
Hydroperoxidation of Docosahexaenoic Acid by Human ALOX12 and pigALOX15-mini-LOX

Abstract
:1. Introduction
2. Results and Discussion
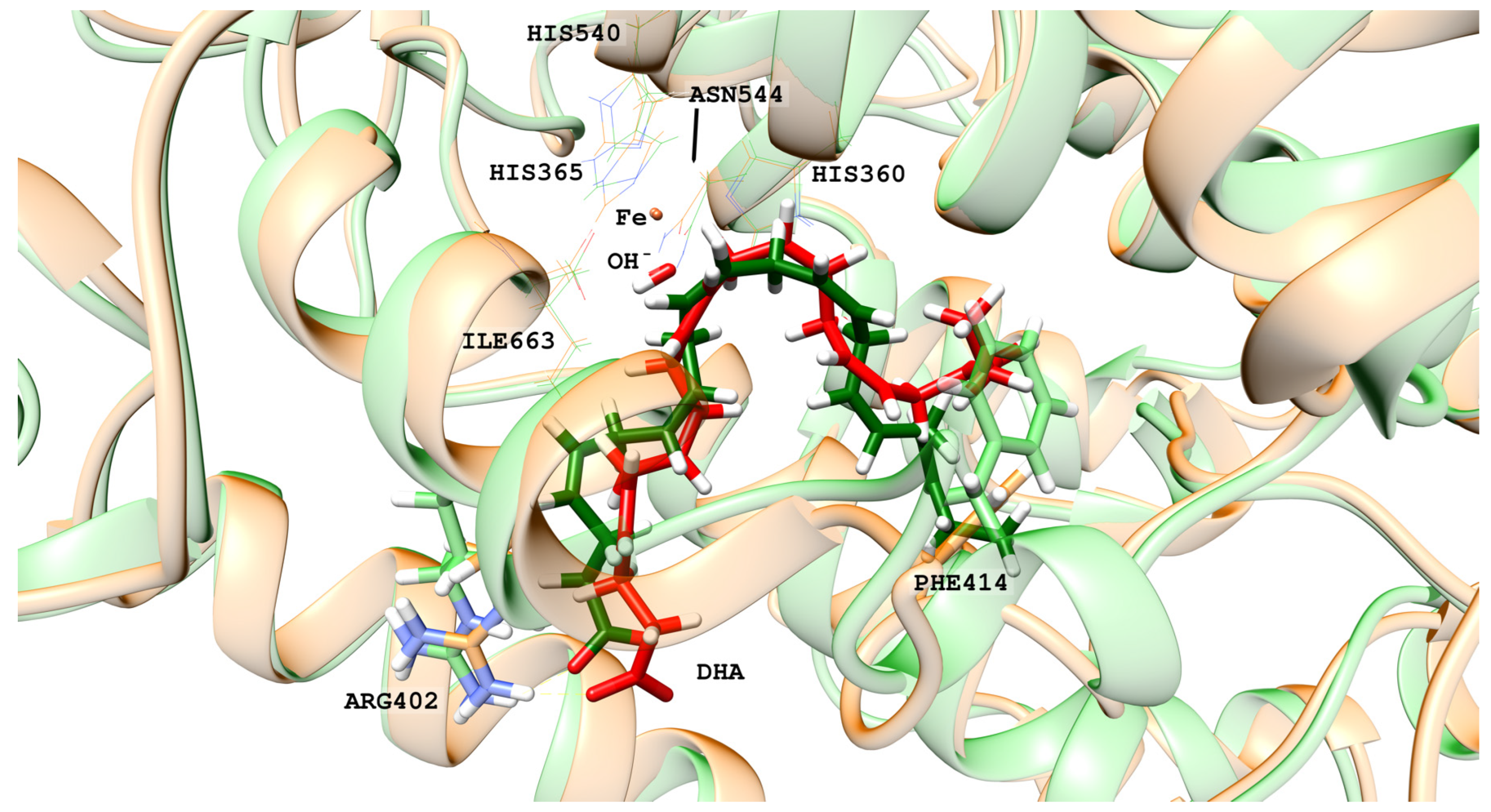
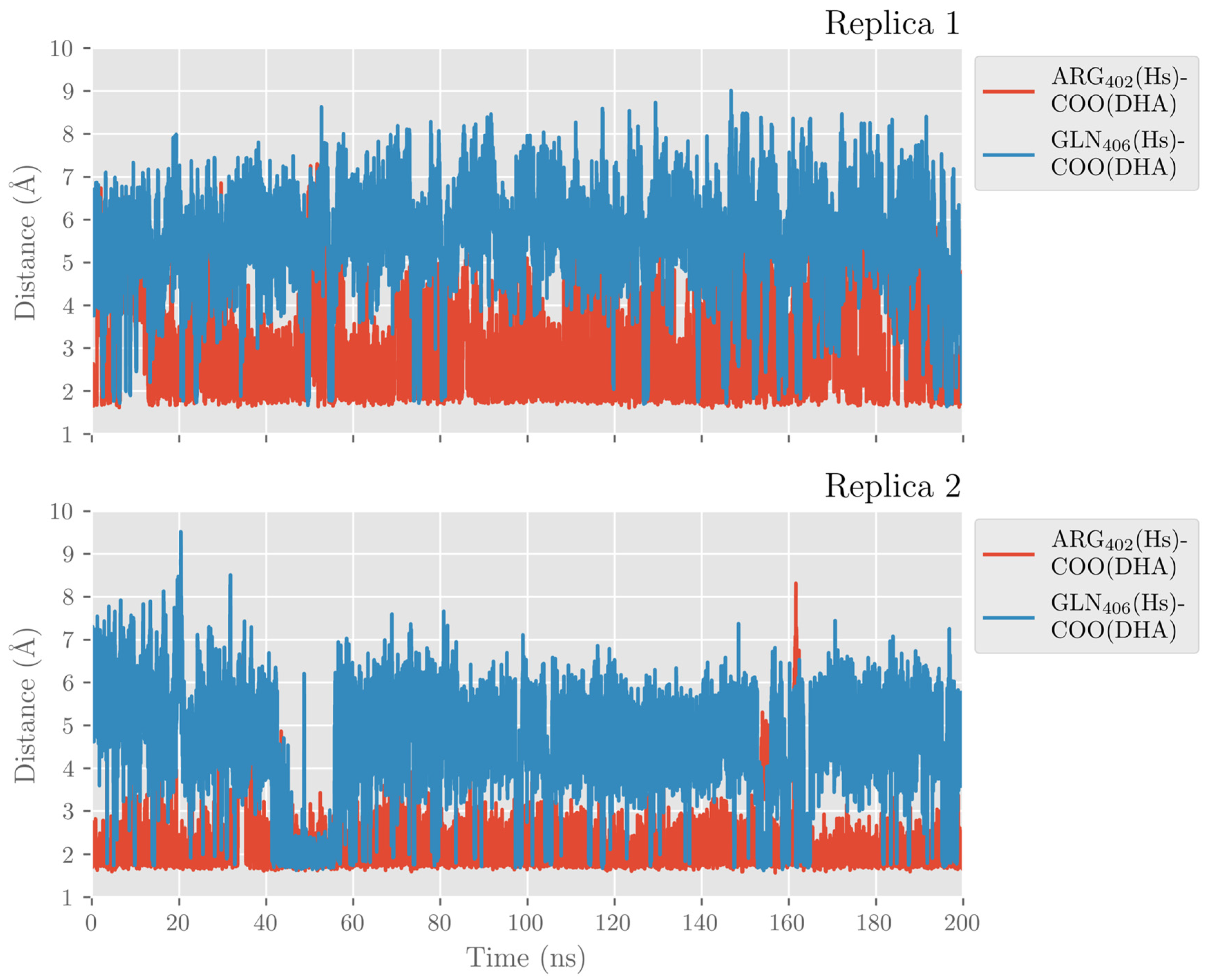
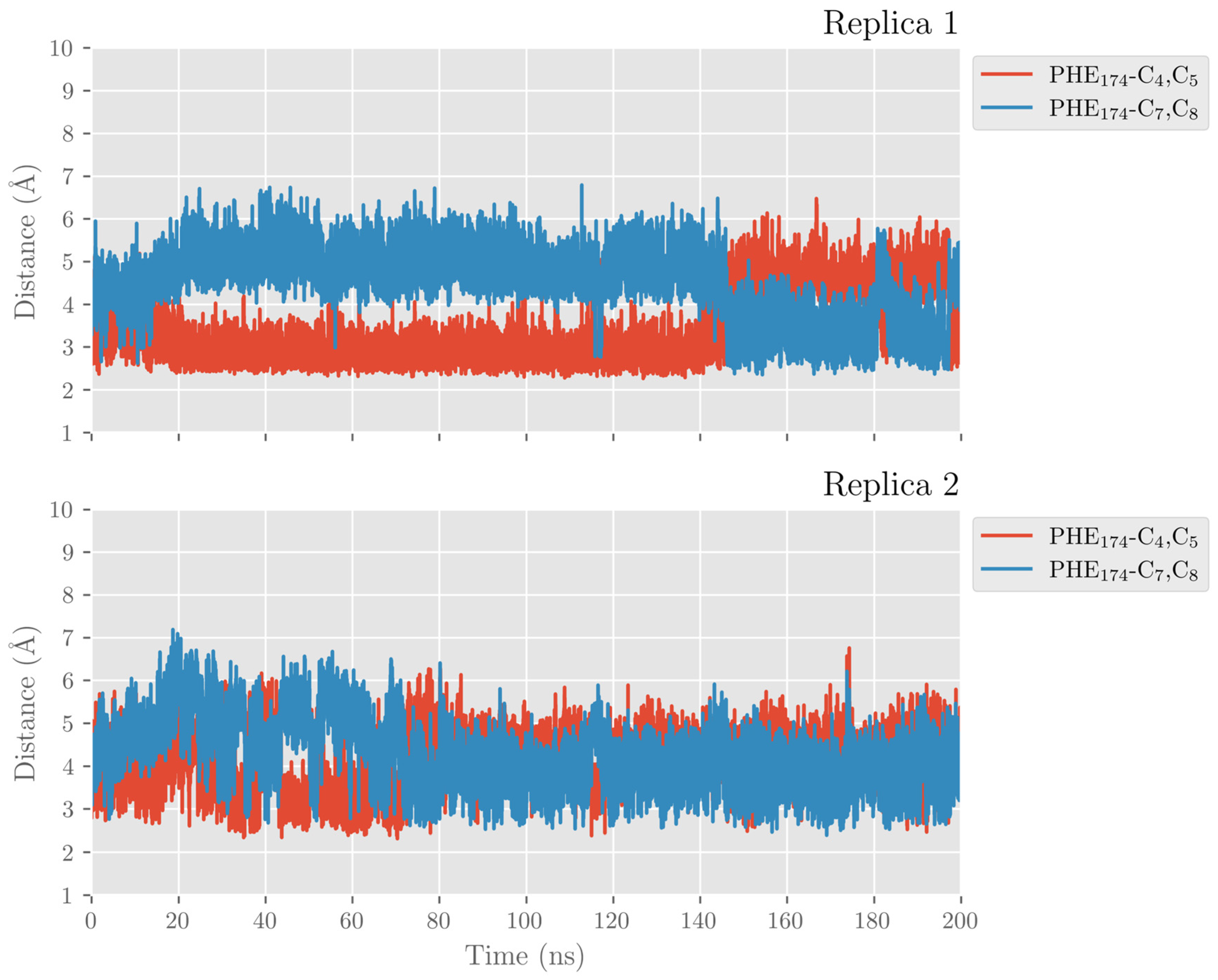
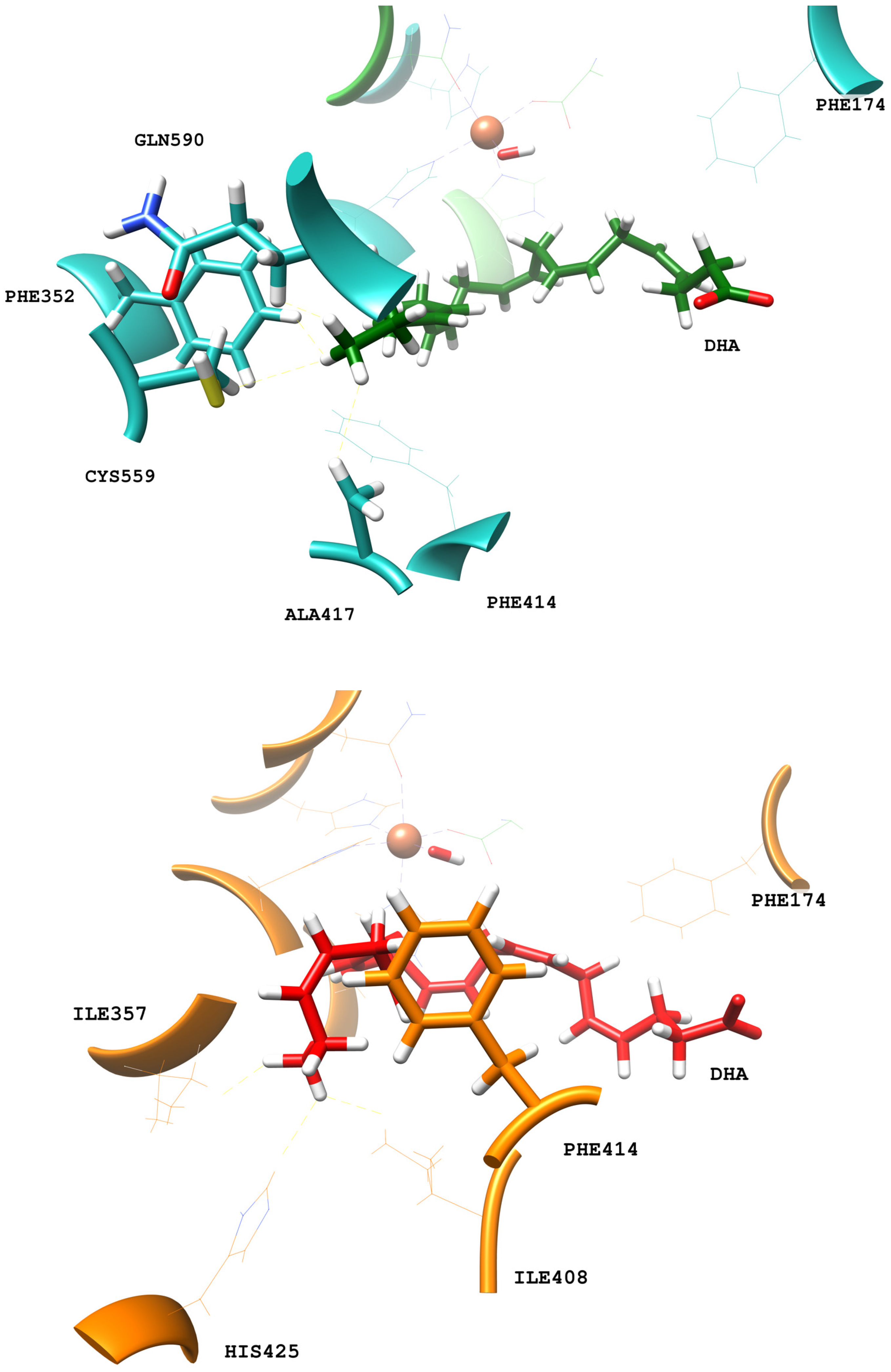
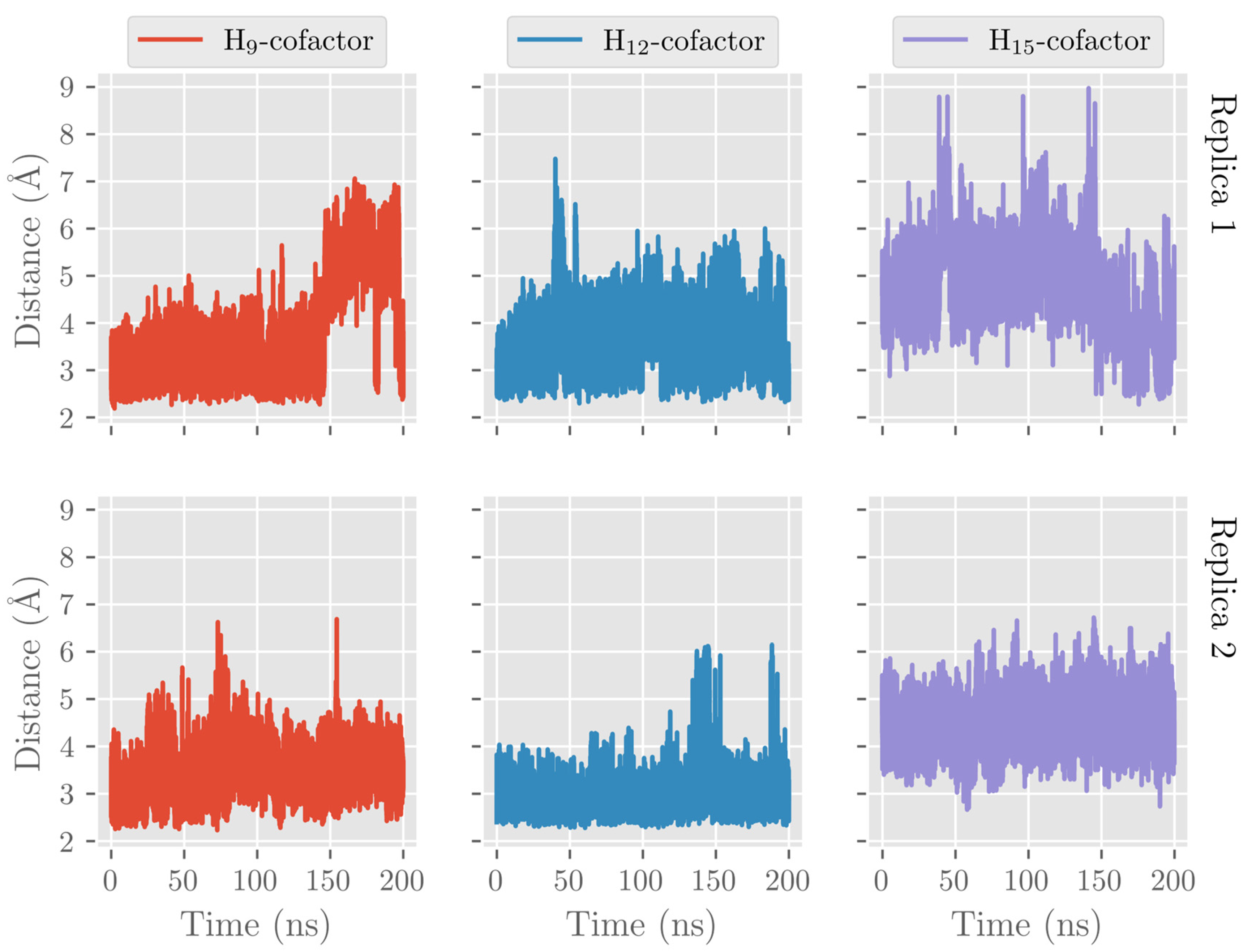
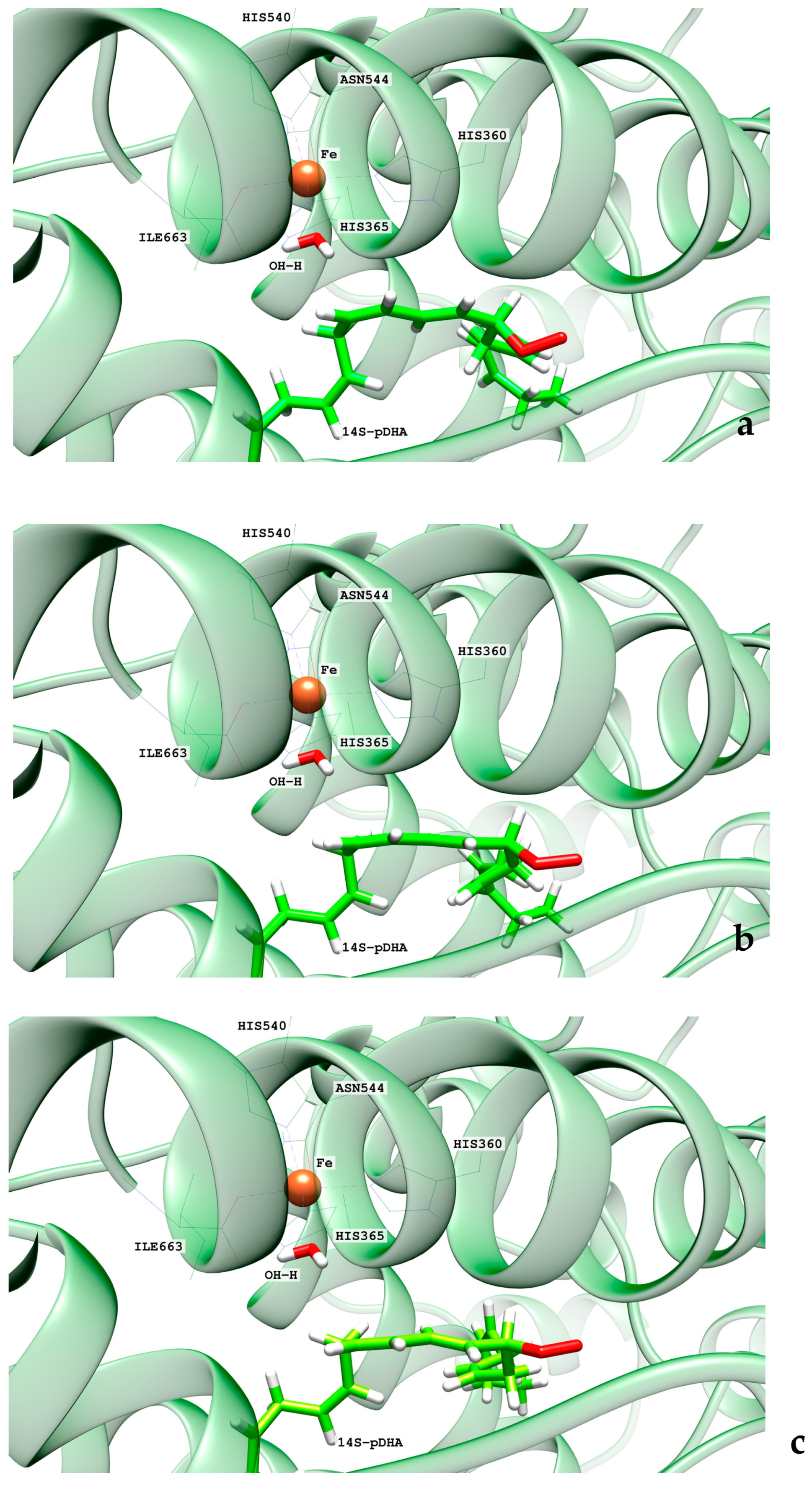
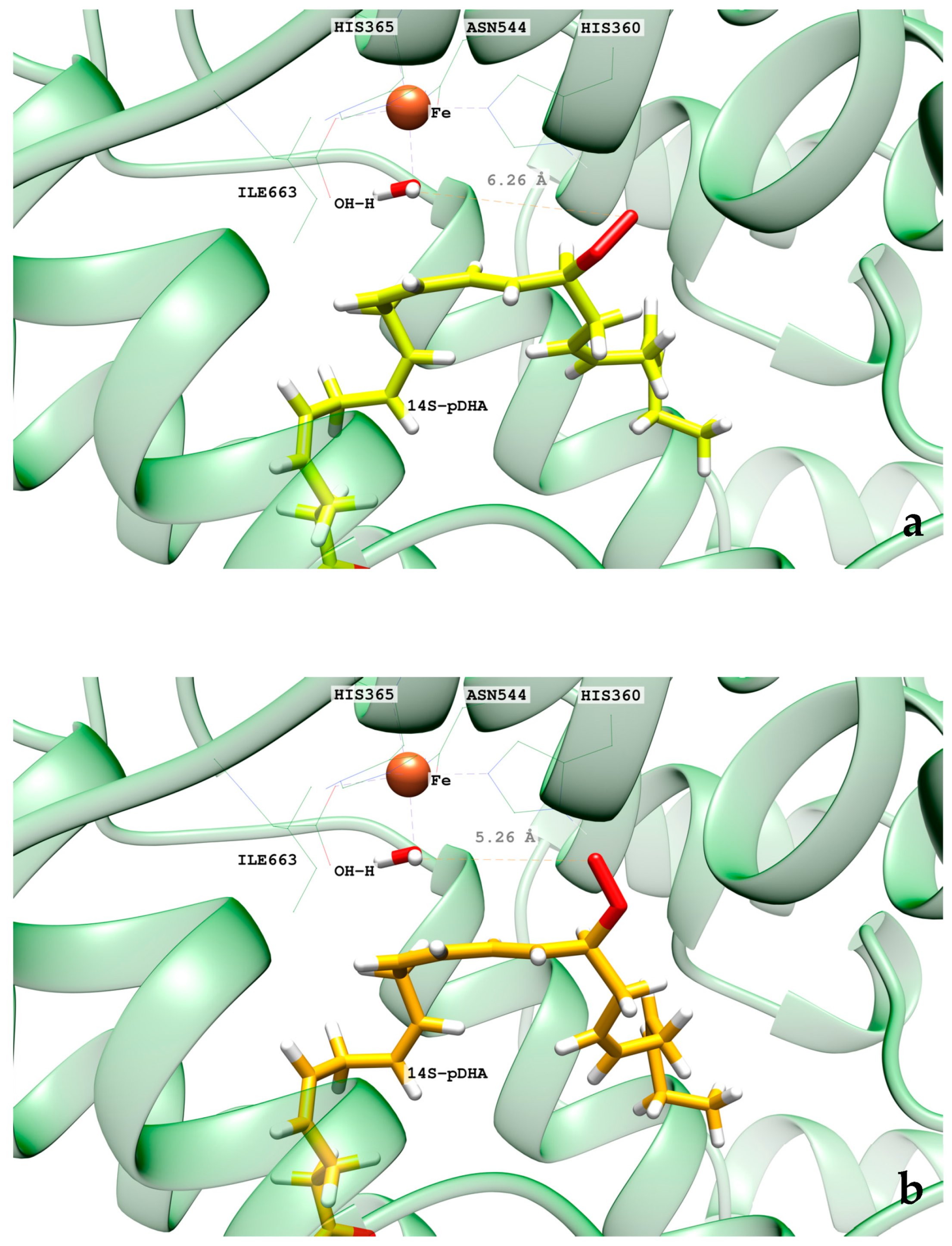
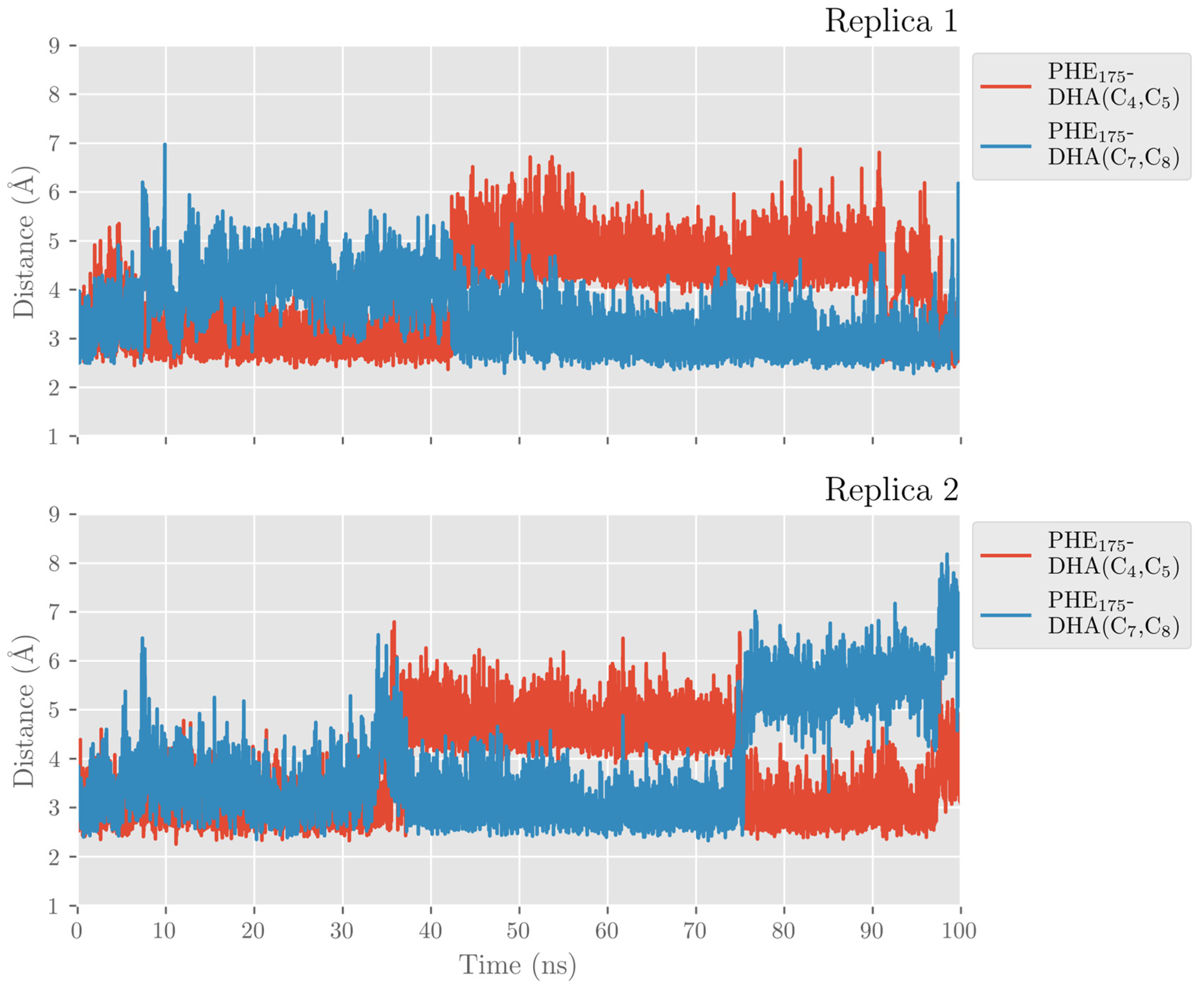
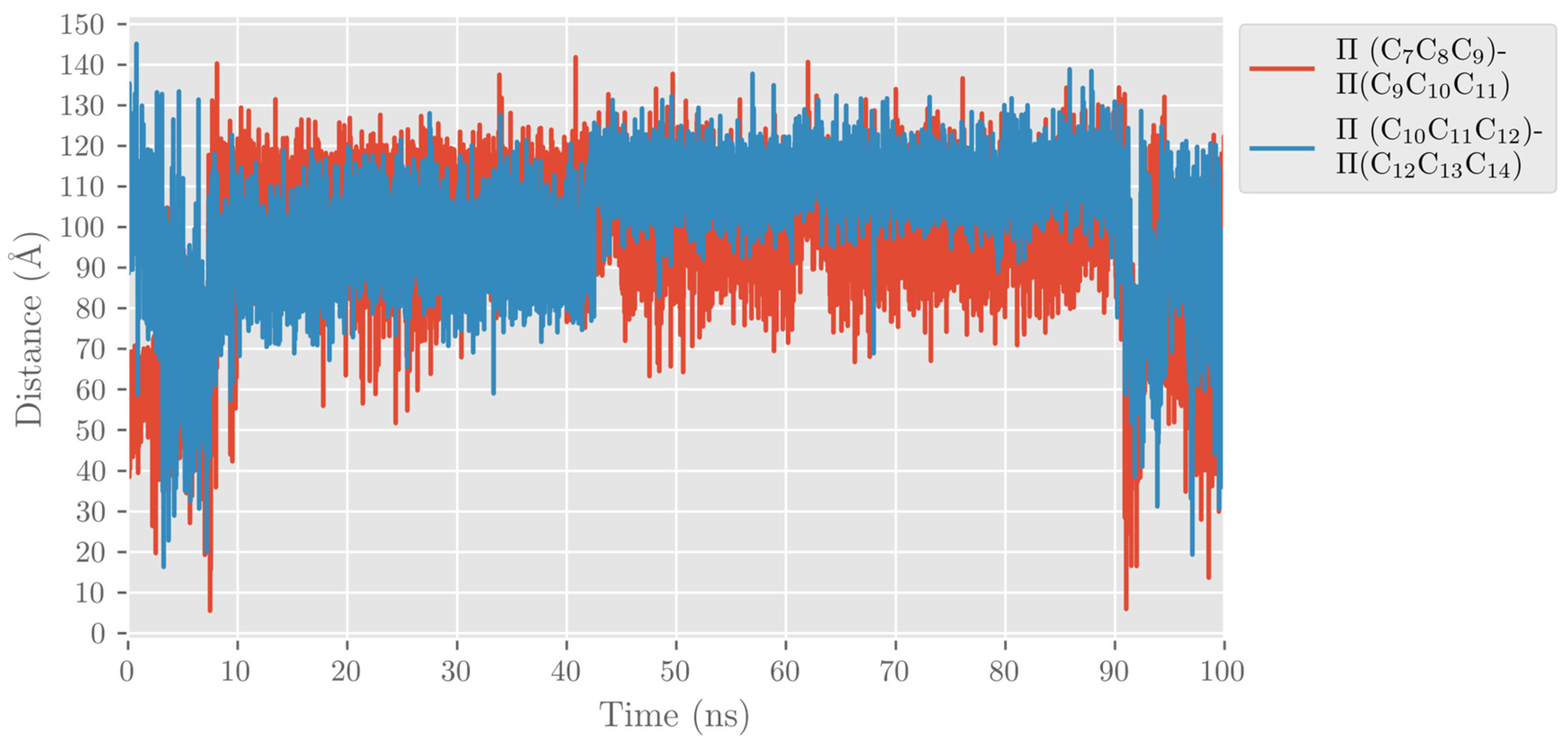
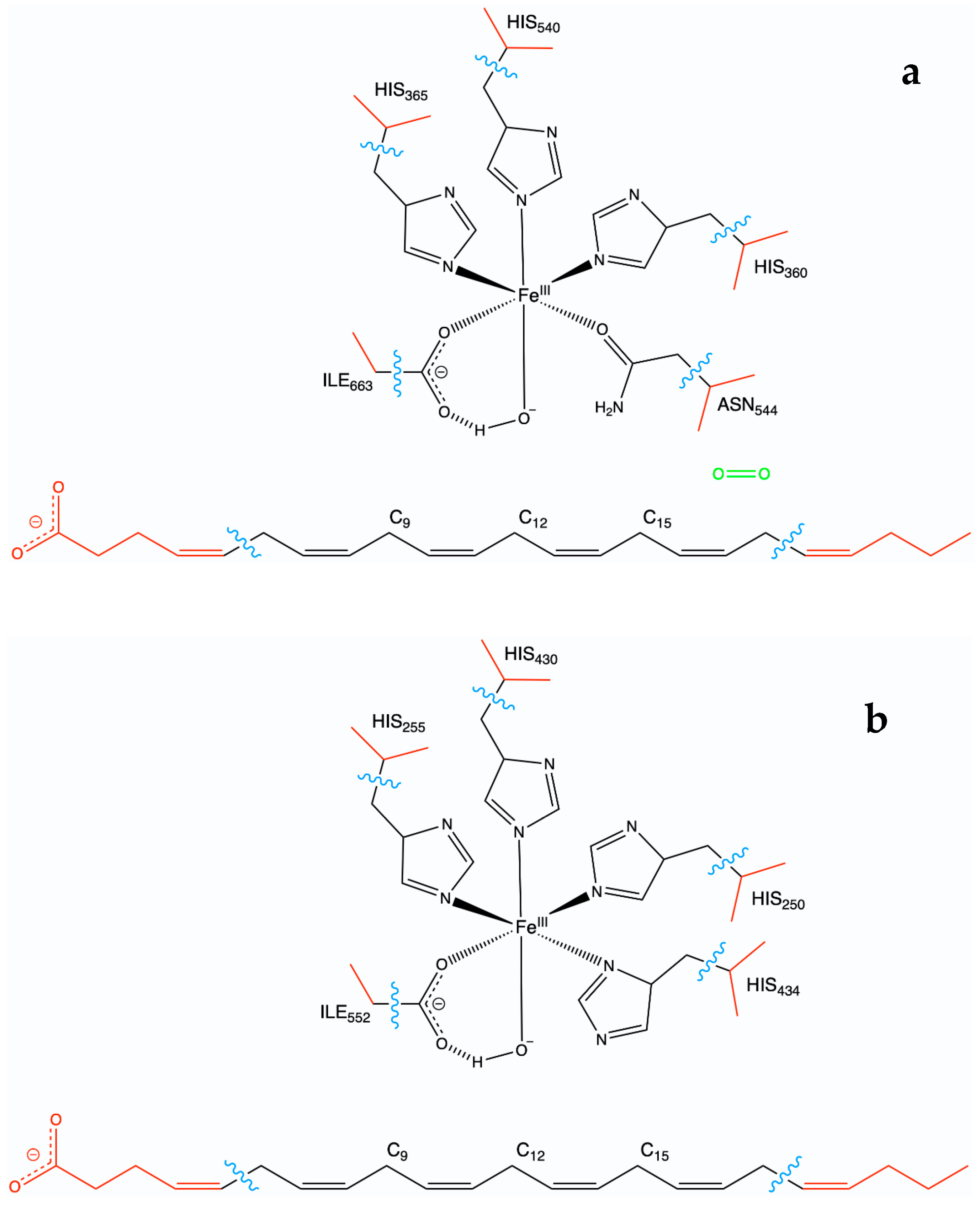
2.1. MD Simulations of the DHA/hALOX12 Complex
2.2. QM/MM Calculations of DHA Hydroperoxidation Catalyzed by hALOX12
2.2.1. Hydrogen Abstraction Reactions Catalyzed by hALOX12
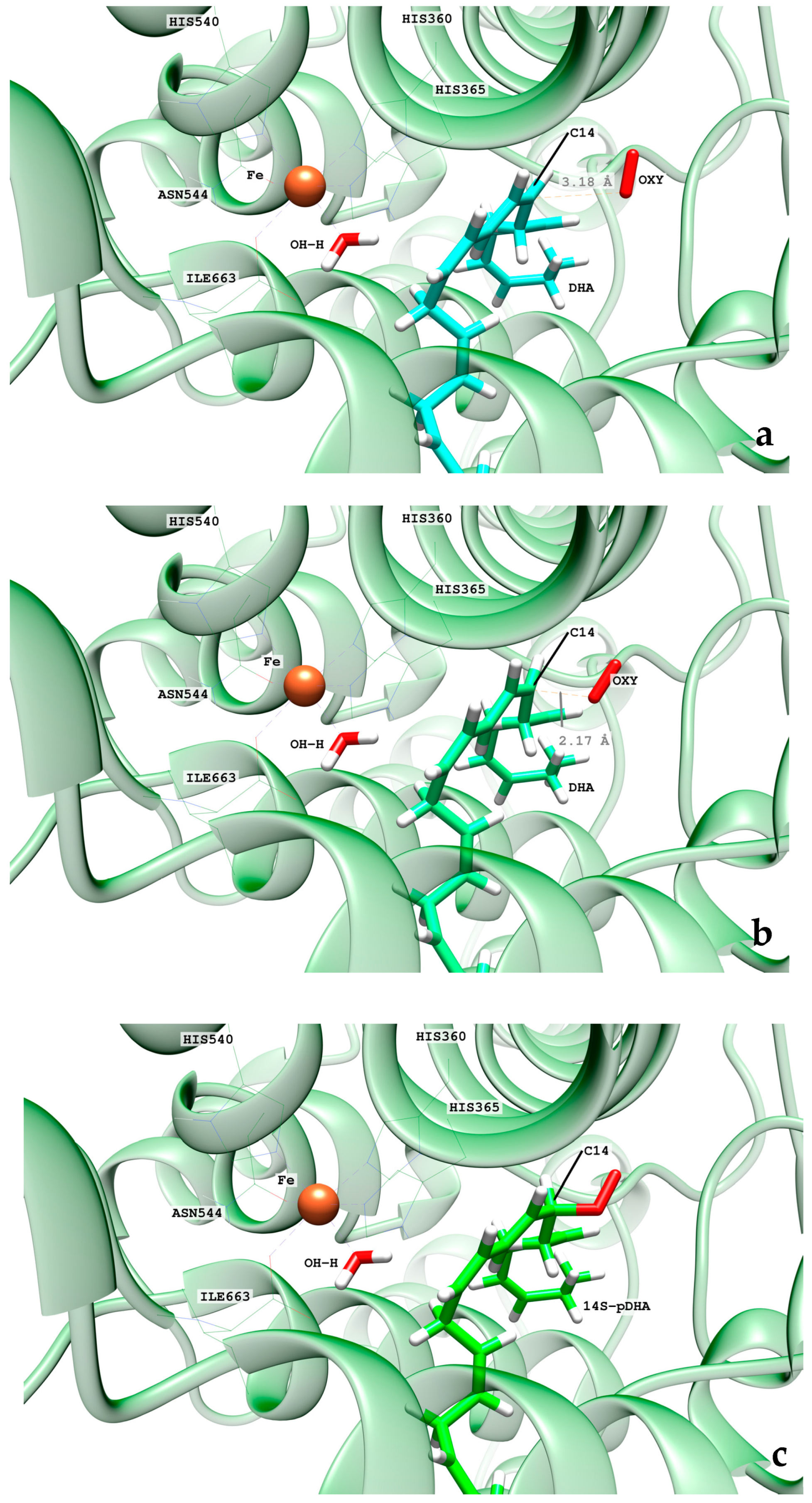
2.2.2. O2 Addition and Retro-Hydrogen Abstraction Reactions Catalyzed by hALOX12
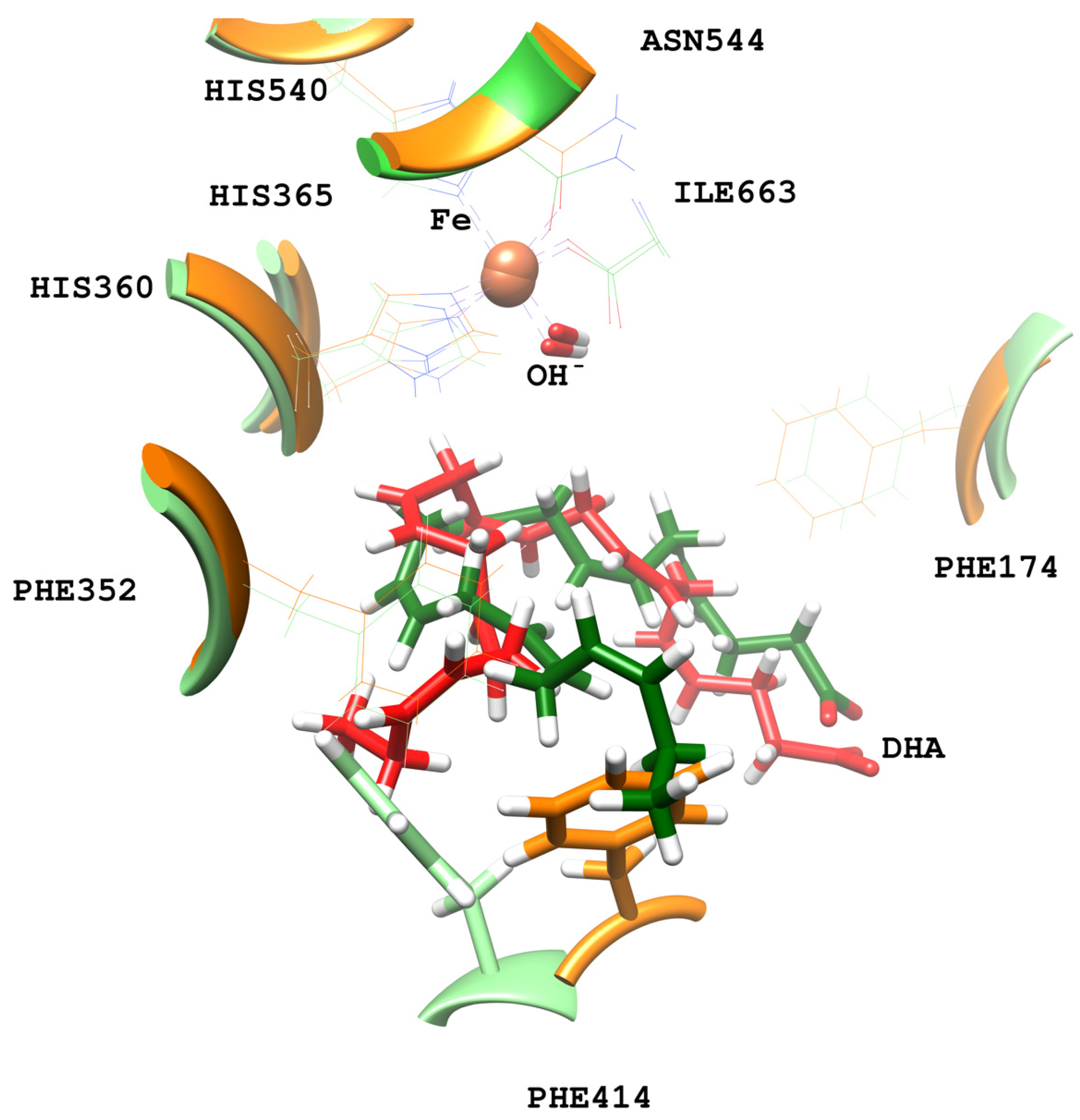
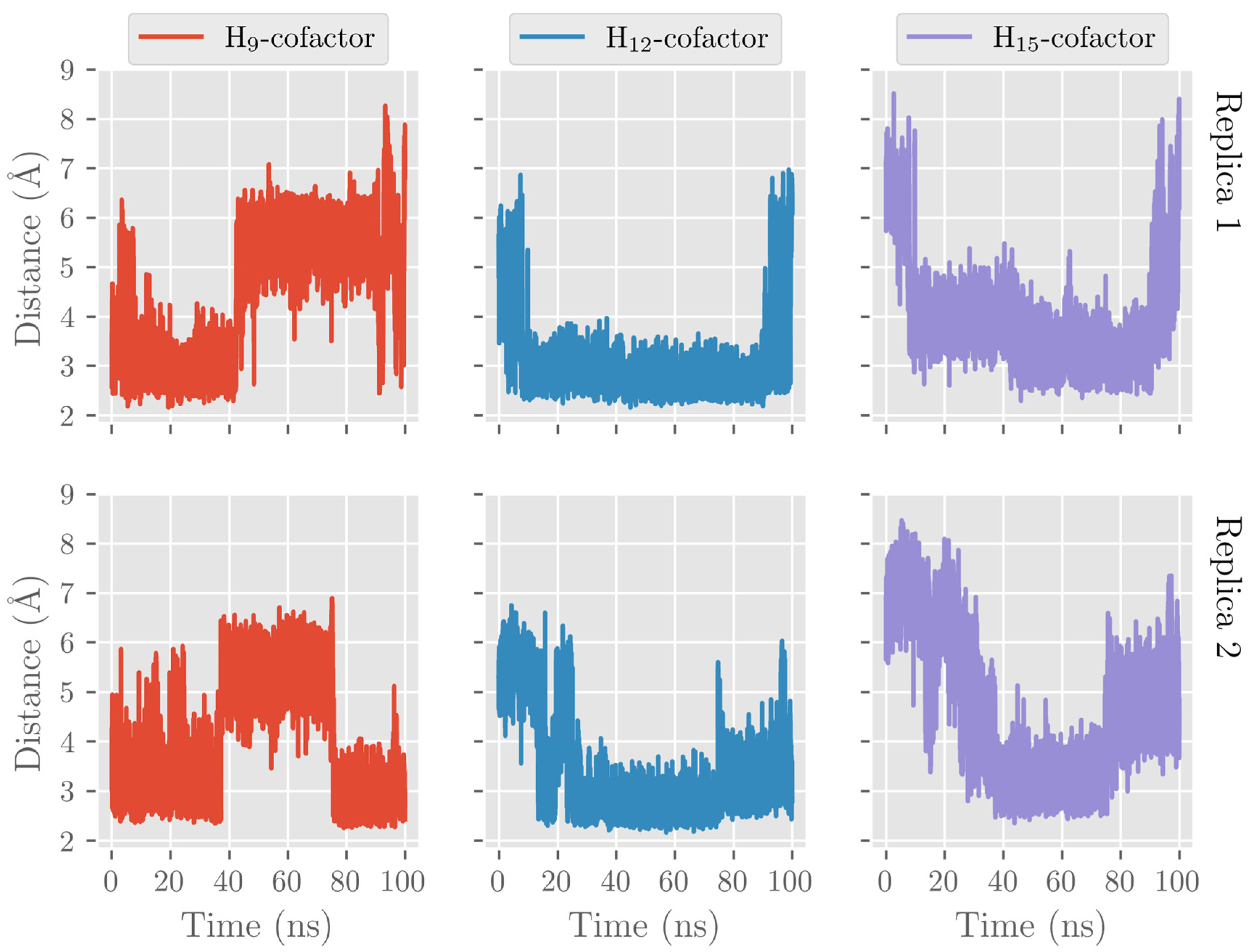
2.3. MD Simulations of the DHA/pigALOX15-mini-LOX Complex
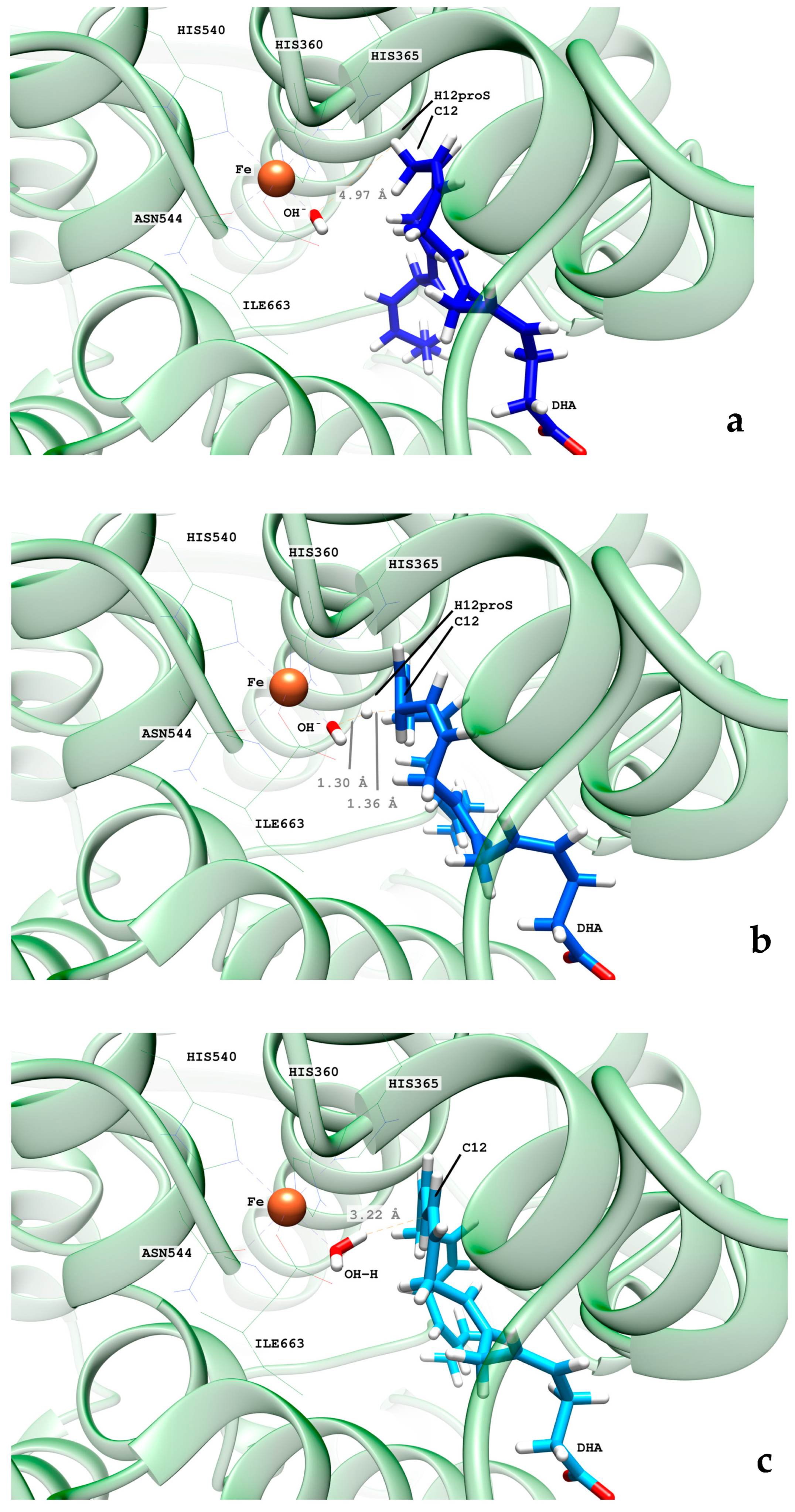
2.4. QM/MM Calculations of H12-Abstraction from DHA Catalyzed by pigALOX15-mini-LOX
3. Materials and Methods
3.1. Protein Setup
3.2. Molecular Docking Simulations
3.3. Molecular Dynamics Simulations
3.4. QM/MM Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steinhilber, D. Lipoxygenases: An Introduction. In Lipoxygenases in Inflammation; Steinhilber, D., Ed.; Springer: Cham, Switzerland, 2016; pp. 1–6. ISBN 978-3-319-27764-6. [Google Scholar]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving Inflammation: Dual Anti-Inflammatory and pro-Resolution Lipid Mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Treating Inflammation and Infection in the 21st Century: New Hints from Decoding Resolution Mediators and Mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [Green Version]
- Hamberg, M.; Samuelsson, B. Prostaglandin Endoperoxides. Novel Transformations of Arachidonic Acid in Human Platelets. Proc. Natl. Acad. Sci. USA 1974, 71, 3400–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikei, K.N.; Yeung, J.; Apopa, P.L.; Ceja, J.; Vesci, J.; Holman, T.R.; Holinstat, M. Investigations of Human Platelet-Type 12-Lipoxygenase: Role of Lipoxygenase Products in Platelet Activation. J. Lipid Res. 2012, 53, 2546–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.; Heydeck, D.; Hofheinz, K.; Roffeis, J.; O’Donnell, V.B.; Kuhn, H.; Walther, M. Molecular Enzymology of Lipoxygenases. Arch. Biochem. Biophys. 2010, 503, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef]
- Nugteren, D.H. Arachidonate Lipoxygenase in Blood Platelets. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1975, 380, 299–307. [Google Scholar] [CrossRef]
- Bürger, F.; Krieg, P.; Marks, F.; Fürstenberger, G. Positional- and Stereo-Selectivity of Fatty Acid Oxygenation Catalysed by Mouse (12S)-Lipoxygenase Isoenzymes. Biochem. J. 2000, 348, 329–335. [Google Scholar] [CrossRef]
- Yu, J.Y.; Lee, J.J.; Jung, J.K.; Min, Y.K.; Ma, J.Y.; Kim, T.J.; Lee, M.Y.; Yun, Y.P. Anti-Platelet Activity of Diacetylated Obovatol through Regulating Cyclooxygenase and Lipoxygenase Activities. Arch. Pharm. Res. 2012, 35, 2191–2198. [Google Scholar] [CrossRef]
- Katoh, A.; Ikeda, H.; Murohara, T.; Haramaki, N.; Ito, H.; Imaizumi, T. Platelet-Derived 12-Hydroxyeicosatetraenoic Acid Plays an Important Role in Mediating Canine Coronary Thrombosis by Regulating Platelet Glycoprotein IIb/IIIa Activation. Circulation 1998, 98, 2891–2898. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Takahashi, Y. Arachidonate 12-Lipoxygenases. Prostaglandins Other Lipid Mediat. 2002, 68–69, 245–262. [Google Scholar] [CrossRef]
- Matsuda, S.; Murakami, J.; Yamamoto, Y.; Konishi, Y.; Yokoyama, C.; Yoshimoto, T.; Yamamoto, S.; Mimura, Y.; Okuma, M. Decreased Messenger RNA of Arachidonate 12-Lipoxygenase in Platelets of Patients with Myeloproliferative Disorders. BBA—Mol. Basis Dis. 1993, 1180, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, F.; Takagi, J.; Sasaki, K.; Kawajiri, K.; Kobayashi, Y.; Sato, F.; Saito, Y. Feedback Regulation of Platelet Function by 12 S-Hydroxyeicosatetraenoic Acid: Inhibition of Arachidonic Acid Liberation from Phospholipids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1990, 1044, 165–168. [Google Scholar] [CrossRef]
- Holm, A.C.B.S.; Grenegård, M.; Öllinger, K.; Lindström, E.G. Inhibition of 12-Lipoxygenase Reduces Platelet Activation and Prevents Their Mitogenic Function. Platelets 2014, 25, 111–117. [Google Scholar] [CrossRef]
- Adili, R.; Tourdot, B.E.; Mast, K.; Yeung, J.; Freedman, J.C.; Green, A.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. First Selective 12-LOX Inhibitor, ML355, Impairs Thrombus Formation and Vessel Occlusion in Vivo with Minimal Effects on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Aleem, A.M.; Tena, J.; Rivera-Velazquez, M.; Brah, H.S.; Tripathi, S.; D’silva, M.; Nadler, J.L.; Kalyanaraman, C.; Jacobson, M.P.; et al. Docking and Mutagenesis Studies Lead to Improved Inhibitor Development of ML355 for Human Platelet 12-Lipoxygenase. Bioorganic Med. Chem. 2021, 46, 116347. [Google Scholar] [CrossRef]
- Contursi, A.; Tacconelli, S.; Hofling, U.; Bruno, A.; Dovizio, M.; Ballerini, P.; Patrignani, P. Biology and Pharmacology of Platelet-Type 12-Lipoxygenase in Platelets, Cancer Cells, and Their Crosstalk. Biochem. Pharmacol. 2022, 205, 115252. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and Pathological Roles of the 12- and 15-Lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Romano, M. Lipoxin Biosynthesis and Actions: Role of the Human Platelet LX-Synthase. J. Lipid Mediat. Cell Signal. 1995, 12, 293–306. [Google Scholar] [CrossRef]
- Kutzner, L.; Goloshchapova, K.; Heydeck, D.; Stehling, S.; Kuhn, H.; Schebb, N.H. Mammalian ALOX15 Orthologs Exhibit Pronounced Dual Positional Specificity with Docosahexaenoic Acid. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2017, 1862, 666–675. [Google Scholar] [CrossRef]
- Aleem, A.M.; Tsai, W.C.; Tena, J.; Alvarez, G.; Deschamps, J.; Kalyanaraman, C.; Jacobson, M.P.; Holman, T. Probing the Electrostatic and Steric Requirements for Substrate Binding in Human Platelet-Type 12-Lipoxygenase. Biochemistry 2019, 58, 848–857. [Google Scholar] [CrossRef]
- Freedman, C.; Tran, A.; Tourdot, B.E.; Kalyanaraman, C.; Perry, S.; Holinstat, M.; Jacobson, M.P.; Holman, T.R. Biosynthesis of the Maresin Intermediate, 13S,14S-Epoxy-DHA, by Human 15-Lipoxygenase and 12-Lipoxygenase and Its Regulation through Negative Allosteric Modulators. Biochemistry 2020, 59, 1832–1844. [Google Scholar] [CrossRef]
- Saura, P.; Suardíaz, R.; Masgrau, L.; Lluch, J.M.; González-Lafont, À. Unraveling How Enzymes Can Use Bulky Residues to Drive Site-Selective c-h Activation: The Case of Mammalian Lipoxygenases Catalyzing Arachidonic Acid Oxidation. ACS Catal. 2014, 4, 4351–4363. [Google Scholar] [CrossRef]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.C.; Kalyanaraman, C.; Tourdot, B.E.; Conrad, W.S.; Akinkugbe, O.; Freedman, J.C.; Holinstat, M.; Jacobson, M.P.; Holman, T.R. 15-Lipoxygenase-1 Biosynthesis of 7S,14S-DiHDHA Implicates 15-Lipoxygenase-2 in Biosynthesis of Resolvin D5. J. Lipid Res. 2020, 61, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Stanger, L.; Freedman, C.J.; Standley, M.; Hoang, T.; Adili, R.; Tsai, W.C.; van Hoorebeke, C.; Holman, T.R.; Holinstat, M. DHA 12-LOX-Derived Oxylipins Regulate Platelet Activation and Thrombus Formation through a PKA-Dependent Signaling Pathway. J. Thromb. Haemost. 2021, 19, 839–851. [Google Scholar] [CrossRef]
- Dalli, J.; Zhu, M.; Vlasenko, N.A.; Deng, B.; Haeggström, J.Z.; Petasis, N.A.; Serhan, C.N. The Novel 13S,14S-Epoxy-Maresinis Converted by Human Macrophages to Maresin 1 (MaR1), Inhibits Leukotriene A4 Hydrolase (LTA4H), and Shifts Macrophage Phenotype. FASEB J. 2013, 27, 2573–2583. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wan, M.; Huang, W.; Stanton, R.C.; Xu, Y. Maresins: Specialized Proresolving Lipid Mediators and Their Potential Role in Inflammatory-Related Diseases. Mediat. Inflamm. 2018, 2018, 2380319. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage Proresolving Mediator Maresin 1 Stimulates Tissue Regeneration and Controls Pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Vogel, R.; Jansen, C.; Roffeis, J.; Reddanna, P.; Forsell, P.; Claesson, H.E.; Kuhn, H.; Walther, M. Applicability of the Triad Concept for the Positional Specificity of Mammalian Lipoxygenases. J. Biol. Chem. 2010, 285, 5369–5376. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-S.; Funk, C.D. Structure-function Properties of Human Platelet 12-lipoxygenase: Chimeric Enzyme and in Vitro Mutagenesis Studies. FASEB J. 1993, 7, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Neau, D.B.; Gilbert, N.C.; Bartlett, S.G.; Boeglin, W.; Brash, A.R.; Newcomer, M.E. The 1.85 Å Structure of an 8R-Lipoxygenase Suggests a General Model for Lipoxygenase Product Specificity. Biochemistry 2009, 48, 7906–7915. [Google Scholar] [CrossRef] [Green Version]
- Neau, D.B.; Bender, G.; Boeglin, W.E.; Bartlett, S.G.; Brash, A.R.; Newcomer, M.E. Crystal Structure of a Lipoxygenase in Complex with Substrate: The Arachidonic Acid-Binding Site of 8R-Lipoxygenase. J. Biol. Chem. 2014, 289, 31905–31913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcomer, M.E.; Brash, A.R. The Structural Basis for Specificity in Lipoxygenase Catalysis. Protein Sci. 2015, 24, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Mueser, T.C.; Marnett, L.J.; Funk, M.O. Crystal Structure of 12-Lipoxygenase Catalytic-Domain-Inhibitor Complex Identifies a Substrate-Binding Channel for Catalysis. Structure 2012, 20, 1490–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, C.; Shinjo, F.; Yoshimoto, T.; Yamamoto, S.; Oates, J.A.; Brash, A.R. Arachidonate 12-Lipoxygenase Purified from Porcine Leukocytes by Immunoaffinity Chromatography and Its Reactivity with Hydroperoxyeicosatetraenoic Acids. J. Biol. Chem. 1986, 261, 16714–16721. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Hiroshima, A.; Natsui, K.; Shinjo, F.; Yoshimoto, T.; Yamamoto, S.; Ii, K.; Gerozissis, K.; Dray, F. Localization of Arachidonate 12-Lipoxygenase in Parenchymal Cells of Porcine Anterior Pituitary. J. Biol. Chem. 1990, 265, 2311–2316. [Google Scholar] [CrossRef]
- Reddy, R.G.; Yoshimoto, T.; Yamamoto, S.; Marnett, L.J. Expression, Purification, and Characterization of Porcine Leukocyte 12-Lipoxygenase Produced in the Methylotrophic Yeast, Pichia Pastoris. Biochem. Biophys. Res. Commun. 1994, 205, 381–388. [Google Scholar] [CrossRef]
- Suzuki, H.; Kishimoto, K.; Yoshimoto, T.; Yamamoto, S.; Kanai, F.; Ebina, Y.; Miyatake, A.; Tanabe, T. Site-Directed Mutagenesis Studies on the Iron-Binding Domain and the Determinant for the Substrate Oxygenation Site of Porcine Leukocyte Arachidonate 12-Lipoxygenase. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1994, 1210, 308–316. [Google Scholar] [CrossRef]
- Saura, P.; Kaganer, I.; Heydeck, D.; Lluch, J.M.; Kühn, H.; González-Lafont, À. Mutagenesis of Sequence Determinants of Truncated Porcine ALOX15 Induces Changes in the Reaction Specificity by Altering the Catalytic Mechanism of Initial Hydrogen Abstraction. Chem.—Eur. J. 2018, 24, 962–973. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Aleem, A.M.; Jankun, J.; Dignam, J.D.; Walther, M.; Kühn, H.; Svergun, D.I.; Skrzypczak-Jankun, E. Human Platelet 12-Lipoxygenase, New Findings about Its Activity, Membrane Binding and Low-Resolution Structure. J. Mol. Biol. 2008, 376, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Ivanov, I.; Svergun, D.I.; Borbulevych, O.Y.; Aleem, A.M.; Stehling, S.; Jankun, J.; Kühn, H.; Skrzypczak-Jankun, E. Probing Dimerization and Structural Flexibility of Mammalian Lipoxygenases by Small-Angle X-Ray Scattering. J. Mol. Biol. 2011, 409, 654–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Téllez, S.; Cruz, A.; Masgrau, L.; González-Lafont, À.; Lluch, J.M. Accounting for the Instantaneous Disorder in the Enzyme-Substrate Michaelis Complex to Calculate the Gibbs Free Energy Barrier of an Enzyme Reaction. Phys. Chem. Chem. Phys. 2021, 23, 13042–13054. [Google Scholar] [CrossRef]
- Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Li, P.; Merz, K.M. MCPB.Py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Seminario, J.M. Calculation of Intramolecular Force Fields from Second-Derivative Tensors. Int. J. Quantum Chem. 1996, 60, 1271–1277. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 20; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R. Molecular Modelling. Principles and Applications, 2nd ed.; Pearson Education: London, UK, 2001; ISBN 978-0-582-38210-7. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Canyelles-Niño, M. MolBioMedUAB/RCBS.Py: 0.2.0bis. GitHub: San Franciso, CA, USA, 2023. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Sherwood, P.; De Vries, A.H.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A General Purpose Implementation of the QM/MM Approach and Its Application to Problems in Catalysis. J. Mol. Struct. THEOCHEM 2003, 632, 1–28. [Google Scholar] [CrossRef]
- Metz, S.; Kästner, J.; Sokol, A.A.; Keal, T.W.; Sherwood, P. ChemShell-a Modular Software Package for QM/MM Simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T.R. DL_POLY_2.0: A General-Purpose Parallel Molecular Dynamics Simulation Package. J. Mol. Graph. 1996, 14, 136–141. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- De Vries, A.H.; Sherwood, P.; Collins, S.J.; Rigby, A.M.; Rigutto, M.; Kramer, G.J. Zeolite Structure and Reactivity by Combined Quantum-Chemical-Classical Calculations. J. Phys. Chem. B 1999, 103, 6133–6141. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the Limited Memory BFGS Method for Large Scale Optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J.; Thiel, S.; Senn, H.M.; Sherwood, P.; Thiel, W. Exploiting QM/MM Capabilities in Geometry Optimization: A Microiterative Approach Using Electrostatic Embedding. J. Chem. Theory Comput. 2007, 3, 1064–1072. [Google Scholar] [CrossRef]
- Billeter, S.R.; Turner, A.J.; Thiel, W. Linear Scaling Geometry Optimisation and Transition State Search in Hybrid Delocalised Internal Coordinates. Phys. Chem. Chem. Phys. 2000, 2, 2177–2186. [Google Scholar] [CrossRef]
- Heyden, A.; Bell, A.T.; Keil, F.J. Efficient Methods for Finding Transition States in Chemical Reactions: Comparison of Improved Dimer Method and Partitioned Rational Function Optimization Method. J. Chem. Phys. 2005, 123, 224101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H12proS | H9proR | |||||||
|---|---|---|---|---|---|---|---|---|
| Frame | Pentadienyl Stereochemistry | Pentadienyl Stereochemistry | ||||||
| 322 | 3.5 | 15.9 | −18.3 | ZE | 3.5 | 36.4 | −13.9 | ZZ |
| 2253 | 5.4 | 20.5 | −19.0 | ZE | 3.5 | 23.4 | −19.1 | ZZ |
| 4986 | 5.4 | 19.3 | −17.0 | ZE | 5.0 | 35.0 | −14.1 | ZZ |
| 6993 | 3.9 | 18.0 | −18.5 | ZE | 3.1 | 29.7 | −14.5 | ZZ |
| 8721 | 5.0 | 21.9 | −17.3 | ZE | 3.3 | 22.7 | −13.6 | ZZ |
| 10,106 | 5.5 | 24.3 | −16.8 | ZE | 3.4 | 25.6 | −17.4 | ZZ |
| 12,860 | 5.2 | 22.9 | −18.5 | ZE | 3.9 | 29.2 | −15.3 | ZZ |
| 14,423 | 4.1 | 21.8 | −19.1 | ZE | 3.4 | 27.5 | −13.6 | ZZ |
| 18,168 | 3.7 | 17.1 | −17.0 | ZE | 3.3 | 31.8 | −13.8 | ZZ |
| 19,729 | 3.4 | 17.1 | −17.0 | ZE | 3.9 | 43.7 | −18.4 | ZZ |
| 17.1 | 23.9 | |||||||
| Frame | Chirality of Product | Geometry of Addition | |||||
|---|---|---|---|---|---|---|---|
| 8721 | 3.2 | 3.0 | S | Antarafacial | 6.6 | 10.2 | 33.3 |
| 10,106 | 3.2 | 4.3 | S | Antarafacial | 5.8 | - | - |
| 18,168 | 3.1 | 3.1 | S | Antarafacial | 5.9 | 13.1 | 20.6 |
| H12proS | ||||
|---|---|---|---|---|
| Frame | Pentadienyl Stereochemistry | |||
| 940 | 3.2 | 20.9 | −12.5 | ZZ |
| 1548 | 3.0 | 19.8 | −11.8 | ZZ |
| 1709 | 2.9 | 24.5 | −12.4 | ZZ |
| 2132 | 2.8 | 22.1 | −9.7 | ZZ |
| 2515 | 3.2 | 27.2 | −8.9 | ZZ |
| 3020 | 2.8 | 19.8 | −10.8 | ZZ |
| 3556 | 3.2 | 24.5 | −9.9 | ZZ |
| 4502 | 2.7 | 15.9 | −16.1 | ZZ |
| 5758 | 2.6 | 20.4 | −12.1 | ZZ |
| 9207 | 2.6 | 22.6 | −13.1 | ZZ |
| 17.6 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canyelles-Niño, M.; González-Lafont, À.; Lluch, J.M. Hydroperoxidation of Docosahexaenoic Acid by Human ALOX12 and pigALOX15-mini-LOX. Int. J. Mol. Sci. 2023, 24, 6064. https://doi.org/10.3390/ijms24076064
Canyelles-Niño M, González-Lafont À, Lluch JM. Hydroperoxidation of Docosahexaenoic Acid by Human ALOX12 and pigALOX15-mini-LOX. International Journal of Molecular Sciences. 2023; 24(7):6064. https://doi.org/10.3390/ijms24076064
Chicago/Turabian StyleCanyelles-Niño, Miquel, Àngels González-Lafont, and José M. Lluch. 2023. "Hydroperoxidation of Docosahexaenoic Acid by Human ALOX12 and pigALOX15-mini-LOX" International Journal of Molecular Sciences 24, no. 7: 6064. https://doi.org/10.3390/ijms24076064