


Autophagy: A Potential Therapeutic Target to Tackle Drug Resistance in Multiple Myeloma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
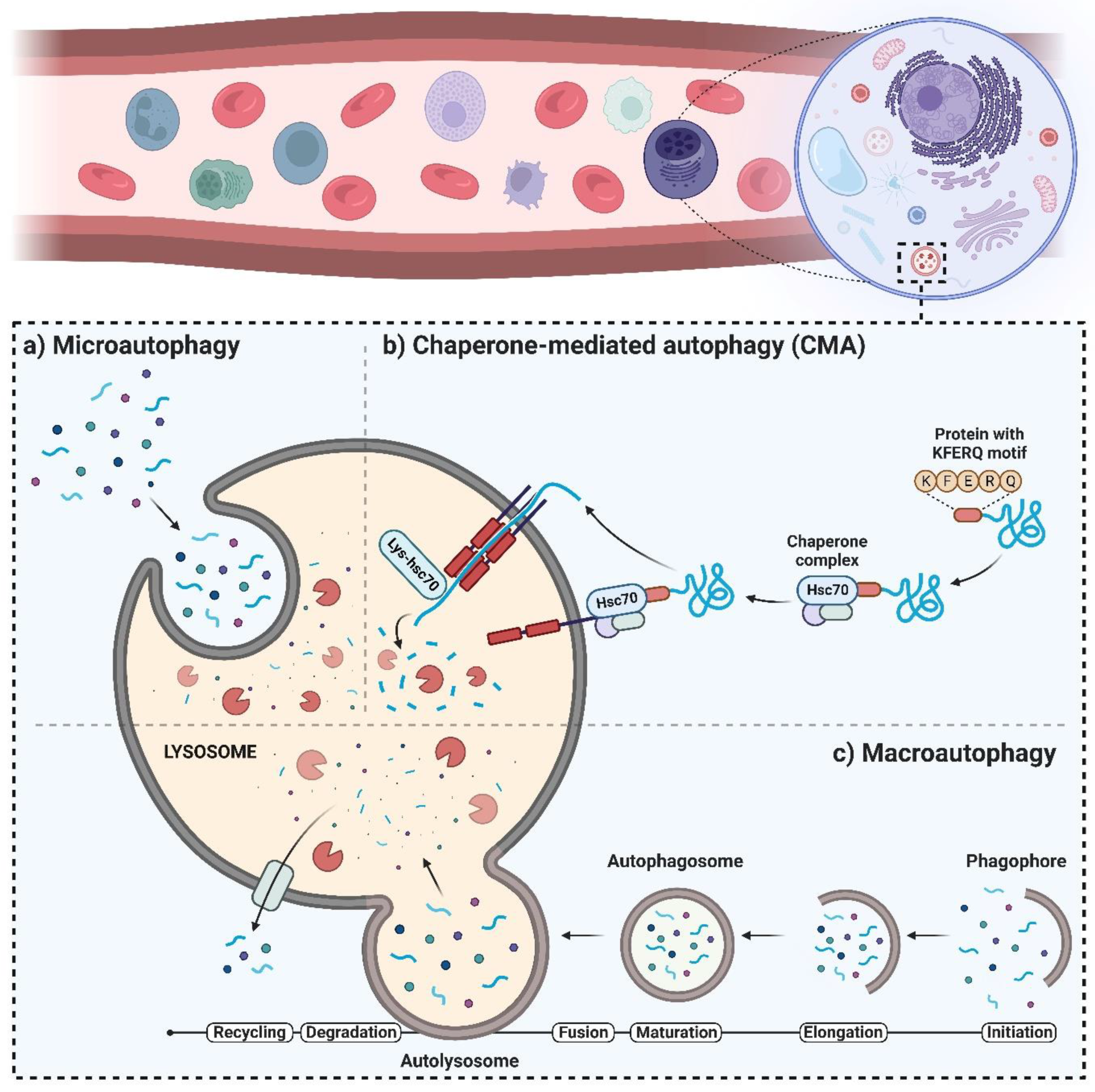
2. The Autophagy Machinery and Its Regulatory Mechanisms
3. Significance of Autophagic Activity in the Survival of MM Cells
4. Multiple Myeloma Drug Resistance and Autophagy
5. Effects of HDAC Inhibitors on the Promotion of Autophagic Activity
6. Effect of CAM, an Autophagy Antagonist, on MM
7. Effects of CAM in Combination with a Proteasome Suppressor or an Antagonist of HDAC6
8. HMGB1-Induced Chemoresistance in MM
9. Rescue from Autophagic Activity, Disrupted Intracellular Signaling Processes, and Apoptosis
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Kunz, J.B.; Schwarz, H.; Mayer, A. Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 2004, 279, 9987–9996. [Google Scholar] [CrossRef] [Green Version]
- Jardon, M.A.; Rothe, K.; Bortnik, S.; Vezenkov, L.; Jiang, X.; Young, R.N.; Lum, J.J.; Gorski, S.M. Autophagy: From Structure to Metabolism to Therapeutic Regulation; Taylor & Francis: Abingdon, UK, 2013. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Arias, E. Methods to study chaperone-mediated autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615. [Google Scholar] [CrossRef] [Green Version]
- Nikesitch, N.; Rebeiro, P.; Ho, L.L.; Pothula, S.; Wang, X.M.; Khong, T.; Quek, H.; Spencer, A.; Lee, C.S.; Roberts, T.L. The role of chaperone-mediated autophagy in Bortezomib resistant multiple myeloma. Cells 2021, 10, 3464. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakifahmetoglu-Norberg, H.; Xia, H.-G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar]
- Rowinsky, E.K. Targeting the molecular target of rapamycin (mTOR). Curr. Opin. Oncol. 2004, 16, 564–575. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Troya, S.; Pérez-Pérez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865. [Google Scholar] [CrossRef]
- Kwon, G.; Marshall, C.A.; Pappan, K.L.; Remedi, M.S.; McDaniel, M.L. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 2004, 53, S225–S232. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 395–401. [Google Scholar]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Liang, S.; Hu, J.; Jin, W.; Zhan, Q.; Zhao, K. Autophagy as a target for hematological malignancy therapy. Blood Rev. 2016, 30, 369–380. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D. Global burden of multiple myeloma: A systematic analysis for the global burden of disease study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [Green Version]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Landgren, O.; Mateos, M.V. Smoldering multiple myeloma. Blood 2015, 125, 3069–3075. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Jelinek, T.; Kryukov, F.; Rihova, L.; Hajek, R. Plasma cell leukemia: From biology to treatment. Eur. J. Haematol. 2015, 95, 16–26. [Google Scholar] [CrossRef]
- Ravi, P.; Kumar, S.K.; Roeker, L.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Go, R.S.; Dispenzieri, A.; Kapoor, P.; Lust, J.A.; et al. Revised diagnostic criteria for plasma cell leukemia: Results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Milan, E.; Fabbri, M.; Cenci, S. Autophagy in plasma cell ontogeny and malignancy. J. Clin. Immunol. 2016, 36, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viabilityAutophagy and Myeloma. Mol. Cancer Ther. 2009, 8, 1974–1984. [Google Scholar] [CrossRef] [Green Version]
- Caro, L.H.P.; Plomp, P.J.; Wolvetang, E.J.; Kerkhof, C.; Meijer, A.J. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur. J. Biochem. 1988, 175, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, P.; Chen, X.; Zhao, L.; Tan, J.; Yang, Y.; Fang, Y.; Zhou, J. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol. Ther. 2017, 18, 584–595. [Google Scholar] [CrossRef]
- Lamy, L.; Ngo, V.N.; Emre, N.T.; Shaffer, A.L.; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358. [Google Scholar] [CrossRef] [Green Version]
- Riz, I.; Hawley, T.S.; Hawley, R.G. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015, 6, 14814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Abstract# 1877: Impact of autophagy on multiple myeloma cell viability. Cancer Res. 2009, 69, 1877. [Google Scholar]
- Fu, Y.-F.; Liu, X.; Gao, M.; Zhang, Y.-N.; Liu, J. Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget 2017, 8, 61093. [Google Scholar] [CrossRef] [Green Version]
- Frassanito, M.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016, 30, 640–648. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood J. Am. Soc. Hematol. 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia-Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.-H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Aquila, S.; Santoro, M.; Caputo, A.; Panno, M.L.; Pezzi, V.; De Amicis, F. The Tumor Suppressor PTEN as Molecular Switch Node Regulating Cell Metabolism and Autophagy: Implications in Immune System and Tumor Microenvironment. Cells 2020, 9, 1725. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggiò, M.; Racanelli, V.; Vacca, A.; Frassanito, M. Autophagy: A new mechanism of prosurvival and drug resistance in multiple myeloma. Transl. Oncol. 2018, 11, 1350–1357. [Google Scholar] [CrossRef]
- Ndoye, A.; Weeraratna, A.T. Autophagy—An emerging target for melanoma therapy. F1000Research 2016, 5, 1888. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2014, 28, 904–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraiso, K.H.; Smalley, K.S. Fibroblast-mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Hamedi, K.R.; Harmon, K.A.; Goodwin, R.L.; Arce, S. Autophagy and the Bone Marrow Microenvironment: A Review of Protective Factors in the Development and Maintenance of Multiple Myeloma. Front. Immunol. 2022, 13, 889954. [Google Scholar] [CrossRef] [PubMed]
- RI, M. Mechanism of action of bortezomib in multiple myeloma therapy. Int. J. Myeloma 2016, 6, 1–6. [Google Scholar]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [Green Version]
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer associated fibroblasts and tumor growth: Focus on multiple myeloma. Cancers 2014, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.Y.; Zaitseva, L.; Auger, M.J.; Craig, J.I.; MacEwan, D.J.; Rushworth, S.A.; Bowles, K.M. Ibrutinib inhibits BTK-driven NF-κB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle 2015, 14, 2367–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S. Autophagy in Multiple Myeloma: What Makes You Stronger Can Also Kill You. Cancer Cell 2013, 23, 425–426. [Google Scholar]
- Bekkers, J.E.; Saris, D.B.; Tsuchida, A.I.; van Rijen, M.H.; Dhert, W.J.; Creemers, L.B. Chondrogenic potential of articular chondrocytes depends on their original location. Tissue Eng. Part A 2014, 20, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: US FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- McBride, A.; Ryan, P.Y. Proteasome inhibitors in the treatment of multiple myeloma. Expert Rev. Anticancer Ther. 2013, 13, 339–358. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Mateos, M.V. Can multiple myeloma become a curable disease? Haematologica 2011, 96, 1246–1248. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.-W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. [Google Scholar] [CrossRef] [Green Version]
- Kirk, C.J. Discovery and development of second-generation proteasome inhibitors. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 207–214. [Google Scholar]
- Dimopoulos, M.A.; San-Miguel, J.F.; Anderson, K.C. Emerging therapies for the treatment of relapsed or refractory multiple myeloma. Eur. J. Haematol. 2011, 86, 1–15. [Google Scholar] [CrossRef]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J. US Food and Drug Administration Approval: Carfilzomib for the Treatment of Multiple MyelomaFDA Approval Summary of Carfilzomib. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome inhibitors in acute leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-S.; Jung, S.-H.; Yang, D.-H.; Bae, S.-Y.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. Patterns of relapse or progression after bortezomib-based salvage therapy in patients with relapsed/refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 389–394. [Google Scholar] [CrossRef]
- Cheriyath, V.; Jacobs, B.S.; Hussein, M.A. Proteasome inhibitors in the clinical setting: Benefits and strategies to overcome multiple myeloma resistance to proteasome inhibitors. Drugs R D 2007, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, M.T.; Giraldo, P.; Corradini, P.; Teixeira, A.; Dimopoulos, M.A.; Blau, I.W.; Drach, J.; Angermund, R.; Allietta, N.; Broer, E. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br. J. Haematol. 2013, 160, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1. [Google Scholar]
- White, E.; DiPaola, R.S. The Double-Edged Sword of Autophagy Modulation in CancerAutophagy in Cancer Therapy. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.-M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Chen, X.; Kang, J.H.; Stolz, D.B.; Liu, J.; Yin, X.-M. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol. Cancer Ther. 2009, 8, 2036–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Gopinathan, G.; Sukumar, J.T.; Gribben, J.G. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS ONE 2012, 7, e32584. [Google Scholar] [CrossRef] [PubMed]
- Montanari, F.; Lu, M.; Marcus, S.; Saran, A.; Malankar, A.; Mazumder, A. A Phase II Trial of Chloroquine in Combination with Bortezomib and Cyclophosphamide in Patients with Relapsed and Refractory Multiple Myeloma. Blood 2014, 124, 5775. [Google Scholar] [CrossRef]
- Harmon, K.A.; Roman, S.; Lancaster, H.D.; Chowhury, S.; Cull, E.; Goodwin, R.L.; Arce, S.; Fanning, S. Structural and Ultrastructural Analysis of the Multiple Myeloma Cell Niche and a Patient-Specific Model of Plasma Cell Dysfunction. Microsc. Microanal. 2022, 28, 254–264. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Corner, G.A.; Augenlicht, L.H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: Comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000, 60, 4561–4572. [Google Scholar]
- Li, G.; Margueron, R.; Hu, G.; Stokes, D.; Wang, Y.H.; Reinberg, D. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell 2010, 38, 41–53. [Google Scholar] [CrossRef] [Green Version]
- McConkey, D.J.; White, M.; Yan, W. HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv. Cancer Res. 2012, 116, 131–163. [Google Scholar] [PubMed]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.-T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood J. Am. Soc. Hematol. 2010, 116, 4906–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and In Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma CellsProteasome Inhibitor MLN9708 as Myeloma Therapy. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.P.; Ban, K.; Dujka, M.E.; McConkey, D.J.; Munsell, M.; Palladino, M.; Chandra, J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood J. Am. Soc. Hematol. 2007, 110, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; Choi, I.K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyên, T.L.-A.; Di Lenardo, T.; Semmes, O.J.; Lin, R. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef] [Green Version]
- Park, M.A.; Reinehr, R.; Häussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef] [Green Version]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Douthwaite, S.; Champney, W.S. Structures of ketolides and macrolides determine their mode of interaction with the ribosomal target site. J. Antimicrob. Chemother. 2001, 48 (Suppl. S2), 1–8. [Google Scholar] [CrossRef]
- Van Nuffel, A.M.; Sukhatme, V.; Pantziarka, P.; Meheus, L.; Sukhatme, V.P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Clarithromycin as an anti-cancer agent. Ecancermedicalscience 2015, 9, 513. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.; Bataille, R. Cytokine network in human multiple myeloma. Hematol. Oncol. Clin. N. Am. 1992, 6, 273–284. [Google Scholar] [CrossRef]
- Klein, B. Cytokine, cytokine receptors, transduction signals, and oncogenes in human multiple myeloma. Semin. Hematol. 1995, 32, 4–19. [Google Scholar] [PubMed]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017, 2017, 1852517. [Google Scholar] [CrossRef] [Green Version]
- Moriya, S.; Che, X.F.; Komatsu, S.; Abe, A.; Kawaguchi, T.; Gotoh, A.; Inazu, M.; Tomoda, A.; Miyazawa, K. Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress-mediated CHOP induction in myeloma cells. Int. J. Oncol. 2013, 42, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Moriya, S.; Komatsu, S.; Yamasaki, K.; Kawai, Y.; Kokuba, H.; Hirota, A.; Che, X.F.; Inazu, M.; Gotoh, A.; Hiramoto, M.; et al. Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress-mediated cell death in multiple myeloma cells. Int. J. Oncol. 2015, 46, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Kikukawa, Y.; Takeya, M.; Mitsuya, H.; Hata, H. Clarithromycin attenuates autophagy in myeloma cells. Int. J. Oncol. 2010, 37, 815–820. [Google Scholar]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef] [Green Version]
- Takemori, N.; Imai, G.; Hoshino, K.; Ooi, A.; Kojima, M. A novel combination of bortezomib, lenalidomide, and clarithromycin produced stringent complete response in refractory multiple myeloma complicated with diabetes mellitus—Clinical significance and possible mechanisms: A case report. J. Med. Case Rep. 2018, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; He, D.; Zhang, E.; Chen, J.; Chen, Q.; Li, Y.; Yang, L.; Yang, Y.; Zhao, Y.; Wang, G. HMGB1 knockdown increases MM cell vulnerability by regulating autophagy and DNA damage repair. J. Exp. Clin. Cancer Res. 2018, 37, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Lv, A.E.; Li, H.P.; Han, D.H.; Zhang, Y.P. LncRNA MALAT-1 elevates HMGB1 to promote autophagy resulting in inhibition of tumor cell apoptosis in multiple myeloma. J. Cell. Biochem. 2017, 118, 3341–3348. [Google Scholar] [CrossRef]
- Roy, M.; Liang, L.; Xiao, X.; Peng, Y.; Luo, Y.; Zhou, W.; Zhang, J.; Qiu, L.; Zhang, S.; Liu, F. Lycorine downregulates HMGB1 to inhibit autophagy and enhances bortezomib activity in multiple myeloma. Theranostics 2016, 6, 2209. [Google Scholar] [CrossRef]
- Pan, B.; Chen, D.; Huang, J.; Wang, R.; Feng, B.; Song, H.; Chen, L. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol. Cancer 2014, 13, 165. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, H.; Sun, M.; Yin, Z.; Qian, J. High mobility group box 1-mediated autophagy promotes neuroblastoma cell chemoresistance. Oncol. Rep. 2015, 34, 2969–2976. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 Promotes Drug Resistance in OsteosarcomaHMGB1 and Osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Sun, X.; Tang, D. HMGB1-dependent and -independent autophagy. Autophagy 2014, 10, 1873–1876. [Google Scholar] [CrossRef] [Green Version]
- Mou, K.; Liu, W.; Han, D.; Li, P. HMGB1/RAGE axis promotes autophagy and protects keratinocytes from ultraviolet radiation-induced cell death. J. Dermatol. Sci. 2017, 85, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Konig, H.; Johnson, D.; Tang, D.; Amaravadi, R.; Boyiadzis, M.; Lotze, M. You eat what you are: Autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015, 29, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Loze, M.T. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011, 7, 112–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yu, Y.; Kang, R.; Yang, M.; Xie, M.; Wang, Z.; Tang, D.; Zhao, M.; Liu, L.; Zhang, H. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk. Lymphoma 2012, 53, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Okazaki, H.; Iwata, H. Dynamin-dependent amino acid endocytosis activates mechanistic target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 2017, 292, 18052–18061. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Chiang, S.-F.; Chen, W.T.-L.; Ke, T.-W.; Chen, T.-W.; You, Y.-S.; Lin, C.-Y.; Chao, K.C.; Huang, C.-Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xie, J.; Li, X.; Fang, J. Poly (ADP-ribosylation) of HMGB1 facilitates its acetylation and promotes HMGB1 translocation-associated chemotherapy-induced autophagy in leukaemia cells. Oncol. Lett. 2020, 19, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.; Banerjee, S.; Friggeri, A.; Bell, C.; Abraham, E.; Zerfaoui, M. Poly (ADP-ribosyl) ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol. Med. 2012, 18, 359–369. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.H.; Ma, M.H.; Vescio, R.A.; Berenson, J.R. Overcoming drug resistance in multiple myeloma: The emergence of therapeutic approaches to induce apoptosis. J. Clin. Oncol. 2003, 21, 4239–4247. [Google Scholar] [CrossRef]
- Lentzsch, S.; Chatterjee, M.; Gries, M.; Bommert, K.; Gollasch, H.; Dörken, B.; Bargou, R.C. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia 2004, 18, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, N.L.; Rudikoff, S. Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood 2000, 96, 2856–2861. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Treon, S.P.; Mitsiades, N.; Shima, Y.; Richardson, P.; Schlossman, R.; Hideshima, T.; Anderson, K.C. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: Therapeutic applications. Blood J. Am. Soc. Hematol. 2001, 98, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsas, P.; López-Royuela, N.; Galán-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouill, S.L.; Podar, K.; Harousseau, J.-L.; Anderson, K.C. Mcl-1 Regulation and Its Role in Multiple Myeloma. Cell Cycle 2004, 3, 1259–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gojo, I.; Fenton, R.G. Myeloid cell factor–1 is a critical survival factor for multiple myeloma. Blood J. Am. Soc. Hematol. 2002, 99, 1885–1893. [Google Scholar] [CrossRef]
- Wuillème-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef] [Green Version]
- Le Gouill, S.; Podar, K.; Amiot, M.; Hideshima, T.; Chauhan, D.; Ishitsuka, K.; Kumar, S.; Raje, N.; Richardson, P.G.; Harousseau, J.-L. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood 2004, 104, 2886–2892. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Veyrune, J.-L.; Vos, J.D.; Redal, N.; Couderc, G.; Klein, B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene 2003, 22, 2950–2959. [Google Scholar] [CrossRef] [Green Version]
- Puthier, D.; Bataille, R.; Amiot, M. IL-6 up-regulates Mcl-1 in human myeloma cells through JAK/STAT rather than Ras/MAP kinase pathway. Eur. J. Immunol. 1999, 29, 3945–3950. [Google Scholar] [CrossRef]
- Spets, H.; Strömberg, T.; Georgii-Hemming, P.; Siljason, J.; Nilsson, K.; Jernberg-Wiklund, H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: Regulation during interleukin-6(IL-6)-induced growth and survival. Eur. J. Haematol. 2002, 69, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gardner, A.; Lichtenstein, A. The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: Roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000, 60, 6763–6770. [Google Scholar] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Du, J.; Zhang, C.; Fu, W.; Xi, H.; Zou, J.; Hou, J. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PLoS ONE 2012, 7, e32215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Nikesitch, N.; Ling, S.C.W. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar]
- Vincenz, L.; Jäger, R.; O’Dwyer, M.; Samali, A. Endoplasmic Reticulum Stress and the Unfolded Protein Response: Targeting the Achilles Heel of Multiple MyelomaUPR in Multiple Myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omedè, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14. [Google Scholar] [CrossRef] [Green Version]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845. [Google Scholar] [CrossRef] [Green Version]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramírez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling chemoresistance in cancer: Root causes and strategies to uproot them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashiri, H.; Tabatabaeian, H. Autophagy: A Potential Therapeutic Target to Tackle Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 6019. https://doi.org/10.3390/ijms24076019
Bashiri H, Tabatabaeian H. Autophagy: A Potential Therapeutic Target to Tackle Drug Resistance in Multiple Myeloma. International Journal of Molecular Sciences. 2023; 24(7):6019. https://doi.org/10.3390/ijms24076019
Chicago/Turabian StyleBashiri, Hamed, and Hossein Tabatabaeian. 2023. "Autophagy: A Potential Therapeutic Target to Tackle Drug Resistance in Multiple Myeloma" International Journal of Molecular Sciences 24, no. 7: 6019. https://doi.org/10.3390/ijms24076019
APA StyleBashiri, H., & Tabatabaeian, H. (2023). Autophagy: A Potential Therapeutic Target to Tackle Drug Resistance in Multiple Myeloma. International Journal of Molecular Sciences, 24(7), 6019. https://doi.org/10.3390/ijms24076019