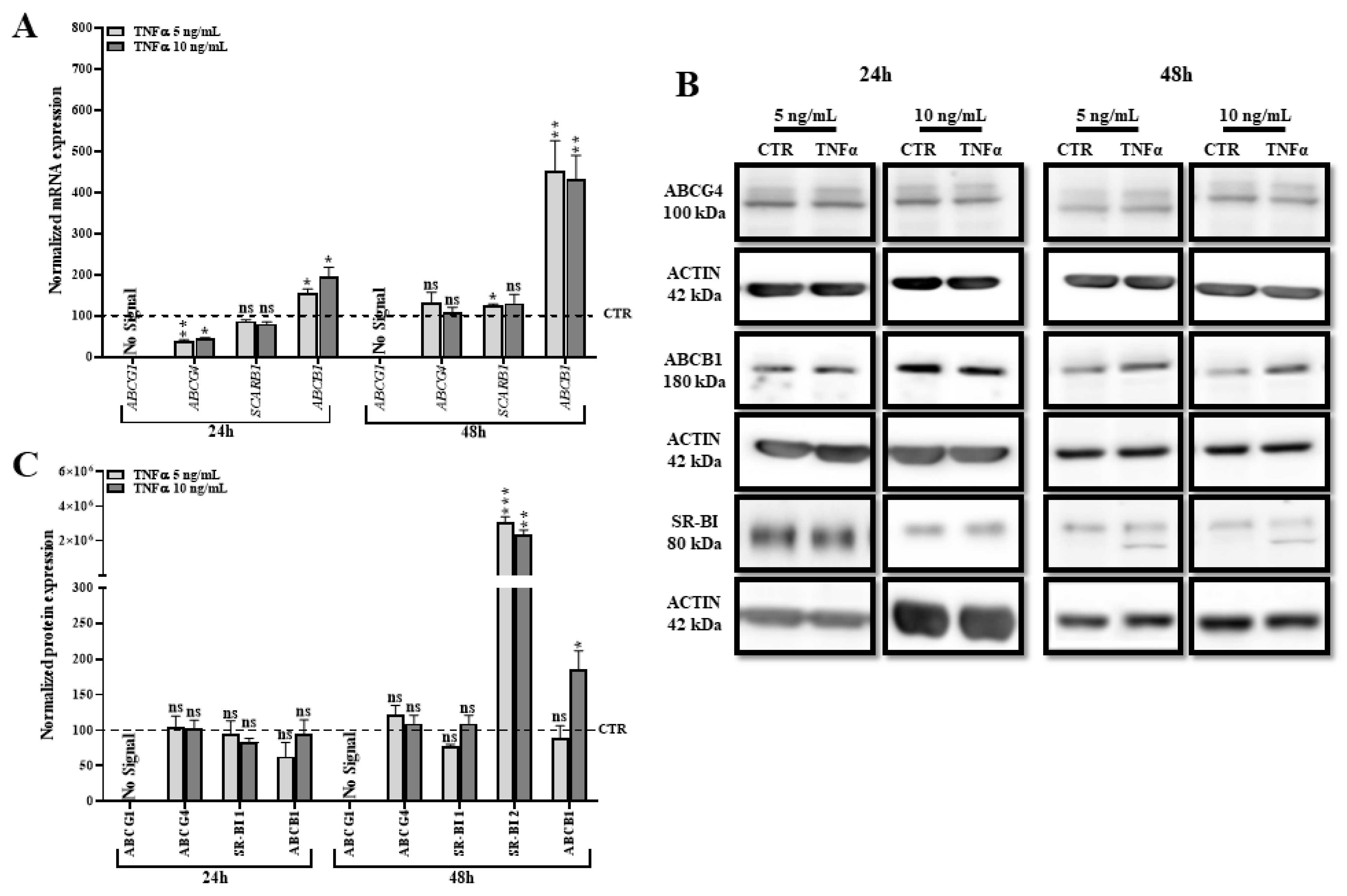
2.7. TNFα Modifies the Expression of Other Transporters Involved in Lipid Efflux
2.8. ABCB1 and SR-BI Inhibition Does Not Rescue the TNFα-Induced Cholesterol Efflux
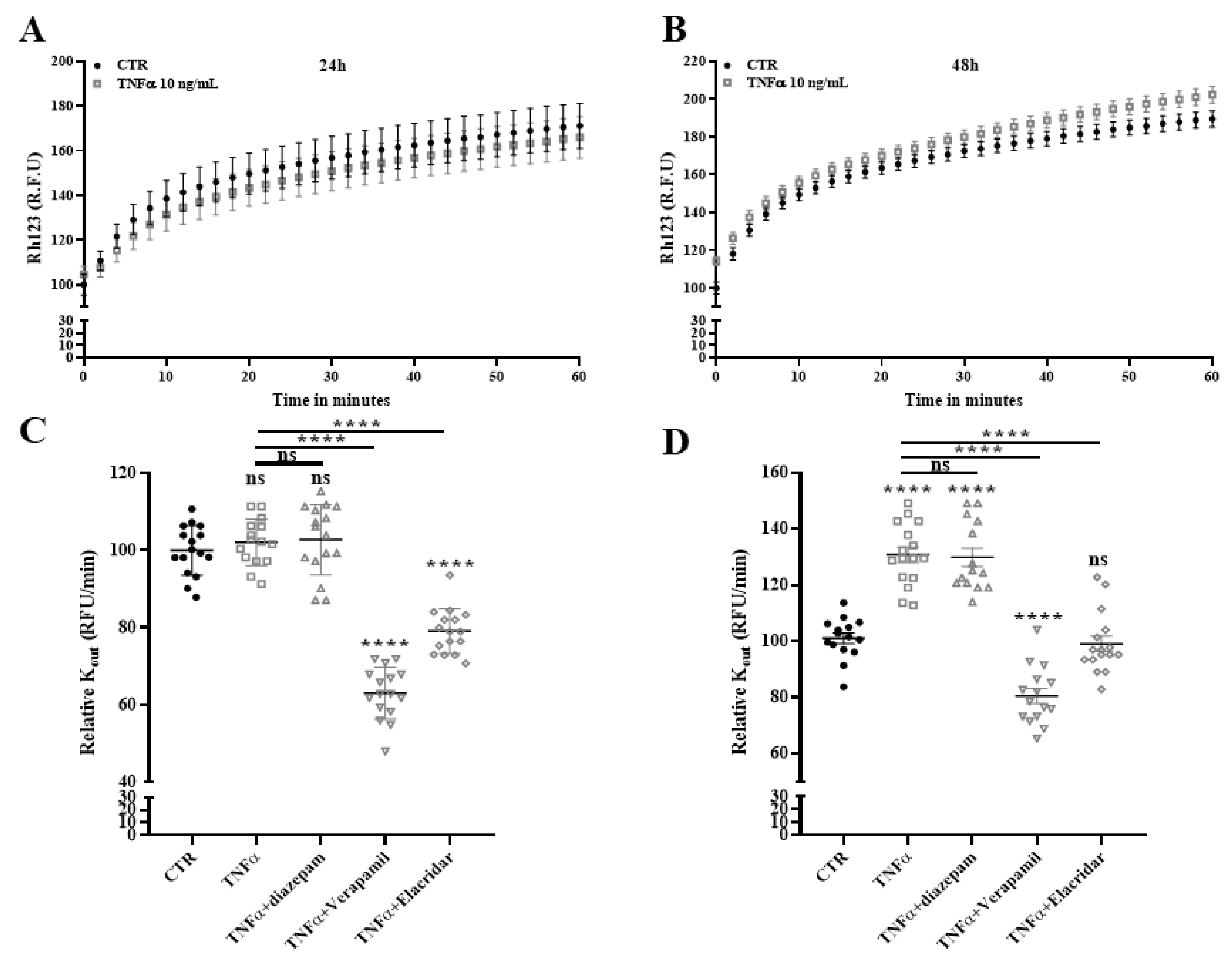
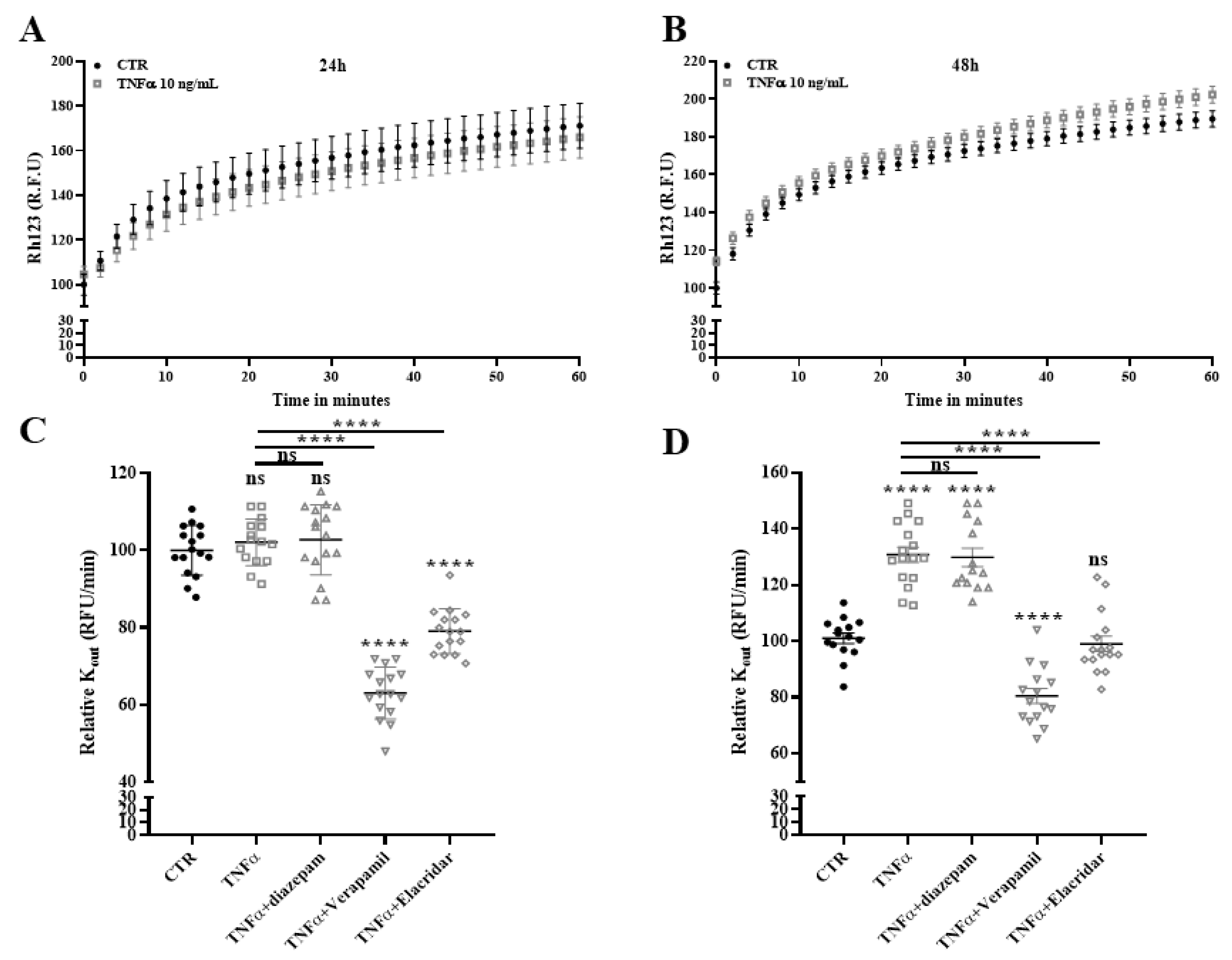
2.9. Drug Efflux Activity of ABCB1 Is Promoted by TNFα
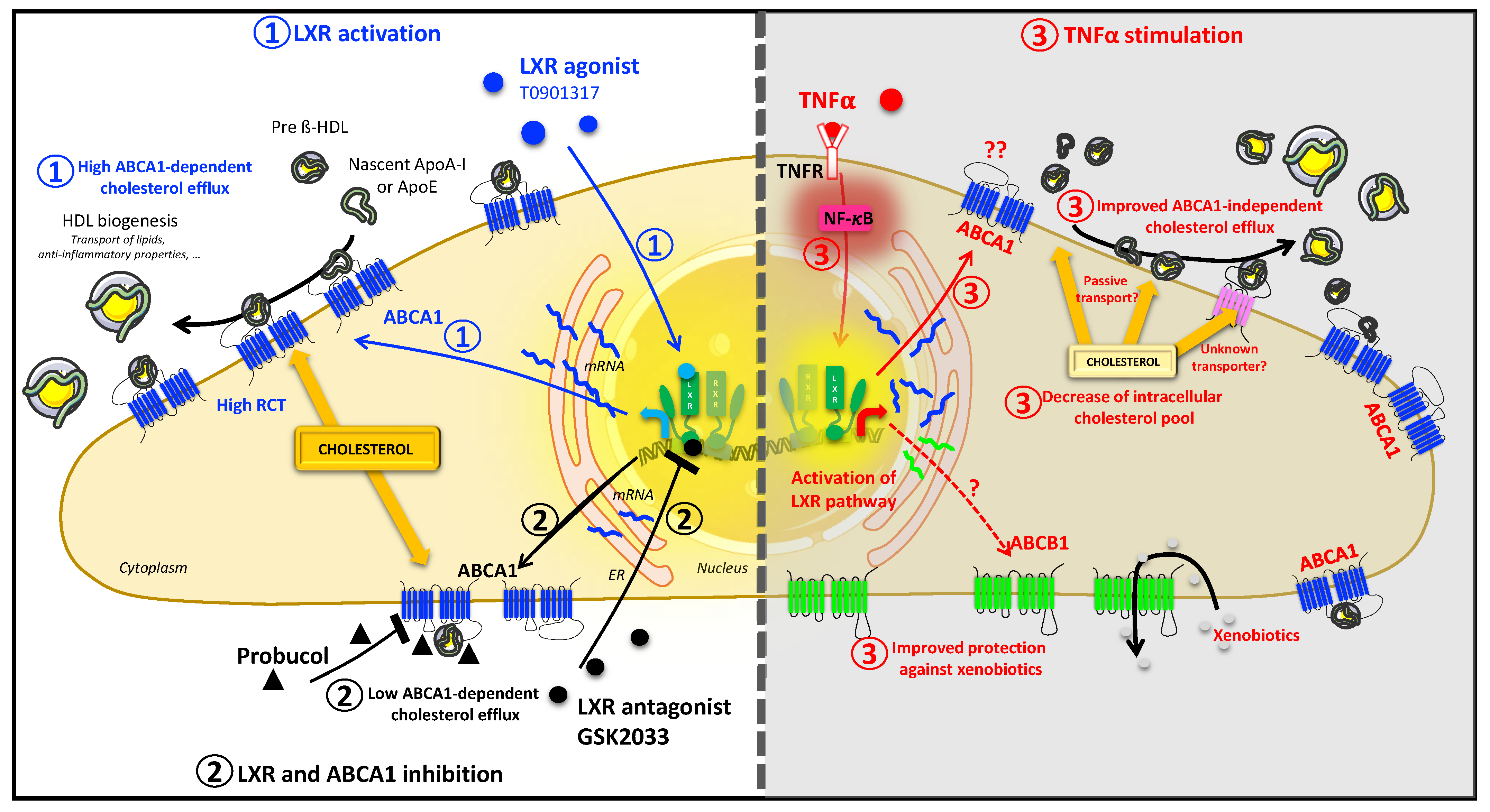
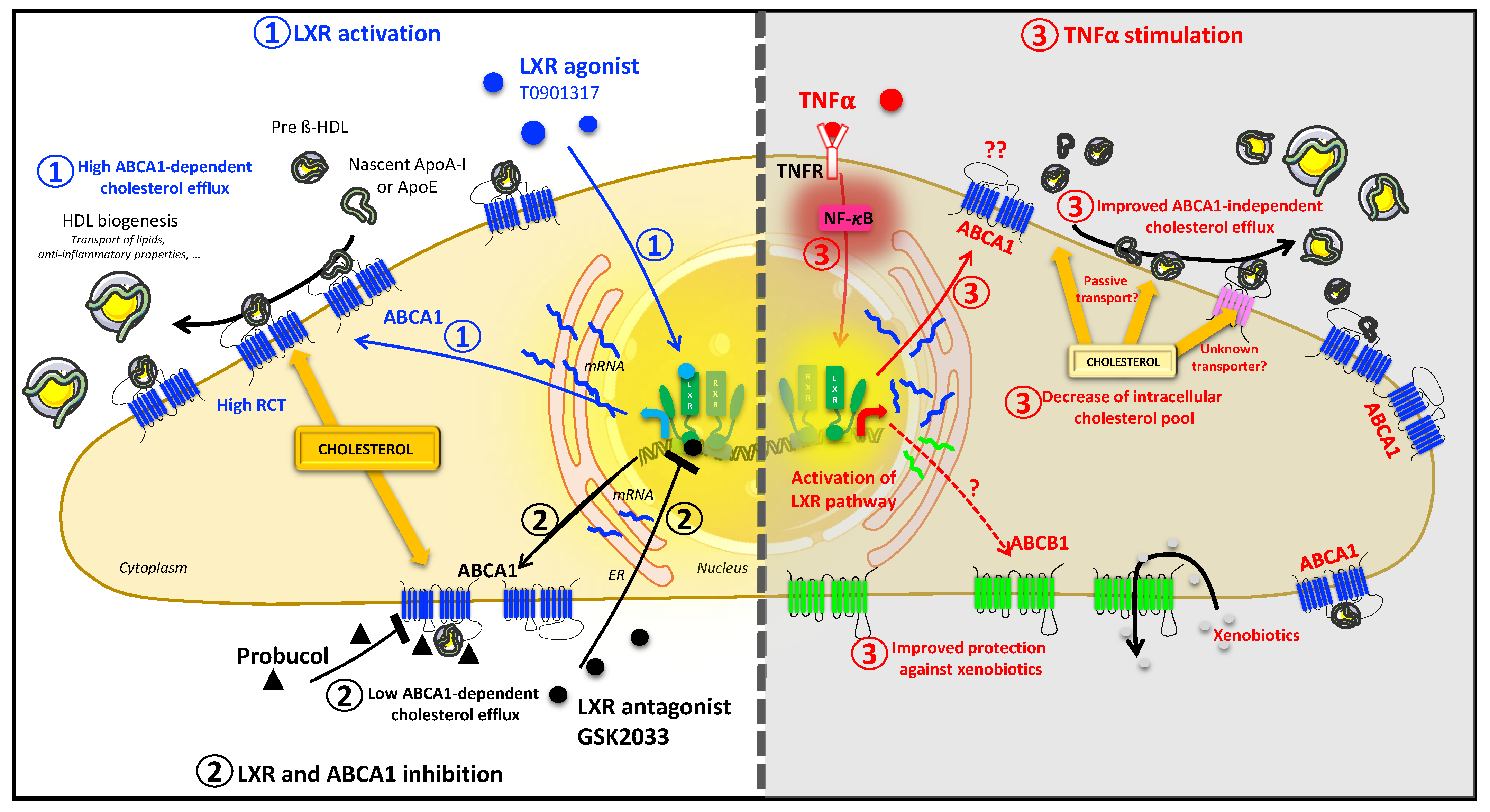
3. Discussion
- The first pool is the accessible or metabolically active pool, which is usually low (around 10% of the total PM cholesterol pool). This pool can be transferred to HDL and apolipoproteins by a process involving a transporter (such ABCA1) or by the aforementioned process called aqueous passive diffusion. If not transferred at external sources, cholesterol molecules of this pool can also be transferred to the intracellular pool of cholesterol located in the endoplasmic reticulum (ER), which senses the total cholesterol pool of the cells. Therefore, this metabolic pool is currently suspected to be a key player in the immune response and defense processes. This pool can be targeted by bacteria and viruses in order to penetrate within cells. Interestingly, it has been suggested that brain pericytes are more permissive for viral and bacterial infection than other CNS cells [61].
- The second pool is the phospholipid-sequestered pool (almost 45% of the total PM pool) representing a strong biophysical basis for the assembly of lipid microdomains or lipid rafts.
- The last cholesterol pool is the essential pool (45% of PM pool also), currently suspected to play a fundamental role in lipid bilayer integrity.
4. Materials and Methods
4.1. Human Brain Pericytes (HBP)
4.2. Treatment of HBP
4.3. Cell Viability and Cytotoxicity Assessment
4.4. RNA Isolation, Reverse Transcription and Quantitative PCR
4.5. Protein Extraction and Immunoblots
4.6. APOE and IL-6 Determination in Cell Culture Supernatant
4.7. Pump out Assay
4.8. Cellular Cholesterol Efflux Assay
4.9. Intracellular Cholesterol Quantification
4.10. Assessment of LXR Pathway Activation
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid. Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Bensinger, S.J. Reprogramming cholesterol metabolism in macrophages and its role in host defense against cholesterol-dependent cytolysins. Cell. Mol. Immunol. 2022, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Gosselet, F.; Miller, F.; Buee-Scherrer, V.; Torpier, G.; Cecchelli, R.; Fenart, L. Physiological pathway for low-density lipoproteins across the blood-brain barrier: Transcytosis through brain capillary endothelial cells in vitro. Endothelium 2008, 15, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid. Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [Green Version]
- Saint-Pol, J.; Gosselet, F. Oxysterols and the NeuroVascular Unit (NVU): A far true love with bright and dark sides. J. Steroid Biochem. Mol. Biol. 2019, 191, 105368. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.P.; Vardanian, L.; Pajoohesh-Ganji, A.; Wang, L.; Cooper, M.; Harris, D.C.; Duff, K.; Rebeck, G.W. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J. Neurochem. 2006, 98, 792–800. [Google Scholar] [CrossRef]
- Lefterov, I.; Bookout, A.; Wang, Z.; Staufenbiel, M.; Mangelsdorf, D.; Koldamova, R. Expression profiling in APP23 mouse brain: Inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener. 2007, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C.L.; Bales, K.R.; et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 671–682. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Hartman, R.E.; Bales, K.R.; Paul, S.M.; Holtzman, D.M. Deletion of ABCA1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2005, 280, 43236–43242. [Google Scholar] [CrossRef] [Green Version]
- Fitz, N.F.; Cronican, A.A.; Saleem, M.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. ABCA1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 2012, 32, 13125–13136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601–10606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassine, H.N.; Feng, Q.; Chiang, J.; Petrosspour, L.M.; Fonteh, A.N.; Chui, H.C.; Harrington, M.G. ABCA1-Mediated Cholesterol Efflux Capacity to Cerebrospinal Fluid Is Reduced in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. J. Am. Heart Assoc. 2016, 5, e002886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.G.J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat. Genet. 2022, 54, 1786–1794. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. TEM 2016, 27, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Gerbod-Giannone, M.C.; Li, Y.; Holleboom, A.; Han, S.; Hsu, L.C.; Tabas, I.; Tall, A.R. TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3112–3117. [Google Scholar] [CrossRef] [Green Version]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.P.; Fenart, L. Modelling of the blood-brain barrier in drug discovery and development. Nat. Rev. Drug. Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolakopoulou, A.M.; Montagne, A.; Kisler, K.; Dai, Z.; Wang, Y.; Huuskonen, M.T.; Sagare, A.P.; Lazic, D.; Sweeney, M.D.; Kong, P.; et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 2019, 22, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chopp, M.; Chen, J. Multifaceted roles of pericytes in central nervous system homeostasis and disease. J. Cereb. Blood. Flow. Metab. 2020, 40, 1381–1401. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Aalderink, M.; Scotter, E.L.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Graham, E.S.; Faull, R.L.; Curtis, M.A.; Park, T.I.; et al. TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J. Neuroinflamm. 2016, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Rustenhoven, J.; Smyth, L.C.; Jansson, D.; Schweder, P.; Aalderink, M.; Scotter, E.L.; Mee, E.W.; Faull, R.L.M.; Park, T.I.; Dragunow, M. Modelling physiological and pathological conditions to study pericyte biology in brain function and dysfunction. BMC Neurosci. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Vandenhaute, E.; Boucau, M.C.; Candela, P.; Dehouck, L.; Cecchelli, R.; Dehouck, M.P.; Fenart, L.; Gosselet, F. Brain Pericytes ABCA1 Expression Mediates Cholesterol Efflux but not Cellular Amyloid-beta Peptide Accumulation. J. Alzheimers Dis. 2012, 30, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Gosselet, F.; Candela, P.; Sevin, E.; Berezowski, V.; Cecchelli, R.; Fenart, L. Transcriptional profiles of receptors and transporters involved in brain cholesterol homeostasis at the blood-brain barrier: Use of an in vitro model. Brain Res. 2009, 1249, 34–42. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Abe, M.A.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Kanda, T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J. Cell Physiol. 2011, 226, 255–266. [Google Scholar] [CrossRef]
- Deligne, C.; Hachani, J.; Duban-Deweer, S.; Meignan, S.; Leblond, P.; Carcaboso, A.M.; Sano, Y.; Shimizu, F.; Kanda, T.; Gosselet, F.; et al. Development of a human in vitro blood-brain tumor barrier model of diffuse intrinsic pontine glioma to better understand the chemoresistance. Fluids Barriers CNS 2020, 17, 37. [Google Scholar] [CrossRef]
- Heymans, M.; Figueiredo, R.; Dehouck, L.; Francisco, D.; Sano, Y.; Shimizu, F.; Kanda, T.; Bruggmann, R.; Engelhardt, B.; Winter, P.; et al. Contribution of brain pericytes in blood-brain barrier formation and maintenance: A transcriptomic study of cocultured human endothelial cells derived from hematopoietic stem cells. Fluids Barriers CNS 2020, 17, 48. [Google Scholar] [CrossRef]
- Fowler, J.W.M.; Zhang, R.; Tao, B.; Boutagy, N.E.; Sessa, W.C. Inflammatory stress signaling via NF-kB alters accessible cholesterol to upregulate SREBP2 transcriptional activity in endothelial cells. eLife 2022, 11, e79529. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral sphingomyelinase 2 regulates inflammatory responses in monocytes/macrophages induced by TNF-α. Sci. Rep. 2020, 10, 16802. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Xiao, X.; Yan, Y.; Zhang, T. Activation of liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain Behav. Immun. 2021, 94, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Candela, P.; Boucau, M.C.; Fenart, L.; Gosselet, F. Oxysterols decrease apical-to-basolateral transport of Abeta peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 2013, 1517, 1–15. [Google Scholar] [CrossRef]
- Favari, E.; Lee, M.; Calabresi, L.; Franceschini, G.; Zimetti, F.; Bernini, F.; Kovanen, P.T. Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J. Biol. Chem. 2004, 279, 9930–9936. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, F.; Farhat, D.; Gursu, G.; Samnani, S.; Lee, J.Y. Snapshots of ABCG1 and ABCG5/G8: A Sterol’s Journey to Cross the Cellular Membranes. Int. J. Mol. Sci. 2022, 24, 484. [Google Scholar] [CrossRef]
- O’Connell, B.J.; Denis, M.; Genest, J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation 2004, 110, 2881–2888. [Google Scholar] [CrossRef] [Green Version]
- Gosselet, F.; Saint-Pol, J.; Candela, P.; Fenart, L. Amyloid-beta Peptides, Alzheimer’s Disease and the Blood-brain Barrier. Curr. Alzheimer Res. 2013, 10, 1015–1033. [Google Scholar] [CrossRef]
- Yancey, P.G.; Bortnick, A.E.; Kellner-Weibel, G.; de la Llera-Moya, M.; Phillips, M.C.; Rothblat, G.H. Importance of different pathways of cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 2008, 104, 1145–1166. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; Kapoor, K.; Tajkhorshid, E. Probing cholesterol binding and translocation in P-glycoprotein. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183090. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Shedding light on drug transport: Structure and function of the P-glycoprotein multidrug transporter (ABCB1). Biochem. Cell Biol. Biochim. Biol. Cell. 2006, 84, 979–992. [Google Scholar] [CrossRef]
- Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M.C.; Boulanger, E.; Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-beta peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimers Dis. 2010, 22, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevin, E.; Dehouck, L.; Versele, R.; Culot, M.; Gosselet, F. A Miniaturized Pump Out Method for Characterizing Molecule Interaction with ABC Transporters. Int. J. Mol. Sci. 2019, 20, 5529. [Google Scholar] [CrossRef] [Green Version]
- Raldúa, D.; Babin, P.J. BLT-1, a specific inhibitor of the HDL receptor SR-BI, induces a copper-dependent phenotype during zebrafish development. Toxicol. Lett. 2007, 175, 1–7. [Google Scholar] [CrossRef]
- Poller, B.; Drewe, J.; Krähenbühl, S.; Huwyler, J.; Gutmann, H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol. Neurobiol. 2010, 30, 63–70. [Google Scholar] [CrossRef]
- Juhl, A.D.; Wüstner, D. Pathways and Mechanisms of Cellular Cholesterol Efflux-Insight from Imaging. Front. Cell Dev. Biol. 2022, 10, 834408. [Google Scholar] [CrossRef]
- Pahnke, J.; Bascuñana, P.; Brackhan, M.; Stefan, K.; Namasivayam, V.; Koldamova, R.; Wu, J.; Möhle, L.; Stefan, S.M. Strategies to gain novel Alzheimer’s disease diagnostics and therapeutics using modulators of ABCA transporters. Free Neuropathol. 2021, 2, 33. [Google Scholar] [CrossRef]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Hsiao, J.H.; Paxinos, G.; Halliday, G.M.; Kim, W.S. ABCA7 Mediates Phagocytic Clearance of Amyloid-beta in the Brain. J. Alzheimers Dis. 2016, 54, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Lamartiniere, Y.; Boucau, M.C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 Downregulation Modifies Cellular Cholesterol Homeostasis and Decreases Amyloid-beta Peptide Efflux in an in vitro Model of the Blood-Brain Barrier. J. Alzheimers Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Erdener, Ş.E.; Küreli, G.; Dalkara, T. Contractile apparatus in CNS capillary pericytes. Neurophotonics 2022, 9, 021904. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Brühl, M.L.; Gärtner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 2013, 14, 41–51. [Google Scholar] [CrossRef]
- Pieper, C.; Marek, J.J.; Unterberg, M.; Schwerdtle, T.; Galla, H.J. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014, 1550, 1–8. [Google Scholar] [CrossRef]
- Hamon, Y.; Broccardo, C.; Chambenoit, O.; Luciani, M.F.; Toti, F.; Chaslin, S.; Freyssinet, J.M.; Devaux, P.F.; McNeish, J.; Marguet, D.; et al. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat. Cell Biol. 2000, 2, 399–406. [Google Scholar] [CrossRef]
- Luciani, M.F.; Chimini, G. The ATP binding cassette transporter ABC1, is required for the engulfment of corpses generated by apoptotic cell death. EMBO J. 1996, 15, 226–235. [Google Scholar] [CrossRef]
- Chen, W.; Li, L.; Wang, J.; Zhang, R.; Zhang, T.; Wu, Y.; Wang, S.; Xing, D. The ABCA1-efferocytosis axis: A new strategy to protect against atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 518, 1–8. [Google Scholar] [CrossRef]
- Ohkawa, R.; Low, H.; Mukhamedova, N.; Fu, Y.; Lai, S.J.; Sasaoka, M.; Hara, A.; Yamazaki, A.; Kameda, T.; Horiuchi, Y.; et al. Cholesterol transport between red blood cells and lipoproteins contributes to cholesterol metabolism in blood. J. Lipid Res. 2020, 61, 1577–1588. [Google Scholar] [CrossRef]
- Salmeri, M.; Motta, C.; Anfuso, C.D.; Amodeo, A.; Scalia, M.; Toscano, M.A.; Alberghina, M.; Lupo, G. VEGF receptor-1 involvement in pericyte loss induced by Escherichia coli in an in vitro model of blood brain barrier. Cell. Microbiol. 2013, 15, 1367–1384. [Google Scholar] [CrossRef] [PubMed]
- Fotakis, P.; Kothari, V.; Thomas, D.G.; Westerterp, M.; Molusky, M.M.; Altin, E.; Abramowicz, S.; Wang, N.; He, Y.; Heinecke, J.W.; et al. Anti-Inflammatory Effects of HDL (High-Density Lipoprotein) in Macrophages Predominate Over Proinflammatory Effects in Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e253–e272. [Google Scholar] [CrossRef] [PubMed]
- Grao-Cruces, E.; Lopez-Enriquez, S.; Martin, M.E.; Montserrat-de la Paz, S. High-density lipoproteins and immune response: A review. Int. J. Biol. Macromol. 2022, 195, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β Modulate Blood-Brain Barrier Permeability and Decrease Amyloid-β Peptide Efflux in a Human Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFα alters occludin and cerebral endothelial permeability: Role of p38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ramirez, M.A.; Male, D.K.; Wang, C.; Sharrack, B.; Wu, D.; Romero, I.A. Cytokine-induced changes in the gene expression profile of a human cerebral microvascular endothelial cell-line, hCMEC/D3. Fluids Barriers CNS 2013, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- da Rocha, G.H.O.; Loiola, R.A.; de Paula-Silva, M.; Shimizu, F.; Kanda, T.; Vieira, A.; Gosselet, F.; Farsky, S.H.P. Pioglitazone Attenuates the Effects of Peripheral Inflammation in a Human In Vitro Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 12781. [Google Scholar] [CrossRef]
- Jennings, P.; Koppelstaetter, C.; Aydin, S.; Abberger, T.; Wolf, A.M.; Mayer, G.; Pfaller, W. Cyclosporine A induces senescence in renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2007, 293, F831–F838. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, M.; Candela, P.; Saint-Pol, J.; Lamartiniere, Y.; Boucau, M.C.; Sevin, E.; Fenart, L.; Gosselet, F. Bexarotene Promotes Cholesterol Efflux and Restricts Apical-to-Basolateral Transport of Amyloid-beta Peptides in an In Vitro Model of the Human Blood-Brain Barrier. J. Alzheimers Dis. 2015, 48, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Nieland, T.J.; Chroni, A.; Fitzgerald, M.L.; Maliga, Z.; Zannis, V.I.; Kirchhausen, T.; Krieger, M. Cross-inhibition of SR-BI- and ABCA1-mediated cholesterol transport by the small molecules BLT-4 and glyburide. J. Lipid. Res. 2004, 45, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Nieland, T.J.; Penman, M.; Dori, L.; Krieger, M.; Kirchhausen, T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc. Natl. Acad. Sci. USA 2002, 99, 15422–15427. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sequence (F/R) | Accession Number |
|---|---|---|
| ABCA1 | F: CAGTGCTTCCTGATTAGCACAC R: AGGCTAGCGAAGATCTTGGTG | NM_005502.4 |
| ABCB1 | F: CAGACAGCAGCTGACAGTCCAAGAACAGGACT R: GCCTGGCAGCTGGAAGACAAATACACAAAATT | NM_001348945.2 |
| ABCG4 | F: GGACATAGAGTTCGTGGAGC R: GGTCTTATAACCCCTTTTGCGCC | NM_001348191.2 |
| SCARB1 | F: ATCCCCTTCTATCTCTCCGTCT R: GTCGTTGTTGTTGAAGGTGATG | NM_001367988.1 |
| HMGCR | F: TGTGTGTGGGACCGTAATGG R: GCTGTCTTCTTGGTGCAAGC | NM_001130996.2 |
| APOE | F: GGTCGCTTTTGGGATTACCT R: CCTTCAACTCCTTCATGGTCTC | NM_001302690.2 |
| LDLR | F: TTCATGGCTTCATGTACTGGAC R: TTTTCAGTCACCAGCGAGTAGA | NM_000527.5 |
| LRP1 | F: AATGAGTGTCTCAGCCGCAA R: AACGGTTCCTCGTCAGTCAC | NM_002332.3 |
| MYLIP | F: TATGTGACGAGGCCGGACG R: TGATTCCCAGTCGCCTGCAC | NM_013262.4 |
| NR1H3 (LXRα) | F: CAGGGCCATGAATGAGCTGC R: TGTGCTGCAGCCTCTCTACC | NM_005693.4 |
| NR1H2 (LXRβ) | F: TCCTACCACGAGTTCCCTGG R: TGGTTCCTCTTCGGGATCTGG | NM_007121.7 |
| TNFRSF1A (TNFR1) | F: ACAAGCCACAGAGCCTAGACACTG R: ACGAATTCCTTCCAGCGCAACG | NM_001065.4 |
| TNFRSF1B (TNFR2) | F: TCTCCAACACGACTTCATCCACGG R: AGACTGCATCCATGCTTGCATTCC | NM_001066.3 |
| GAPDH | F: GATGACATCAAGAAGGTGGTGA R: GCTGTTGAAGTCAGAGGAGACC | NM_001357943.2 |
| Target | kDa | Reference | Supplier | Conditions | Dilution |
|---|---|---|---|---|---|
| ABCA1 | 254 | ab18180 | Abcam | non-heated/reduced | 1:1000 |
| ABCB1 (P-gp) | 180 | GTX23364 | Genetex | heated/reduced | 1:500 |
| ABCG2 (BCRP) | 72 | Ab207732 | Abcam | non-heated/reduced | 1:1000 |
| ABCG4 | 100 | PA5-34855 | Invitrogen | heated/reduced | 1:1000 |
| SR-BI | 80 | ab52629 | Abcam | heated/reduced | 1:1000 |
| HMGCR | 100 | Mab90619 | Sigma | non-heated/reduced | 1:1000 |
| LRP1 | 80 | sc57351 | Santa Cruz | heated/reduced | 1:500 |
| LDLR | 140 | ab52818 | Abcam | heated/reduced | 1:1000 |
| APOE | 34 | ab1906 | Abcam | heated/reduced | 1:1000 |
| COX2 | 70 | NBD100-689SS | Novus | heated/reduced | 1:1000 |
| NLRP3 | 118 | ab263899 | Abcam | heated/reduced | 1:1000 |
| VCAM1 | 90–100 | ab134047 | Abcam | heated/reduced | 1:1000 |
| IL-6 | 22–28 | NBD600-1131SS | Novus | heated/reduced | 1:1000 |
| PhosphoNF-κB | 66–75 | Mab72261 | R&D | heated/reduced | 1:1000 |
| NF-κB | 66–75 | ab32536 | Abcam | heated/reduced | 1:1000 |
| ACTIN | 42 | A5441 | Sigma | heated/reduced | 1:10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dib, S.; Loiola, R.A.; Sevin, E.; Saint-Pol, J.; Shimizu, F.; Kanda, T.; Pahnke, J.; Gosselet, F. TNFα Activates the Liver X Receptor Signaling Pathway and Promotes Cholesterol Efflux from Human Brain Pericytes Independently of ABCA1. Int. J. Mol. Sci. 2023, 24, 5992. https://doi.org/10.3390/ijms24065992
Dib S, Loiola RA, Sevin E, Saint-Pol J, Shimizu F, Kanda T, Pahnke J, Gosselet F. TNFα Activates the Liver X Receptor Signaling Pathway and Promotes Cholesterol Efflux from Human Brain Pericytes Independently of ABCA1. International Journal of Molecular Sciences. 2023; 24(6):5992. https://doi.org/10.3390/ijms24065992
Chicago/Turabian StyleDib, Shiraz, Rodrigo Azevedo Loiola, Emmanuel Sevin, Julien Saint-Pol, Fumitaka Shimizu, Takashi Kanda, Jens Pahnke, and Fabien Gosselet. 2023. "TNFα Activates the Liver X Receptor Signaling Pathway and Promotes Cholesterol Efflux from Human Brain Pericytes Independently of ABCA1" International Journal of Molecular Sciences 24, no. 6: 5992. https://doi.org/10.3390/ijms24065992
APA StyleDib, S., Loiola, R. A., Sevin, E., Saint-Pol, J., Shimizu, F., Kanda, T., Pahnke, J., & Gosselet, F. (2023). TNFα Activates the Liver X Receptor Signaling Pathway and Promotes Cholesterol Efflux from Human Brain Pericytes Independently of ABCA1. International Journal of Molecular Sciences, 24(6), 5992. https://doi.org/10.3390/ijms24065992