Abstract
The emergence of the SARS-CoV-2 coronavirus has garnered global attention due to its highly pathogenic nature and the resulting health crisis and economic burden. Although drugs such as Remdesivir have been considered a potential cure by targeting the virus on its RNA polymerase, the high mutation rate and unique 3’ to 5’ exonuclease with proofreading function make it challenging to develop effective anti-coronavirus drugs. As a result, there is an increasing focus on host–virus interactions because coronaviruses trigger stress responses, cell cycle changes, apoptosis, autophagy, and the dysregulation of immune function and inflammation in host cells. The p53 tumor suppressor molecule is a critical regulator of cell signaling pathways, cellular stress responses, DNA repair, and apoptosis. However, viruses can activate or inhibit p53 during viral infections to enhance viral replication and spread. Given its pivotal role in cell physiology, p53 represents a potential target for anti-coronavirus drugs. This review aims to summarize the relationship between p53 and coronaviruses from various perspectives, to shed light on potential targets for antiviral drug development and vaccine design.
1. Introduction
Coronaviruses (CoVs) are a diverse family of single-stranded positive-sense enveloped RNA viruses that can induce a range of issues, including the stress response of host cells, cell cycle changes, cell apoptosis, and host immune dysregulation and inflammation [1]. They can infect humans and various vertebrates, leading to severe public health problems and economic losses worldwide [2,3]. Coronaviruses were first isolated from chickens in 1937. Usually, the host range of CoVs is very narrow, and there are six coronaviruses other than the 2019 coronavirus disease (COVID-19) that can infect humans [4]. However, the multiple coronavirus pandemics that have occurred in recent years, such as the 2019 SARS-CoV-2, the 2012 middle east respiratory syndrome (MERS), and the 2002 severe acute respiratory syndrome (SARS) outbreaks, have demonstrated the potential for the zoonotic and human-to-human transmission of emerging coronaviruses [5,6]. Coronaviruses use various strategies to create an optimal environment for replication, including inducing cell cycle arrest, evading host immune proteins, and modulating cellular processes such as apoptosis and autophagy. Inducing cell cycle arrest slows down or halts the cell’s replication machinery, allowing the virus to redirect cellular resources toward viral replication [7]. Coronaviruses also employ multiple mechanisms to evade the host’s natural immune defenses, including blocking immune signaling molecules and altering the expression of host immune proteins [8]. Moreover, coronaviruses can manipulate apoptosis and autophagy by inducing or inhibiting these processes, as needed, to promote their survival and replication [9]. The recurrence of these viruses and human endemic coronaviruses indicate that future outbreaks are likely, making it essential to understand their pathogenesis and find safe and effective treatment methods.
The p53 tumor suppressor is a crucial molecule attending to the regulation of various basic cell signaling, including cell cycle, DNA repair, apoptosis, and cellular stress response [10,11,12]. Moreover, apart from its role in regulating cell growth and apoptosis, p53 also plays a crucial role in various cellular processes, including metabolism, autophagy, and innate immunity [13]. This protein can directly or indirectly influence cellular metabolism, impacting the production and consumption of energy within the cell [14]. Additionally, p53 is involved in the process of autophagy, which plays a key role in removing damaged cellular components and recycling cellular building blocks [15]. In addition to its role in regulating cell growth and division, p53 also plays a role in regulating the innate immune response, such as interferon production [16]. p53 is a protein that can be found in both the nucleus and cytoplasm of a cell. In the nucleus, p53 specifically binds to DNA. Normally p53 checks for DNA damage spots in the G1 phase, monitoring the integrity of the genome [17,18]. Upon detecting DNA damage, the p53 protein activates a signaling pathway that can halt cell division and initiate DNA repair mechanisms. If the damage is severe and cannot be repaired, p53 can induce apoptosis, a process of programmed cell death. [19]. If both copies of the p53 gene are mutated, cell proliferation goes out of control, and the cell becomes cancerous [20,21]. In normal physiological mammalian cells, p53 is maintained at low levels, usually by continued ubiquitination and subsequent degradation by murine double minute-2 (MDM2) [10,22,23]. Different amounts of MDM2 can inactivate p53 in different ways. The polyubiquitination and degradation of p53 in the nucleus are caused by high levels of MDM2, and p53 monoubiquitination and nuclear exclusion are caused by low levels of MDM2 [24]. The p53 protein is involved in a negative feedback loop with MDM2, which targets p53 for destruction. Therefore, the activation of p53 is usually accompanied by an inhibition of MDM2 levels. However, when cells encounter stressors such as DNA damage, hypoxia, or viral infection, p53 ubiquitylation is inhibited, allowing it to accumulate in the nucleus. In this location, p53 undergoes multiple covalent modifications, including phosphorylation and acetylation, which activate and stabilize the protein [25]. p53 is intricately linked to viral infection, with both beneficial and detrimental effects. This review summarizes the relationship between p53 and coronavirus development, providing insights into treating human and animal coronaviruses (Figure 1).
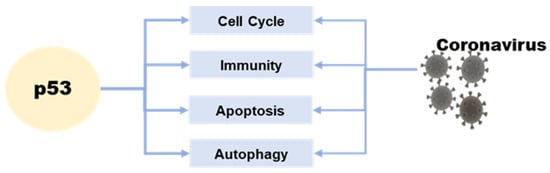
Figure 1.
The relationship between p53 and coronavirus.
2. p53 Affects Coronaviruses by Regulating the Cell Cycle
Cell cycle progression may be an advanced and orderly regulated method. Throughout this method, multiple units work alongside to make sure of smart division and proliferation [26]. Cyclin-dependent kinases (CDK) and cyclins mediate the cell cycle progression [27]. Inhibition or absence of CDK activity can result in cells’ arrest in the G1 phase and their entry into a quiescent state [28]. The G1-S and G2-M phases of the cell cycle provide protection against both exogenous and endogenous agents that can cause DNA damage [29]. The molecular level changes in these two stages are complex and susceptible to environmental conditions caused by coronaviruses [30]. P21 is an inhibitor of CDKs and regulates the cell cycle by interfering with cyclins [31]. The transition from the S phase to the G1 phase is facilitated by cyclins. CDKs are inhibited by p21WAF1/CIP1, which causes low phosphorylation of retinoblastoma (Rb) and prevents E2F release, thereby blocking the transformation of the G1-S and G2-M phases. G2-M arrest is mainly regulated by the p53-p21-DREAM pathway, which indirectly inhibits the transcription of cell cycle genes such as CCNB2, KIF23, and PLK4 [32]. In addition to inducing G2 arrest through 14-3-3 stimulation that sequesters cyclin B1-CDK1 complexes outside the nucleus, p53 can also cause cell cycle arrest by transactivating the GADD45 and 14-3-3 genes. These are among the various functions of p53 [30].
Regulation of the host cell cycle could be a common strategy several viruses use for infectious agent replication (creating an excellent cellular environment). Coronavirus infection not only induces oxidative stress and DNA damage in host cells, but it can also interfere with the host cell cycle by directly altering the activity of p53 or its upstream and downstream proteins [33,34,35,36,37]. SARS-CoV-2 N protein can induce acute kidney injury by arresting the cell’s G1 cycle [38]. With the deepening of coronavirus–host interaction research, the p53-DREAM pathway provides a novel antiviral strategy. Porcine epidemic diarrhea virus (PEDV) N protein induces S-phase arrest in host cells by activating p53 and binding downstream protein DREAM to CHR [39]. Mouse hepatitis virus (MHV) infection reduces G1 cyclin–CDK complexes and induces G0-G1 cell cycle arrest [40]. Infectious bronchitis virus (IBV) induces G2-M phase perturbation in both asynchronous and synchronous replication cells, thereby inducing G2-M phase cell cycle arrest to promote progeny virus replication and reproduction [41]. Research has shown that the SARS coronavirus can hinder the activity of cyclin–CDK complexes, leading to reduced phosphorylation of retinoblastoma protein and decreased E2F1-mediated transcriptional activation. This, in turn, can impede S-phase progression in mammalian cells [42]. Infection with PEDV can modify the expression of proteins involved in cell cycle regulation, such as p21, CDK1, CDK2, CDK4, cyclin A, and cyclin E. The viral infection can cause cell cycle arrest in the G0-G1 phase, which can be restored by inhibiting the p53 signaling pathway. This leads to the downregulation of p21 and associated cyclin/CDK proteins [43] (Figure 2).

Figure 2.
Role of p53 on the cell cycle during coronavirus infection. During viral infection, p53 is activated and initiates a cascade of events that result in cell cycle arrest. Specifically, activated p53 mediates the protein p21 to form complexes with CDKs, resulting in the inhibition of cyclin–CDK activity. This also leads to a reduction in Rb phosphorylation, thereby maintaining the stability of the RB–E2F complex and inhibiting the transcription of cell cycle genes. Additionally, p53 mediates the proteins 14-3-3 and GADD45 to inhibit cyclin–CDK1 activity, resulting in cell arrest in the G2-M phase.
3. The Interaction between p53 and Interferon
IFNs possess diverse biological activities, which include antiviral effects, antiproliferative effects, and the activation of immune cell cytotoxicity. IFNs are central to antiviral immunity and can inhibit coronaviruses’ replication. Cells produce type I IFN (primarily IFN-α and IFN-β) in response to viral infections, which is critical for immunity against many types of viruses. The transcription of antiviral-related genes is induced by IFN-I, which can be triggered by viral recognition sensors such as toll-like receptors and RNA helicases such as the RNA helicase retinoic acid-inducible gene I (RIG-I). The activation of interferon regulatory factor (IRF) leads to the production of type I interferon [44]. IFNAR1 and IFNAR2 form a type I IFN receptor that recognizes and binds to type I IFN, giving bystander cells antiviral effects. The phosphorylation of STAT-1, STAT-2, and IRF9 transcription factors is increased by the activation of the JAK-STAT signaling pathway. These factors then form a heterotrimeric complex called IFN-stimulated gene factor 3 (ISGF3) and translocate to the nucleus [45]. This complex has been shown to activate p53 transcription but is not associated with p53 phosphorylation [46].
p53 also contributes to the increased release of IFN-1 from virus-infected cells [47]. IRF9 was confirmed to be a p53 target gene, suggesting that type I IFN can not only increase the expression of p53 by activating IRF9 but also that p53 can activate IRF9. IRF9 continues to activate retinoic acid inducer 1 (RIG-I) and ISRE-dependent genes such as IRF7 [47,48]. IRF3 and IRF7 play a crucial role in inducing the expression of type I interferon genes downstream of pattern recognition receptors. These transcription factors bind to the promoters of IFN-α and IFN-β through homologous or heterologous interactions, thereby controlling their expression [49]. In addition, recent studies have revealed the relationship between p53 and the cGAS/STING innate immune system pathway. p53 induces the ubiquitination of three prime repair exonuclease 1 (TREX1) through the ubiquitin ligase TRIM24. The degradation of TREX1 prevents the timely removal of cytoplasmic DNA, thus activating the cGAS/STING pathway and increasing the synthesis of IFN [50].
p53 can inhibit the replication of coronaviruses and shows antiviral activity in vivo; this effect may be due to its ability to activate natural immune pathways. Previous studies have shown that several coronaviruses, such as SARS-CoV-2, SAR-CoV, and human coronavirus NL63 infections, can only induce deficient levels of IFN-I [51,52,53], which is likely to lead to uninhibited viral replication and damage to the immune system. Low-level IFN responses may be a means by which coronaviruses evade immunity. The lack of adequate IFN response may be due to the decrease of p53 hydrolyzed by the coronavirus papain-like proteases (PLPs). PLPs are a class of cysteine proteases that inhibit innate immunity by stabilizing the binding of MDM2 and p53 to cause the ubiquitination of p53 [54]. The SARS-unique domain (SUD) and papain-like proteases (PLPs) interact with cell E3 ubiquitin ligase and CHY zinc-finger domain-containing 1 (RCHY1) to facilitate their activities. SUD and PLPs target p53 with E3 ubiquitin ligase RCHY1 to degrade p53. The degradation of p53 decreases the level of earthly IFN [55]. In other words, coronaviruses can evade the host’s natural immune defenses by degrading p53 through their own proteins. At the same time, the application of small molecule inhibitors of MDM2, such as nutlin-3 and idasanutlin, can promote the stable presence of p53 in cells, help regulate the IFN signaling pathway, and inhibit the replication of coronaviruses [56] (Figure 3).

Figure 3.
Role of p53 in the regulation of interferon during coronavirus infection. The toll-like receptor and RNA helicase retinoic acid-inducing gene I (RIG-I) are activated after the RNA virus invasion of cells. IRF3 is phosphorylated to produce type I interferon. The JAK-STAT signaling pathway is subsequently activated, and the transcription factors STAT-1, STAT-2, and IFN regulatory factor 9 (IRF9) bind to induce p53 expression. IRF9 is also a p53 target gene and is regulated by p53. Virus infection activates p53-mediated TLR3, phosphorylates downstream IRF3 and IRF7 to form dimers, and promotes the synthesis of type I interferon. p53 also activates IRF3 through the p53-cgas-STING pathway. In addition, coronaviruses can evade the host’s natural immune defenses by degrading p53 through their papain-like proteases (PLPs).
Interferon is a common clinical strategy for treating coronavirus infection [57]. In the case of MERS-CoV, delayed treatment with IFN-β not only failed to effectively inhibit virus replication but also increased the expression of pro-inflammatory cytokines, resulting in mouse mortality [58]. In addition, delayed IFN-I treatment promoted SARS-CoV infection and caused severe acute pneumonia in SARS-CoV-infected mice [59]. Recent studies have shown that IFN treatment reduces epithelial cell proliferation and differentiation, exacerbating COVID-19 disease and susceptibility to bacterial co-infection [53]. Therefore, the use of IFN in treating various coronavirus infections should be considered with caution due to time and other issues.
4. p53 Affects Coronavirus-Associated Apoptosis
Apoptosis is a programmed death mechanism evolved by cells in response to the complex external environment and self-injury. This mechanism is dangerous but effective for living organisms. Due to the particularity and importance of apoptosis, there is a complicated regulatory mechanism network, and p53 is one of the links. Apoptosis is primarily controlled by intrinsic and extrinsic pathways. The intrinsic pathway of apoptosis involves a complex downstream signaling network that is regulated by p53 [60]. The TNFR family, including the death receptor and Fas, initiates the extrinsic pathway of apoptosis by activating the formation of the death-inducing signaling complex (DISC). This leads to the activation of caspases, including caspase-8 and caspase-3, ultimately resulting in apoptosis [61]. The Bcl-2 family of proteins regulates the intrinsic apoptotic pathway, and it includes anti-apoptotic and pro-apoptotic members. Anti-apoptotic proteins, such as Bcl-XL, can perform anti-apoptotic functions due to their structural similarity to Bcl-2. In contrast, pro-apoptotic proteins such as Bax and Bak can antagonize the anti-apoptotic function of Bcl-2 because they share a similar structure with Bcl-2 and Bcl-XL. The external apoptotic pathway involves the activation of caspases, including caspase-8 and caspase-3, through the death-inducing signaling complex (DISC) formation initiated by the tumor necrosis factor receptor (TNFR) family (such as death receptor and Fas), leading to apoptosis. P53 plays a crucial role in regulating the intrinsic pathway of apoptosis [62]. Interestingly, Bax, Noxa, PUMA (p53-up-regulated apoptosis regulator), and BH3 interaction domain death agonists (BID) are important targets for p53. p53 can upregulate the expression level of these proteins, thus inducing apoptosis [63]. Other studies have shown that p53 can directly stimulate mitochondria to release high ROS and cause cell apoptosis [64] (Figure 4).

Figure 4.
Role of p53 in the regulation of apoptosis during coronavirus infection. The virus induces ROS accumulation in cells and activates p53 protein, which activates the pro-apoptotic proteins of the Bcl-2 family -Bax, Noxa, PUMA, and BID, antagonizing the anti-apoptotic effect of BCL-2. Then, cytochrome C from mitochondria spills over, triggering a cascade of caspase proteins, leading to apoptosis. p53 can also act on the tumor necrosis factor receptor (TNFR) family to activate caspase-3 and caspase-8 through the exogenous apoptotic pathway and cause apoptosis.
In many traditional views, virus-induced apoptosis is regarded as an antiviral strategy for the host. The relationship between apoptosis and viruses is very subtle and complex. For some viruses, inhibiting host cell apoptosis can increase the replication time in the cell, and rapid cell death may reduce the replication level. The human cytomegalovirus (HCMV)–encoded UL37 exon-1 protein (UL37 × 1) can increase HCMV replication and infection by inhibiting host cell apoptosis proteins. Meanwhile, the immunosuppressive and anti-apoptotic activity of UL37 × 1 is essential for HCMV replication in vivo [65]. Flaviviruses, such as the Zika virus (ZIKV), produce subgenomic flavivirus RNA (sfRNA) using host mRNA degradation mechanisms. sfRNA can modulate the expression of genes involved in cell death pathways in the mosquito genome, leading to the inhibition of cell apoptosis in mosquito tissue. This process can prolong viral genome replication and promote virion assembly, which, in turn, facilitates viral infection [66]. Hepatitis C virus (HCV) NS5A proteins form complexes with inositol triphosphate receptor type 3 (IP3R3) and F-box and leucine 2 (FBXL2), promoting the FBXL2-mediated degradation of IP3R3. Degradation of IP3R3 inhibits calcium flux, reduces mitochondrial calcium overload, and leads to apoptosis. Therefore, the anti-apoptotic effect of NS5A prolongs the replication time of HCV and is conducive to HCV infection [67]. However, in some cases, activation of the apoptotic pathway can promote viral replication and spread. The ORF3a protein of SARS-CoV-2 has been shown to induce apoptosis in infected cells [68]. Porcine deltacoronavirus (PDCoV) infection triggers the release of apoptotic cells into the cytoplasm by stimulating mitochondrial outer membrane permeabilization (MOMP) via either Bax recruitment or mitochondrial permeability transition pore (MPTP) opening. This results in the activation of an intrinsic caspase-dependent apoptotic pathway that promotes viral replication in vitro [69]. PEDV induces apoptosis in Vero cells through a series of events that include increasing intracellular ROS accumulation, which, in turn, increases MDM2 and CBP expression. This process further stimulates the phosphorylation of p53 at serine 20, promotes the nuclear translocation of p53, and activates p53, ultimately leading to apoptosis [64]. Because of the inextricable link between coronavirus infection and apoptosis, p53 is a link that cannot be bypassed.
5. p53 Affects Coronavirus-Associated Autophagy
Autophagy is a process by which cells recycle cellular components and eliminate damaged organelles or misfolded proteins. It is also a critical mechanism for the innate immune response against viral infection. p53 regulates autophagy through transcription-dependent and transcription-independent mechanisms. It can stimulate autophagy by upregulating genes such as damage-regulated autophagy modulator (DRAM) and sestrin2. Additionally, p53 can interact directly with autophagy-related proteins, such as LC3 [70,71]. However, p53 can also inhibit autophagy by upregulating genes such as tuberous sclerosis complex 2 (TSC2) and PTEN, which negatively regulate the mTOR pathway, a key regulator of autophagy [72].
However, some viruses, including coronaviruses, can hijack the autophagy pathway to promote their own replication and spread. p53 has a dual role in regulating autophagy during coronavirus infection. On the one hand, it can stimulate autophagy as a host defense mechanism against viral infection [73]. For instance, p53 can induce autophagy in response to SARS-CoV-2 infection, leading to the degradation of viral particles and the suppression of viral replication. Similarly, p53-mediated autophagy inhibits the replication of other coronaviruses, such as HCoV-229E and MERS-CoV [74]. On the other hand, some coronaviruses can manipulate the p53 pathway to promote their own replication by inhibiting autophagy. For instance, the SARS-CoV-2 virus downregulates p53 expression and induces MDM2 expression. This leads to the degradation of p53, which inhibits p53-mediated autophagy and promotes viral replication [55,75]. Similarly, the MERS-CoV virus also inhibits autophagy by downregulating the expression of p53 [76].
In conclusion, the relationship between p53 and autophagy during coronavirus infection is complex and context-dependent. While p53 can promote autophagy as a host defense mechanism against viral infection, some coronaviruses can also manipulate the p53 pathway to inhibit autophagy and promote their own replication (Figure 5). Further research is necessary to fully comprehend the interplay between p53 and autophagy during coronavirus infection and identify potential targets for therapeutic intervention.
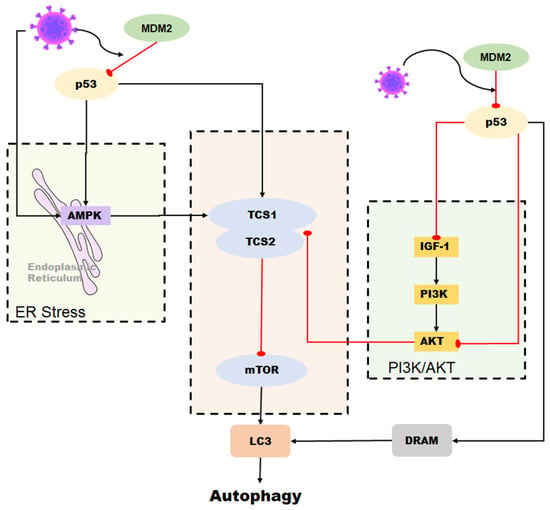
Figure 5.
Role of p53 in the regulation of autophagy during coronavirus infection. Autophagy can be induced by p53 through the DRAM protein. In some coronaviruses, autophagy is activated by p53-mediated DRAM protein as well as through the AMPK pathway and the PI3K/AKT pathway. However, some coronaviruses can reduce the stability of p53 in cells and inhibit p53-mediated autophagy by upregulating the expression of MDM2.
8. Conclusions
In conclusion, despite not being considered a primary target for developing anti-coronavirus drugs, p53 presents a promising avenue for therapeutic interventions against coronaviruses. The multifaceted role of p53 in regulating cellular responses to stress and DNA damage makes it an essential player in maintaining host homeostasis, and its intricate interactions with coronaviruses highlight its potential as a critical drug target. Furthermore, recent advancements in understanding the molecular mechanisms underlying p53-mediated signaling have provided valuable insights into the development of small molecule inhibitors that selectively target p53 activity, offering a potential avenue for the development of novel anti-coronavirus therapeutics. Thus, exploring the therapeutic potential of p53 in the context of coronavirus infection presents a compelling opportunity to combat the ongoing global health crisis caused by COVID-19 and future emerging viral diseases.
Author Contributions
Writing—original draft preparation, X.W. and Y.L.; data curation, X.W.; methodology, Y.L. and K.L.; supervision, Z.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the State Key Research and Development Plan, China (No. 2022YFD1801105).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article. The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare that they have no competing interest.
References
- Enjuanes, L.; Almazán, F.; Sola, I.; Zuñiga, S. Biochemical aspects of coronavirus replication and virus-host interaction. Annu. Rev. Microbiol. 2006, 60, 211–230. [Google Scholar] [CrossRef]
- Amarilla, A.A.; Sng, J.D.J.; Parry, R.; Deerain, J.M.; Potter, J.R.; Setoh, Y.X.; Rawle, D.J.; Le, T.T.; Modhiran, N.; Wang, X.; et al. A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat. Commun. 2021, 12, 3431. [Google Scholar] [CrossRef]
- Wang, L.-F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Infectious diseases. Calling all coronavirologists. Science 2003, 300, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Sui, L.; Li, L.; Zhao, Y.; Zhao, Y.; Hao, P.; Guo, X.; Wang, W.; Wang, G.; Li, C.; Liu, Q. Host cell cycle checkpoint as antiviral target for SARS-CoV-2 revealed by integrative transcriptome and proteome analyses. Signal Transduct. Target. Ther. 2023, 8, 21. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Wang, Z.; Gutiérrez-Castrellón, P.; Shi, H. Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19. Signal Transduct. Target. Ther. 2022, 7, 186. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef]
- Bensaad, K.; Vousden, K.H. p53: New roles in metabolism. Trends Cell Biol. 2007, 17, 286–291. [Google Scholar] [CrossRef]
- Levine, B.; Abrams, J. p53: The Janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef]
- Schwartz, D.; Rotter, V. p53-dependent cell cycle control: Response to genotoxic stress. Semin. Cancer Biol. 1998, 8, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Offer, H.; Zurer, I.; Banfalvi, G.; Reha’k, M.; Falcovitz, A.; Milyavsky, M.; Goldfinger, N.; Rotter, V. p53 modulates base excision repair activity in a cell cycle-specific manner after genotoxic stress. Cancer Res. 2001, 61, 88–96. [Google Scholar]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Shmueli, A.; Oren, M. Regulation of p53 by Mdm2: Fate is in the numbers. Mol. Cell 2004, 13, 4–5. [Google Scholar] [CrossRef]
- Guo, G.; Cui, Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J. Immunother. Cancer 2015, 3, 9. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Seo, H.R.; Lee, D.H.; Lee, H.J.; Baek, M.; Bae, S.; Soh, J.W.; Lee, S.J.; Kim, J.; Lee, Y.S. Cyclin G1 overcomes radiation-induced G2 arrest and increases cell death through transcriptional activation of cyclin B1. Cell Death Differ. 2006, 13, 1475–1484. [Google Scholar] [CrossRef]
- Braithwaite, A.W.; Prives, C.L. p53: More research and more questions. Cell Death Differ. 2006, 13, 877–880. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Grams, T.R.; Bloom, D.C.; Toth, Z. Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity. Viruses 2022, 14, 1039. [Google Scholar] [CrossRef] [PubMed]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Chander, Y.; Kumar, R.; Khandelwal, N.; Singh, N.; Shringi, B.N.; Barua, S.; Kumar, N. Role of p38 mitogen-activated protein kinase signalling in virus replication and potential for developing broad spectrum antiviral drugs. Rev. Med. Virol. 2021, 31, 1–16. [Google Scholar] [CrossRef]
- Kyriakopoulos, A.M.; Nigh, G.; McCullough, P.A.; Seneff, S. Mitogen Activated Protein Kinase (MAPK) Activation, p53, and Autophagy Inhibition Characterize the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Spike Protein Induced Neurotoxicity. Cureus 2022, 14, e32361. [Google Scholar] [CrossRef]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA Damage Response by RNA Viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Hu, D.; Pan, P.; Liang, L.; Wu, W.; Tang, Y.; Huang, X.R.; Yu, X.; Wu, J.; et al. SARS-CoV-2 N Protein Induces Acute Kidney Injury via Smad3-Dependent G1 Cell Cycle Arrest Mechanism. Adv. Sci. 2022, 9, e2103248. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Shi, D.; Xing, X.; Qi, S.; Yang, D.; Zhang, J.; Han, Y.; Zhu, Q.; Sun, H.; Wang, X.; et al. Coronavirus Porcine Epidemic Diarrhea Virus Nucleocapsid Protein Interacts with p53 to Induce Cell Cycle Arrest in S-Phase and Promotes Viral Replication. J. Virol. 2021, 95, e0018721. [Google Scholar] [CrossRef]
- Chen, C.-J.; Makino, S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2004, 78, 5658–5669. [Google Scholar] [CrossRef]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the G0/G1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Macip, S.; Martínez-Sobrido, L.; Brown, L.; Ashour, J.; García-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, X.; Guo, L.; Qiu, Y.; Li, X.; Yu, H.; Xiang, H.; Tong, G.; Ma, Z. Influenza A virus induces p53 accumulation in a biphasic pattern. Biochem. Biophys. Res. Commun. 2009, 382, 331–335. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-α/β Gene Induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e266. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.L.M.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.-H.; Yuen, K.-Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef]
- Zhao, X.-N.; You, Y.; Cui, X.-M.; Gao, H.-X.; Wang, G.-L.; Zhang, S.-B.; Yao, L.; Duan, L.-J.; Zhu, K.-L.; Wang, Y.-L.; et al. Single-cell immune profiling reveals distinct immune response in asymptomatic COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 342. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Harford, J.B.; Kim, S.S.; Pirollo, K.F.; Chang, E.H. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses 2022, 14, 739. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Bortot, B.; Romani, A.; Ricci, G.; Biffi, S. Exploiting Extracellular Vesicles Strategies to Modulate Cell Death and Inflammation in COVID-19. Front. Pharmacol. 2022, 13, 877422. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Rudin, C.M.; Thompson, C.B. Apoptosis and disease: Regulation and clinical relevance of programmed cell death. Annu. Rev. Med. 1997, 48, 267–281. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Fornace, A.J. Death and decoy receptors and p53-mediated apoptosis. Leukemia 2000, 14, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 FAMILY: Regulators of Cell Death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. Living with p53, Dying of p53. Cell 2007, 130, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, Y.; Zhang, Q.; Yang, F.; Yin, Z.; Wang, L.; Li, Q. Porcine epidemic diarrhea virus infections induce apoptosis in Vero cells via a reactive oxygen species (ROS)/p53, but not p38 MAPK and SAPK/JNK signalling pathways. Vet. Microbiol. 2019, 232, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, A.; Wu, D.; Wang, C.; Huang, M.; Xiong, X.; Jin, L.; Zhou, W.; Qiu, Y.; Zhou, X. Dual inhibition of innate immunity and apoptosis by human cytomegalovirus protein UL37x1 enables efficient virus replication. Nat. Microbiol. 2022, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Saeed, M.; Giorgi, C.; Li, J.; Hoffmann, H.H.; Pinton, P.; Rice, C.M.; Pagano, M. NS5A Promotes Constitutive Degradation of IP3R3 to Counteract Apoptosis Induced by Hepatitis C Virus. Cell Rep. 2018, 25, 833–840.e833. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, C. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- Lodi, G.; Gentili, V.; Casciano, F.; Romani, A.; Zauli, G.; Secchiero, P.; Zauli, E.; Simioni, C.; Beltrami, S.; Fernandez, M.; et al. Cell cycle block by p53 activation reduces SARS-CoV-2 release in infected alveolar basal epithelial A549-hACE2 cells. Front. Pharmacol. 2022, 13, 1018761. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mlcochova, P. Cyclin D3 restricts SARS-CoV-2 envelope incorporation into virions and interferes with viral spread. EMBO J. 2022, 41, e111653. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Draczkowski, P.; Wang, Y.S.; Chang, C.Y.; Chien, Y.C.; Cheng, Y.H.; Wu, Y.M.; Wang, C.H.; Chang, Y.C.; Chang, Y.C.; et al. In Situ structure and dynamics of an alphacoronavirus spike protein by cryo-ET and cryo-EM. Nat. Commun. 2022, 13, 4877. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Huang, Y.; Du, Q.; Dong, F.; Zhao, X.; Zhang, W.; Xu, X.; Tong, D. TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling. Biochem. Biophys. Res. Commun. 2014, 445, 497–503. [Google Scholar] [CrossRef]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar] [CrossRef]
- Chen, C.J.; Sugiyama, K.; Kubo, H.; Huang, C.; Makino, S. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 2004, 78, 10410–10419. [Google Scholar] [CrossRef]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Burch, C.L.; Chao, L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 2000, 406, 625–628. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus-host interaction networks as new antiviral drug targets for IAV and SARS-CoV-2. Emerg. Microbes Infect. 2022, 11, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Podyminogin, R.L.; Diercks, A.H.; Treuting, P.M.; Peschon, J.J.; Rodriguez, D.; Gundapuneni, M.; Weiss, M.J.; Aderem, A. miR-144 attenuates the host response to influenza virus by targeting the TRAF6-IRF7 signaling axis. PLoS Pathog. 2017, 13, e1006305. [Google Scholar] [CrossRef]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2020, 53, 100721. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, A.P.G. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future (‡). Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef]
- Alzhanova, D.; Corcoran, K.; Bailey, A.G.; Long, K.; Taft-Benz, S.; Graham, R.L.; Broussard, G.S.; Heise, M.; Neumann, G.; Halfmann, P.; et al. Novel modulators of p53-signaling encoded by unknown genes of emerging viruses. PLoS Pathog. 2021, 17, e1009033. [Google Scholar] [CrossRef]
- Singh, N.; Bharara Singh, A. S2 subunit of SARS-nCoV-2 interacts with tumor suppressor protein p53 and BRCA: An in silico study. Transl. Oncol. 2020, 13, 100814. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, Z.; Song, S.; Wang, S.; Tian, C.; Xing, G.; Chen, X.; Xiao, Z.X.; He, F.; Zhang, L. p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J. Biol. Chem. 2015, 290, 3172–3182. [Google Scholar] [CrossRef]
- Khalil, H.; Abd El Maksoud, A.I.; Roshdey, T.; El-Masry, S. Guava flavonoid glycosides prevent influenza A virus infection via rescue of P53 activity. J. Med. Virol. 2019, 91, 45–55. [Google Scholar] [CrossRef]
- Liu, Y.; Song, X.; Li, C.; Hu, H.; Li, W.; Wang, L.; Hu, J.; Liao, C.; Liang, H.; He, Z.; et al. Chrysin Ameliorates Influenza Virus Infection in the Upper Airways by Repressing Virus-Induced Cell Cycle Arrest and Mitochondria-Dependent Apoptosis. Front. Immunol. 2022, 13, 872958. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Liu, Y.; Xie, J.; Hu, C.; Wang, X. Role of p53 in pseudorabies virus replication, pathogenicity, and host immune responses. Vet. Res. 2019, 50, 9. [Google Scholar] [CrossRef]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential Role for p53 in the Permissive Life Cycle of Human Cytomegalovirus. J. Virol. 2006, 80, 8390–8401. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; AlHilali, S.; Al-Swailem, S.; Secchiero, P.; Voltan, R. Therapeutic potential of the MDM2 inhibitor Nutlin-3 in counteracting SARS-CoV-2 infection of the eye through p53 activation. Front. Med. 2022, 9, 902713. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Wang, J.; Zhang, J.; Duan, C.; Wang, J. Ergosterol Peroxide Inhibits Porcine Epidemic Diarrhea Virus Infection in Vero Cells by Suppressing ROS Generation and p53 Activation. Viruses 2022, 14, 402. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Li, K.; Yang, M.; Wang, Q.; Hao, Z. Triacetyl Resveratrol Inhibits PEDV by Inducing the Early Apoptosis In Vitro. Int. J. Mol. Sci. 2022, 23, 14499. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).