
Polymorphism of Butyl Ester of Oleanolic Acid—The Dominance of Dispersive Interactions over Electrostatic

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
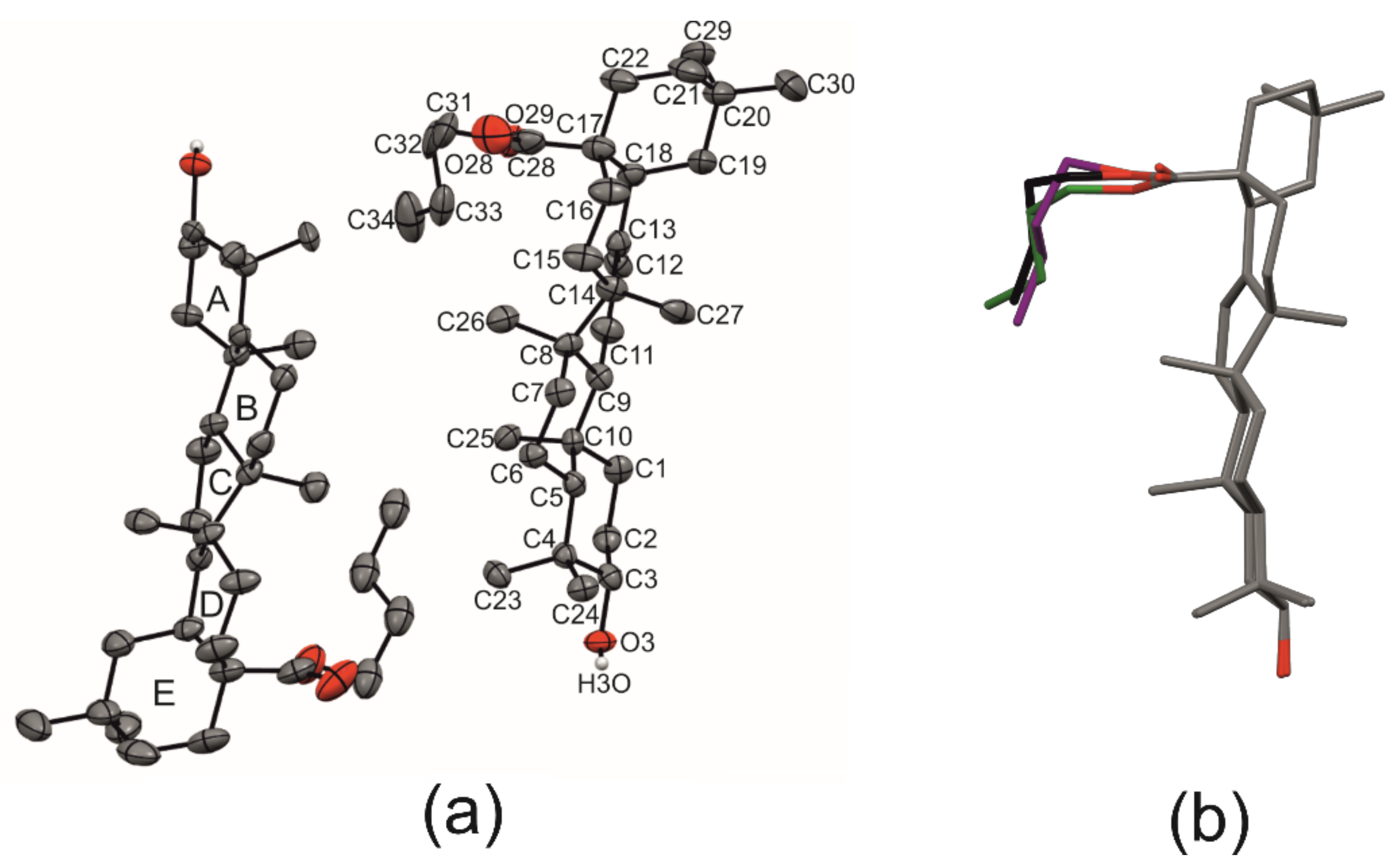
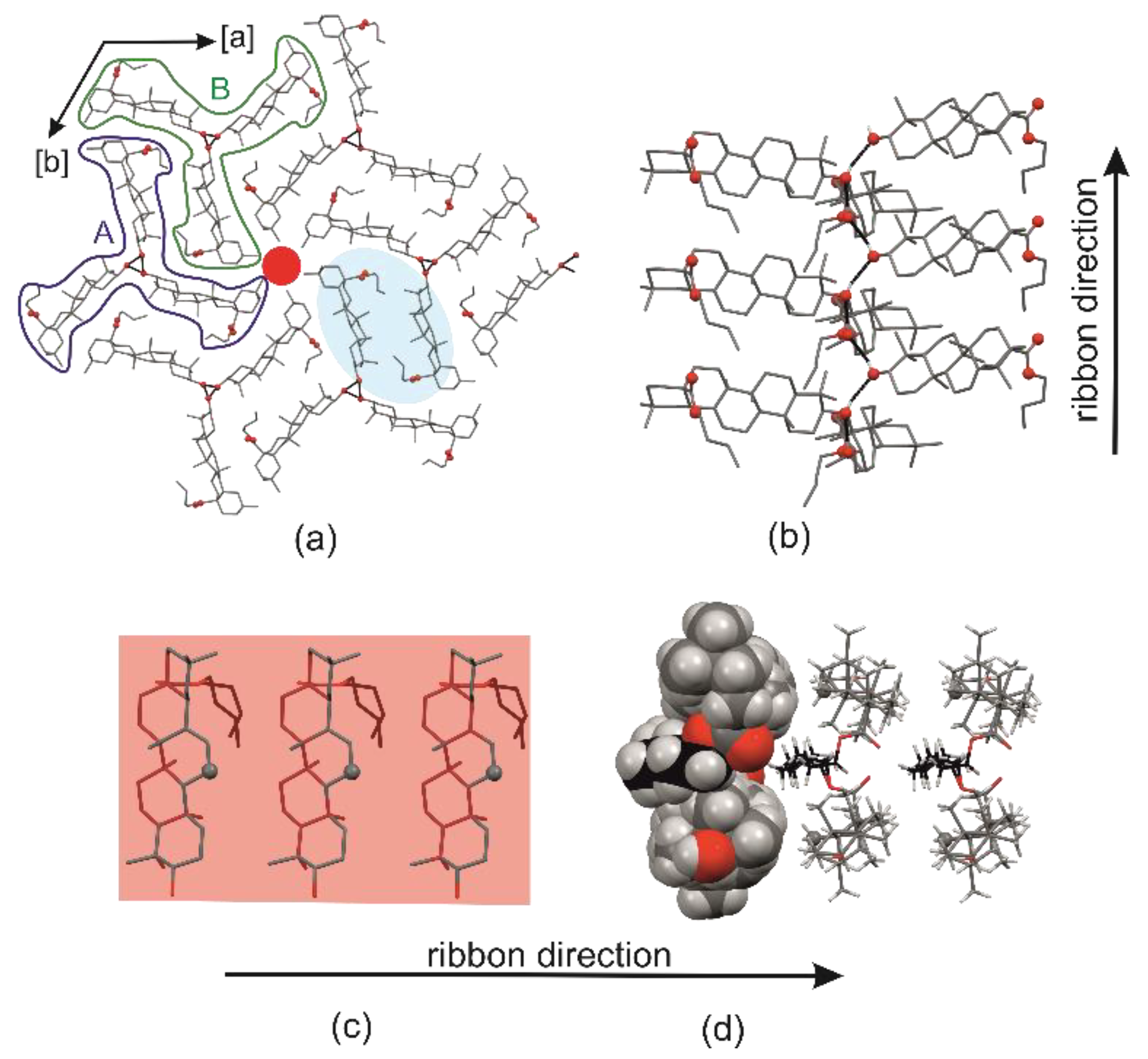
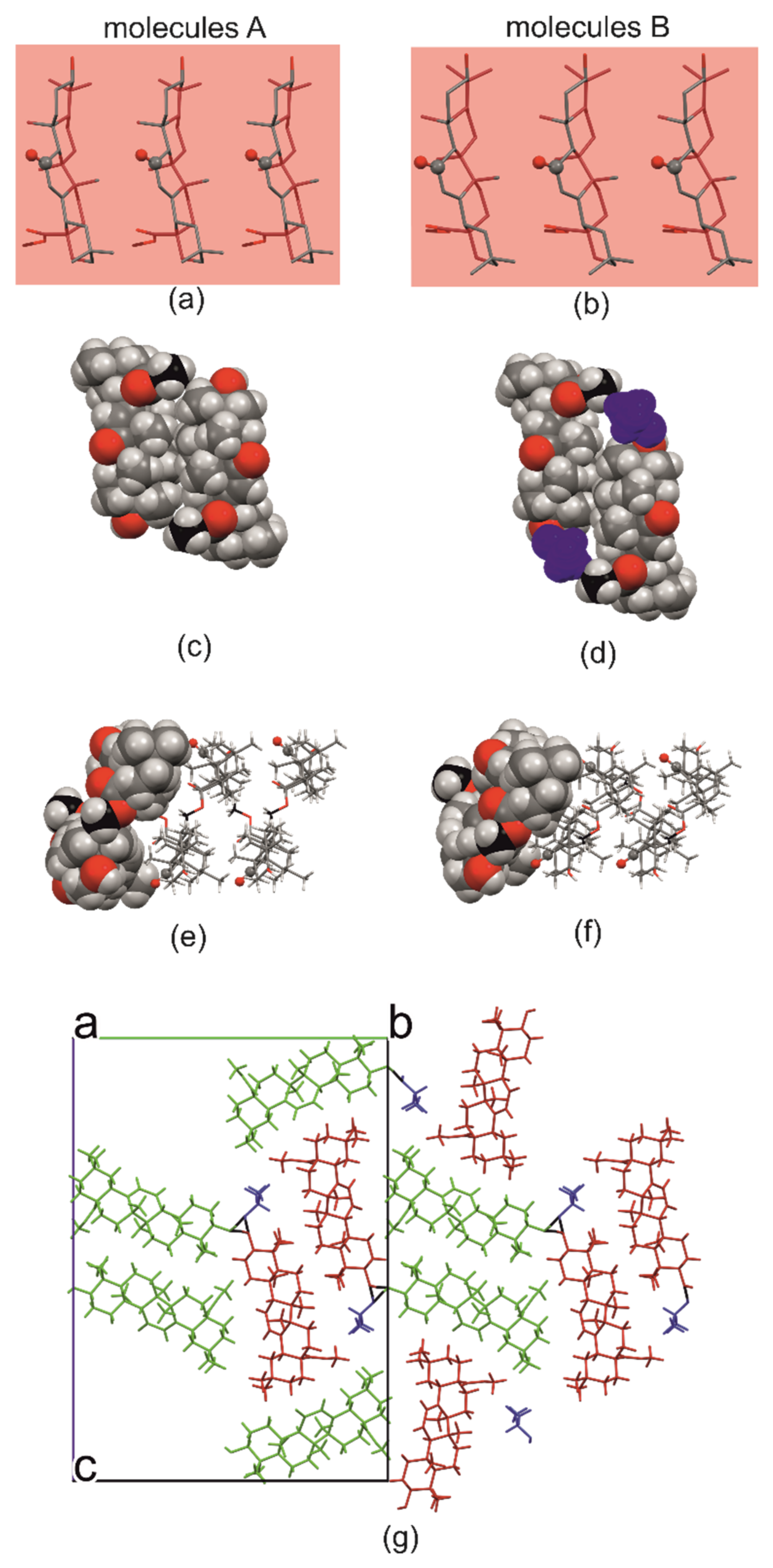
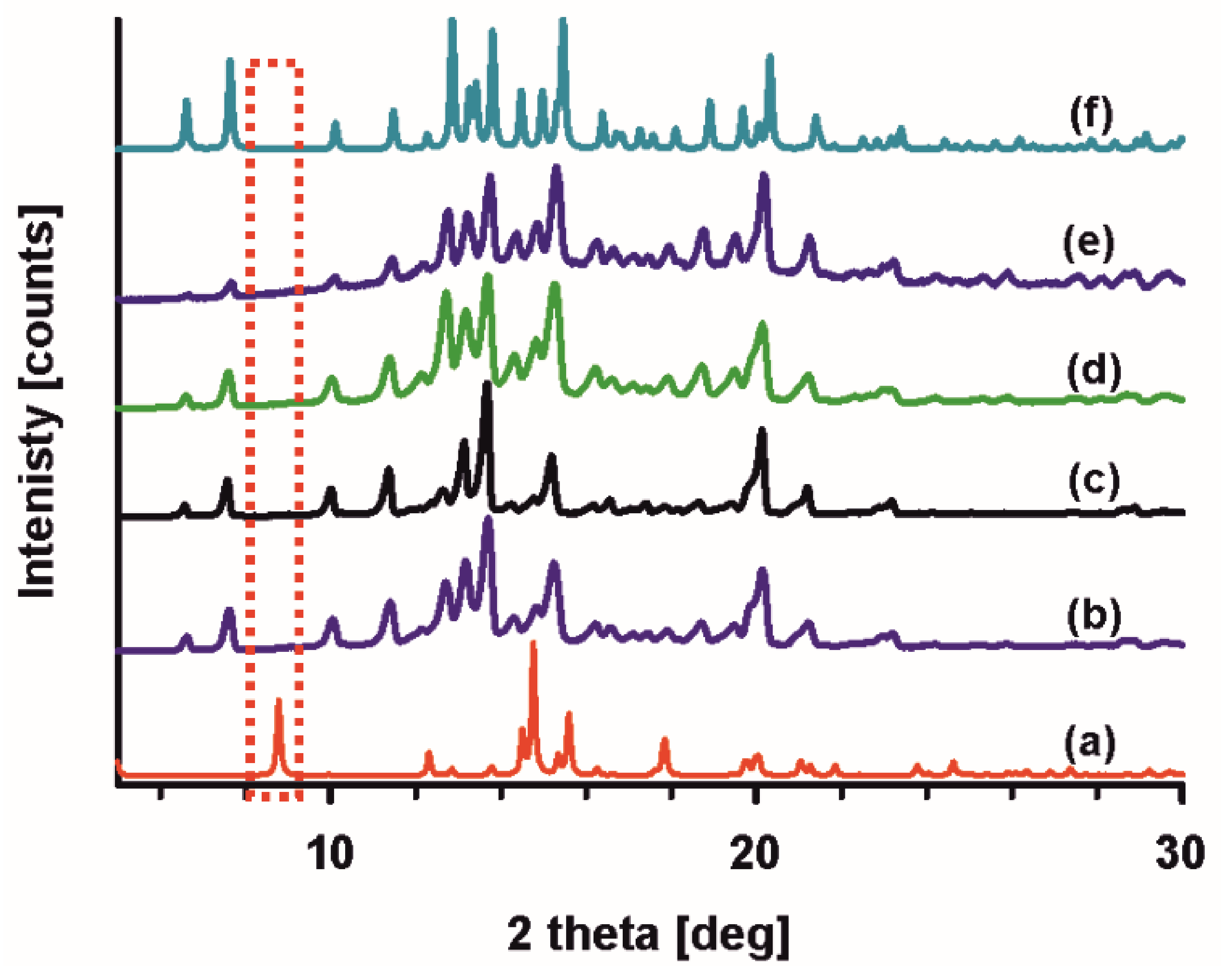
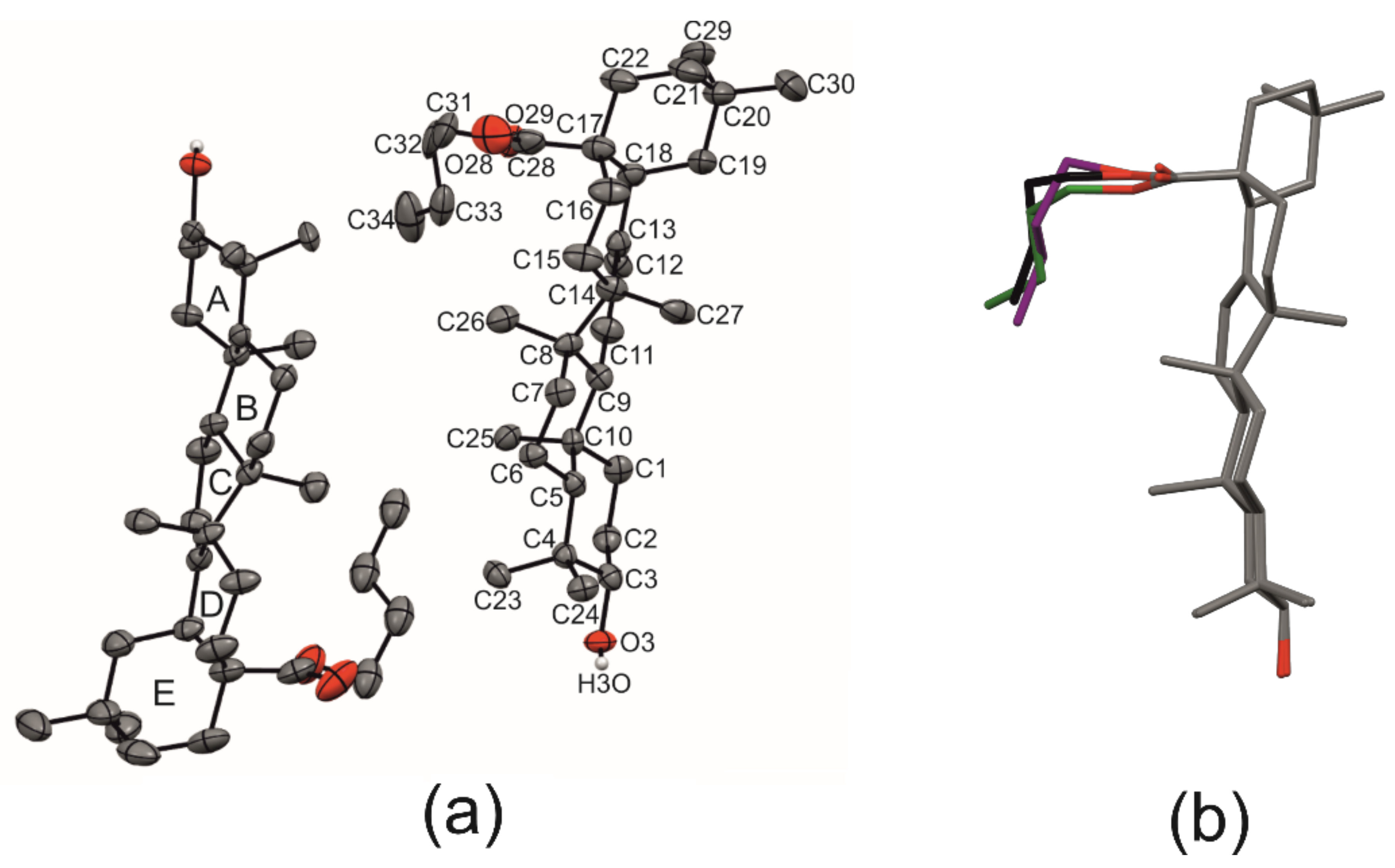
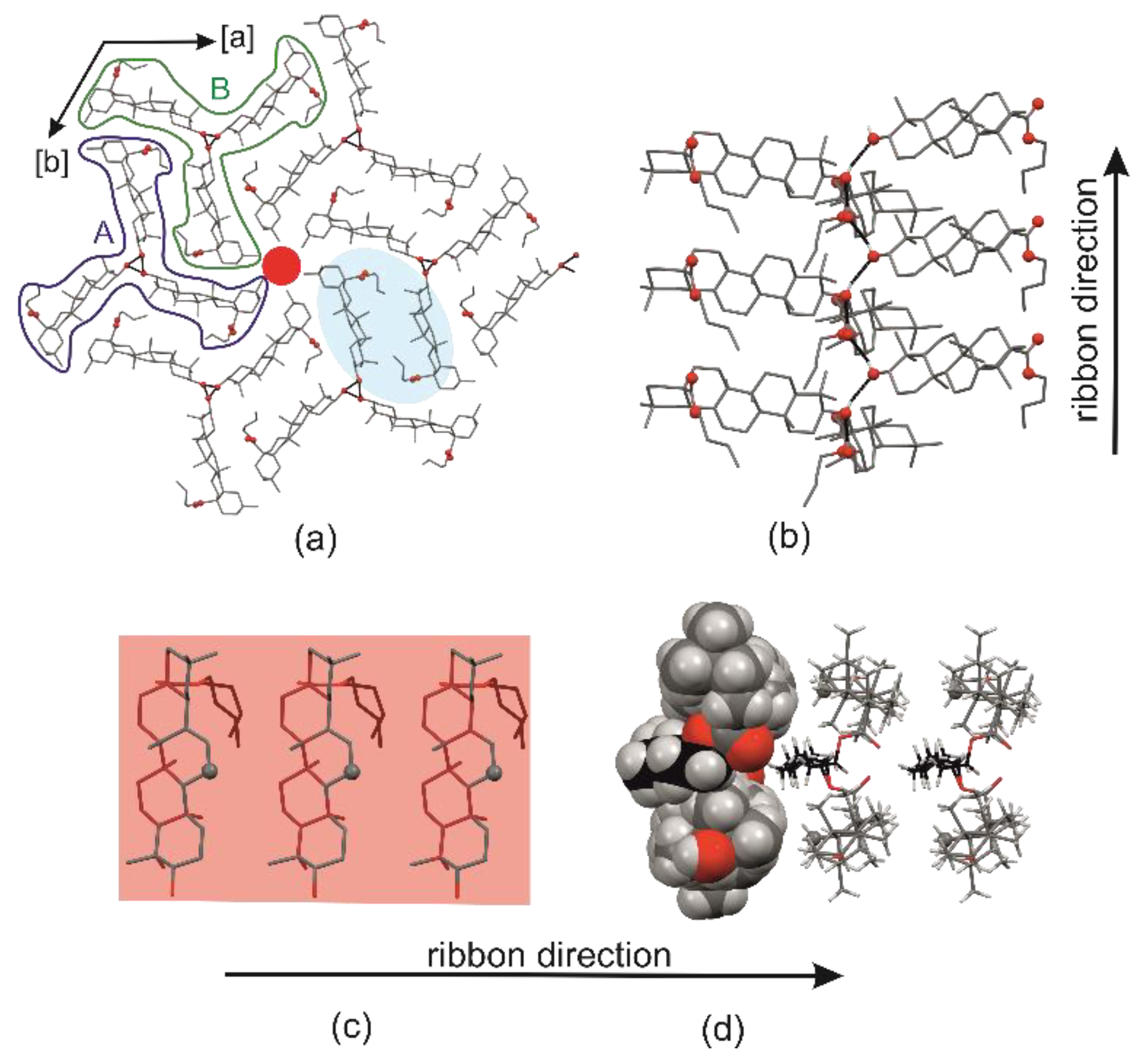
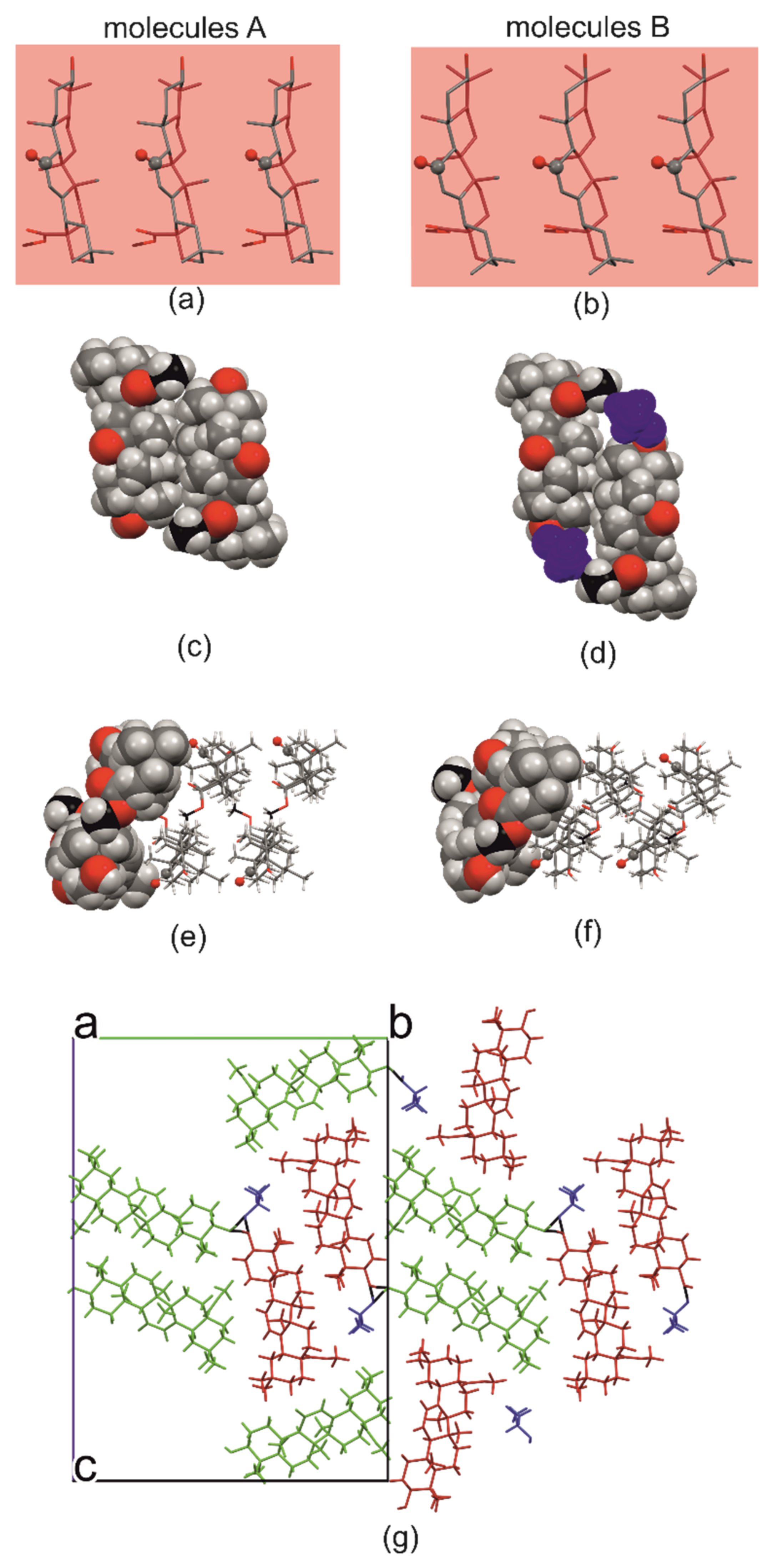
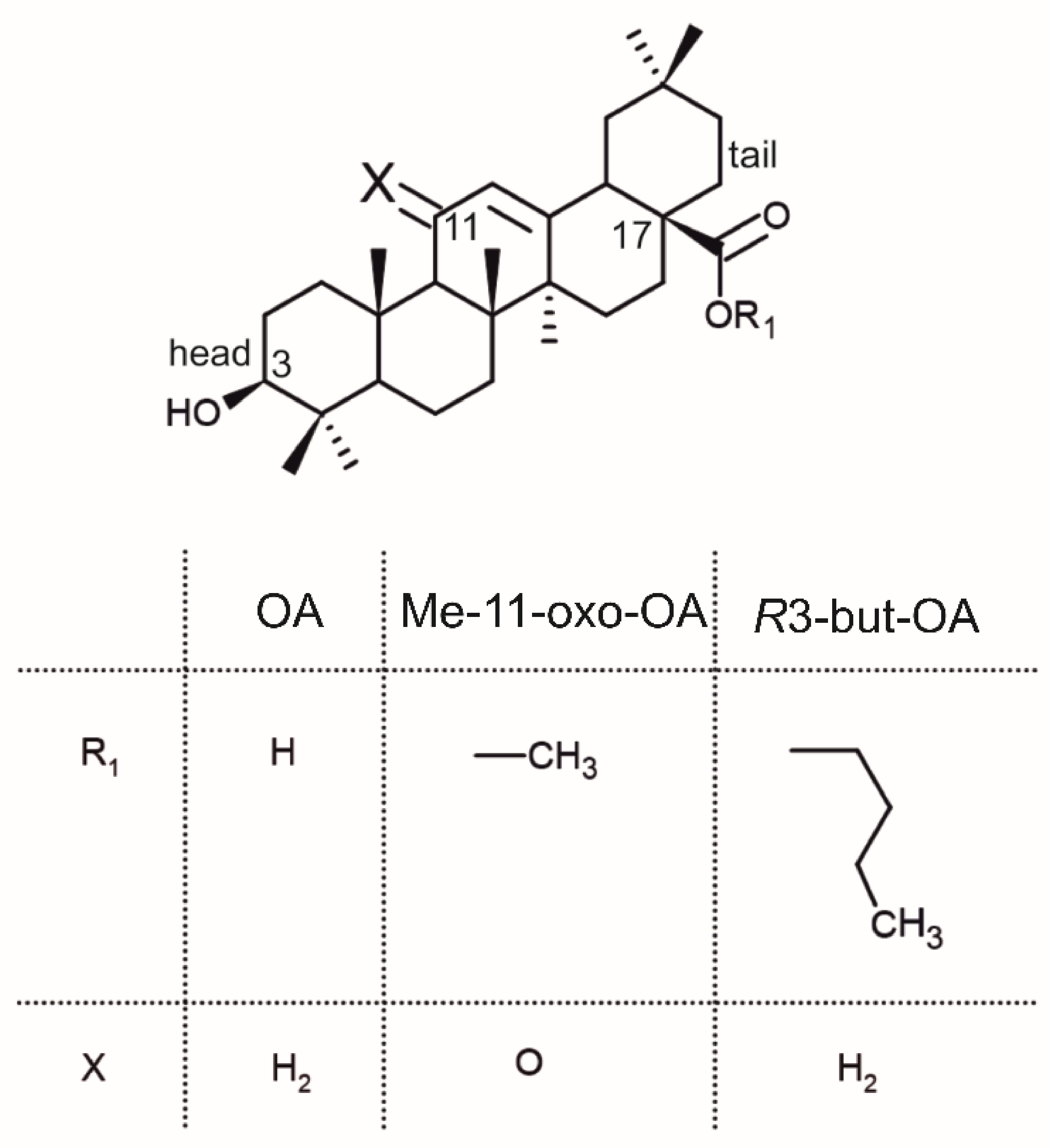
2.1. Crystal Structure of R3-but-OA versus P21-but-OA
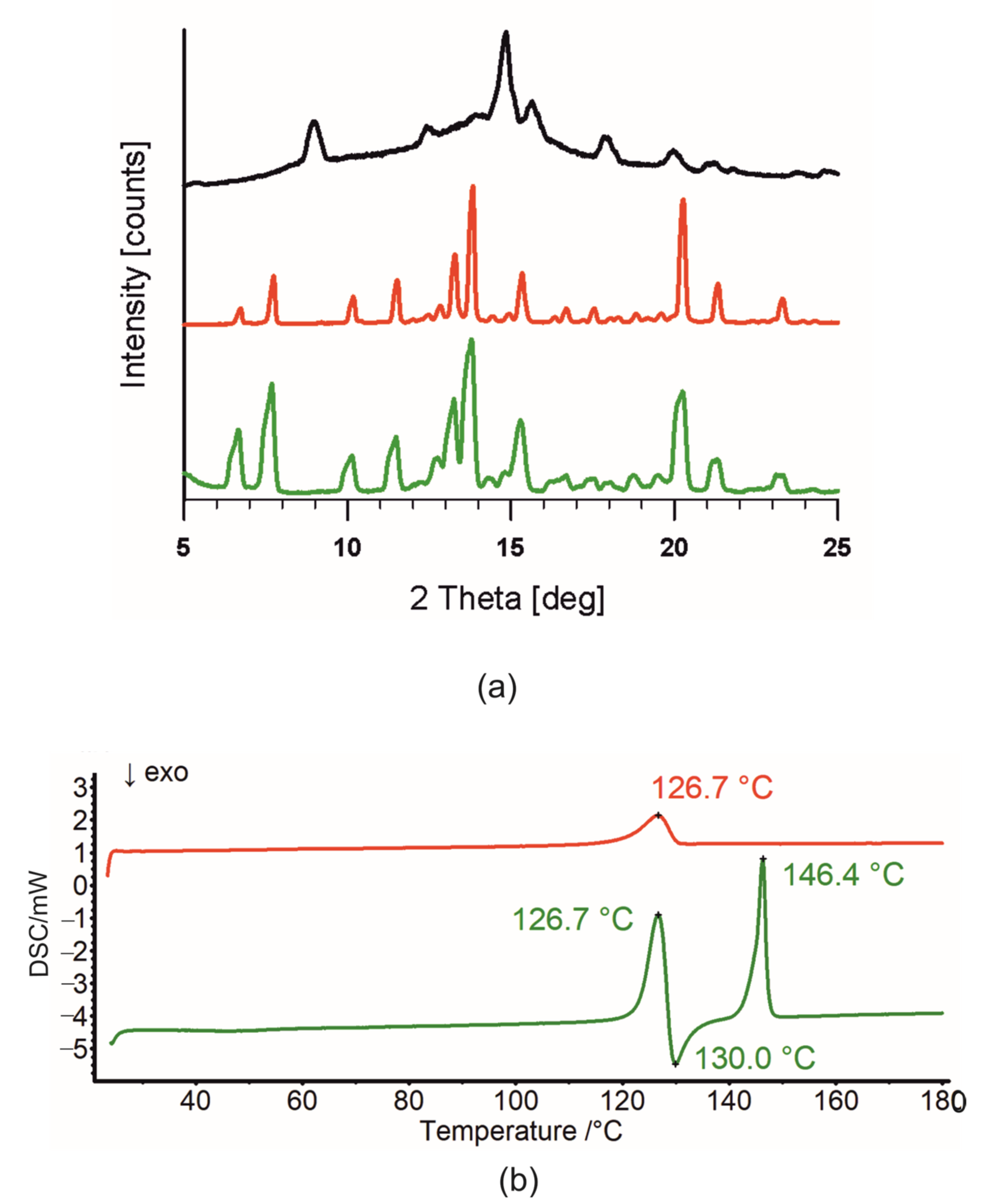
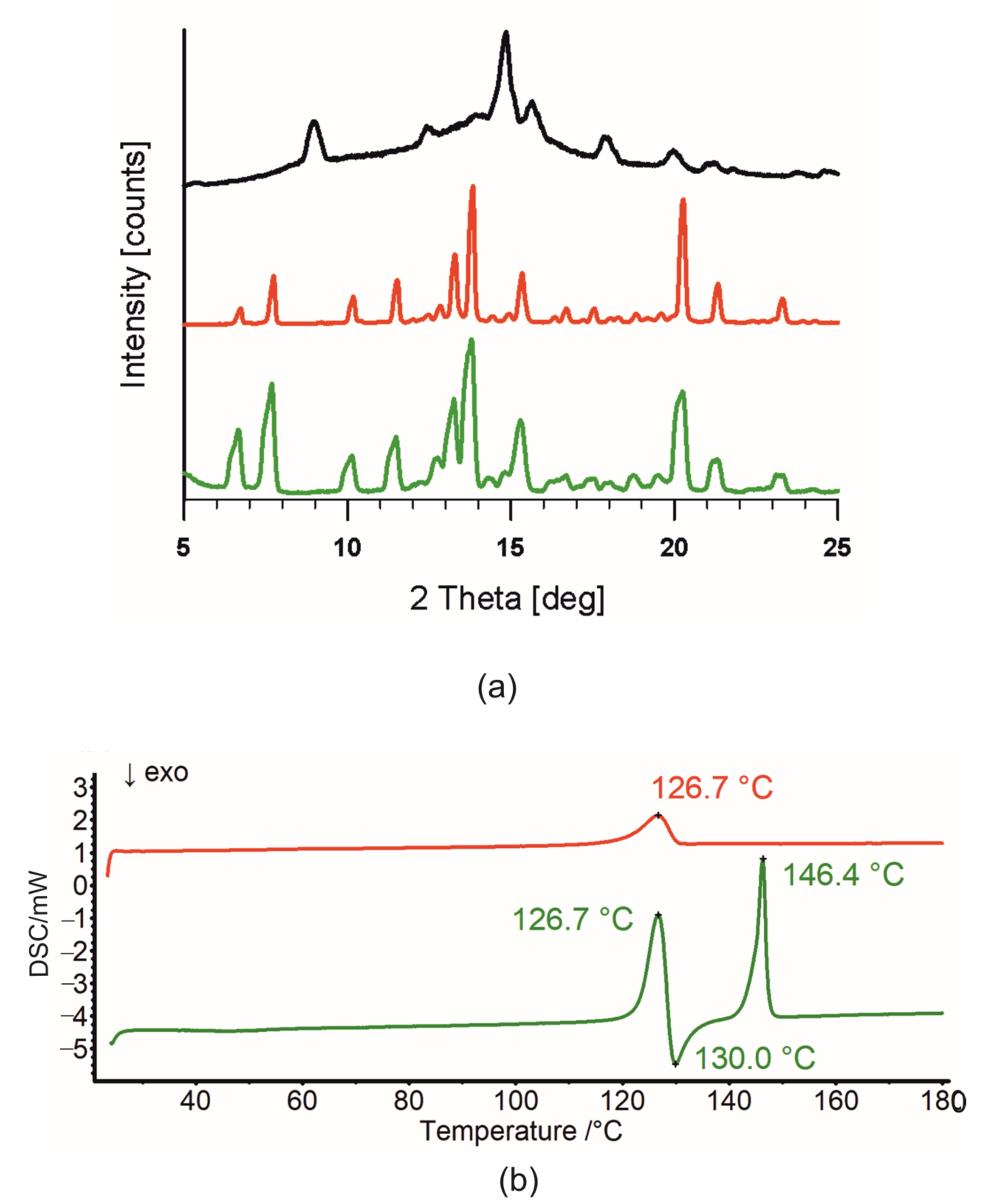
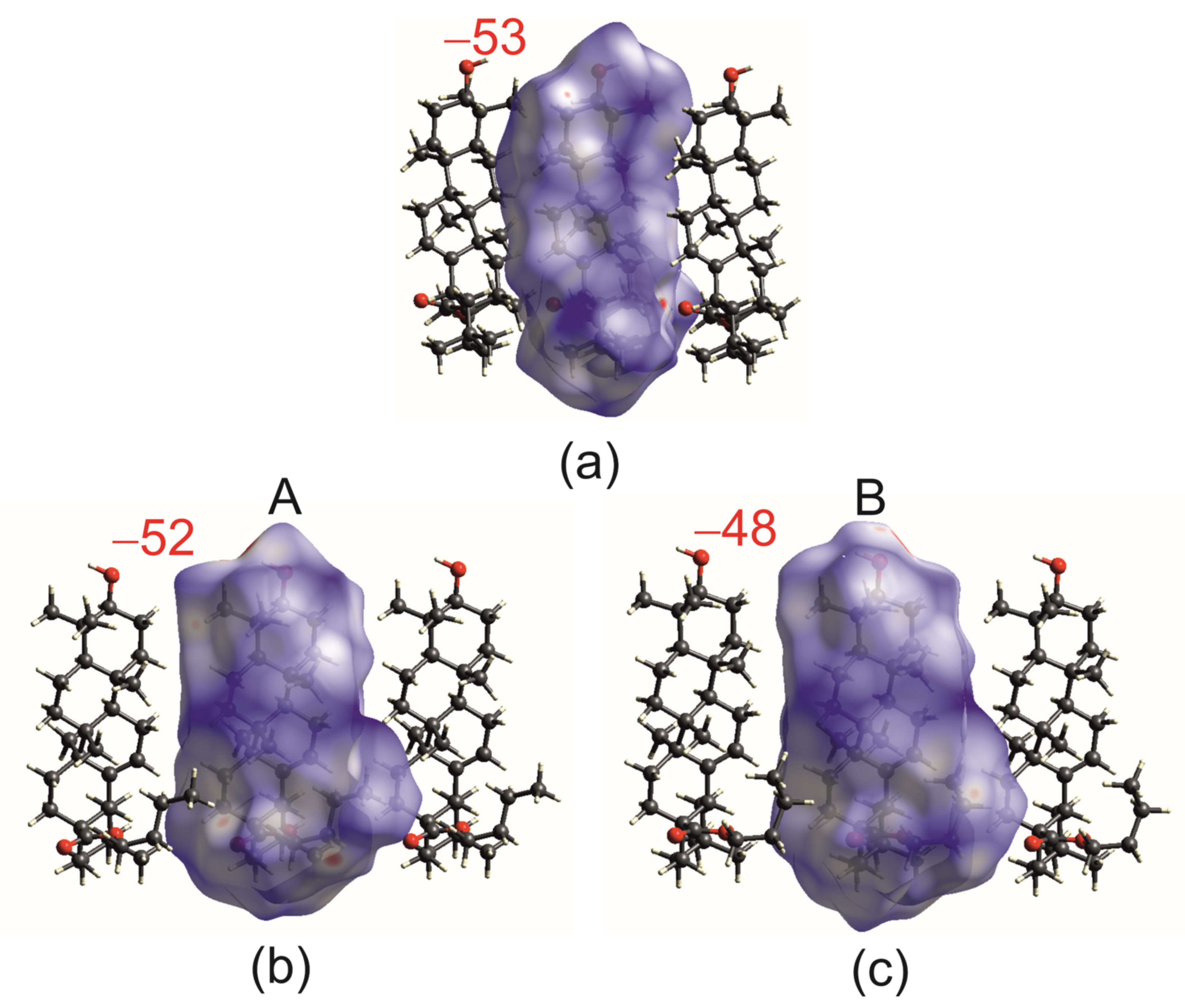
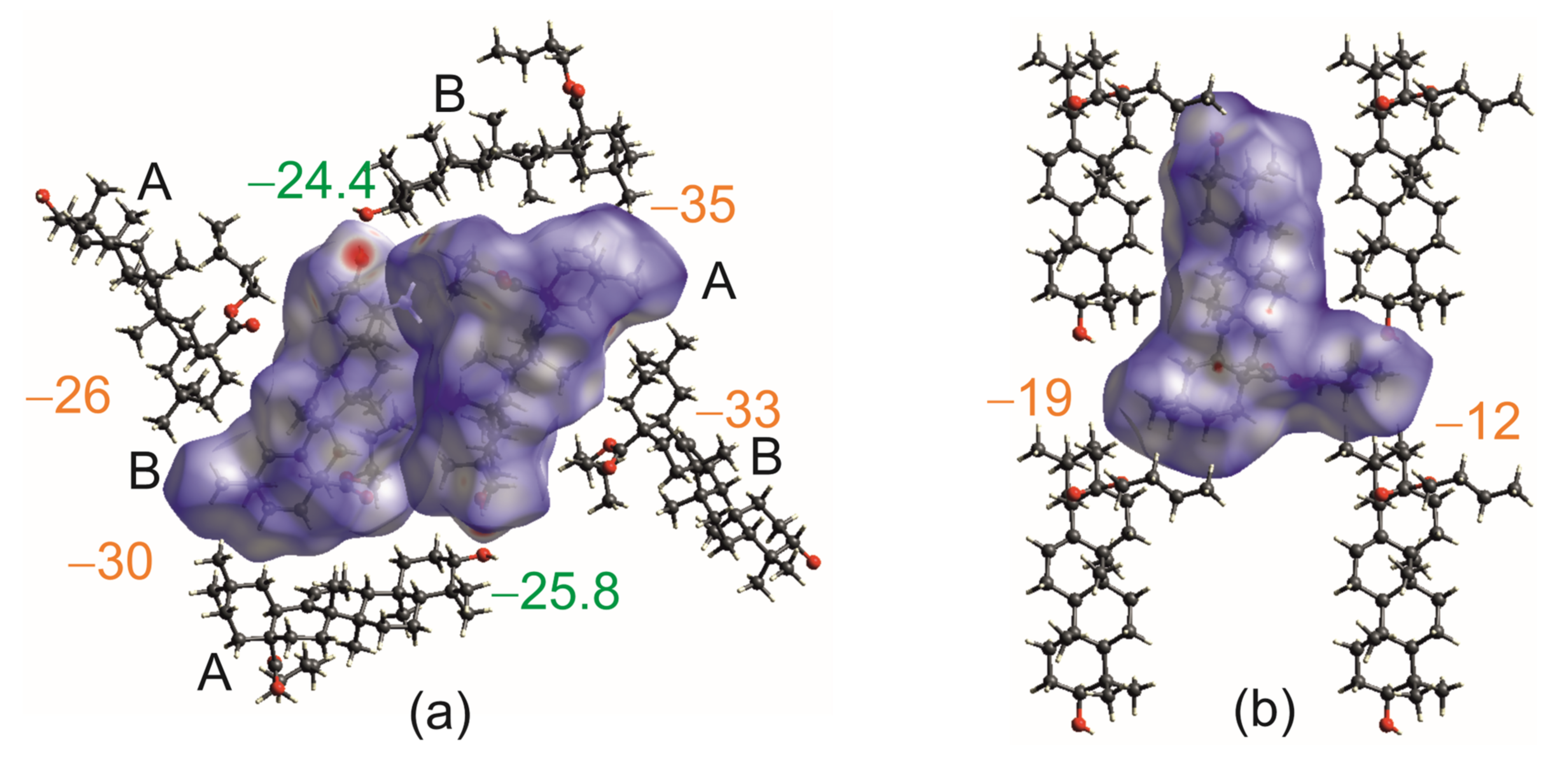
2.2. Differential Scanning Calorimetry (DSC) and Intermolecular Interaction Energy of OA Butyl Ester Polymorphs
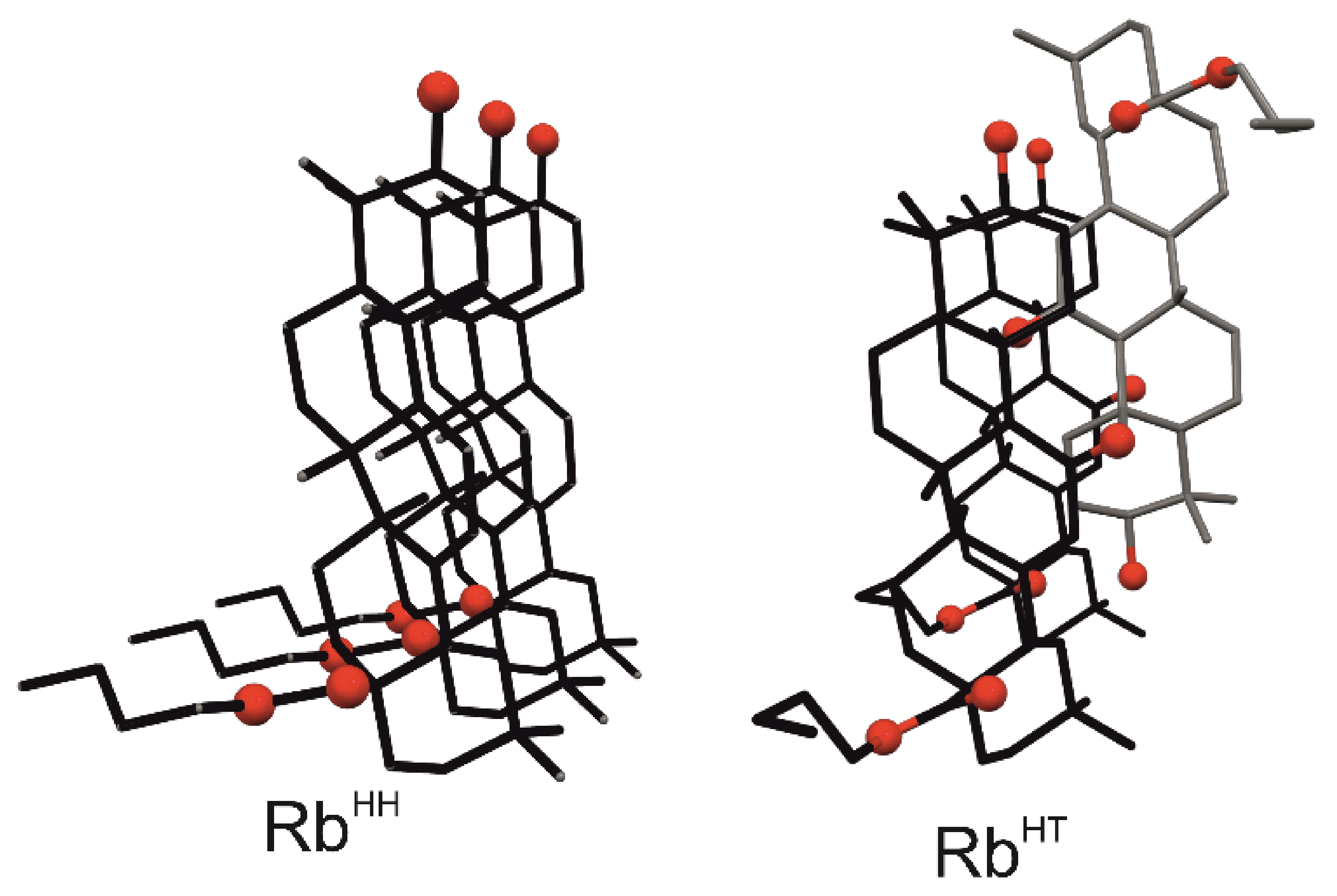
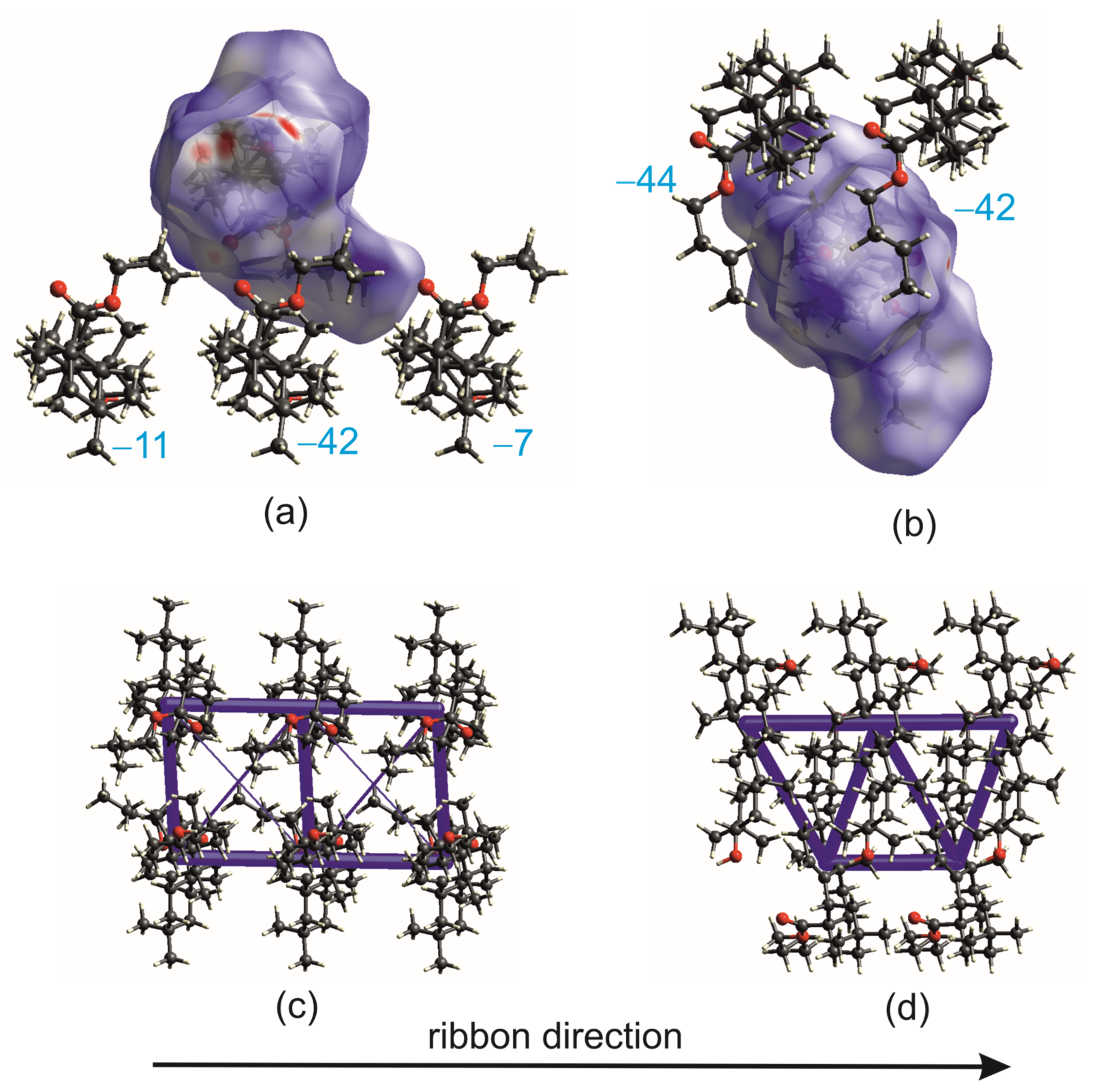
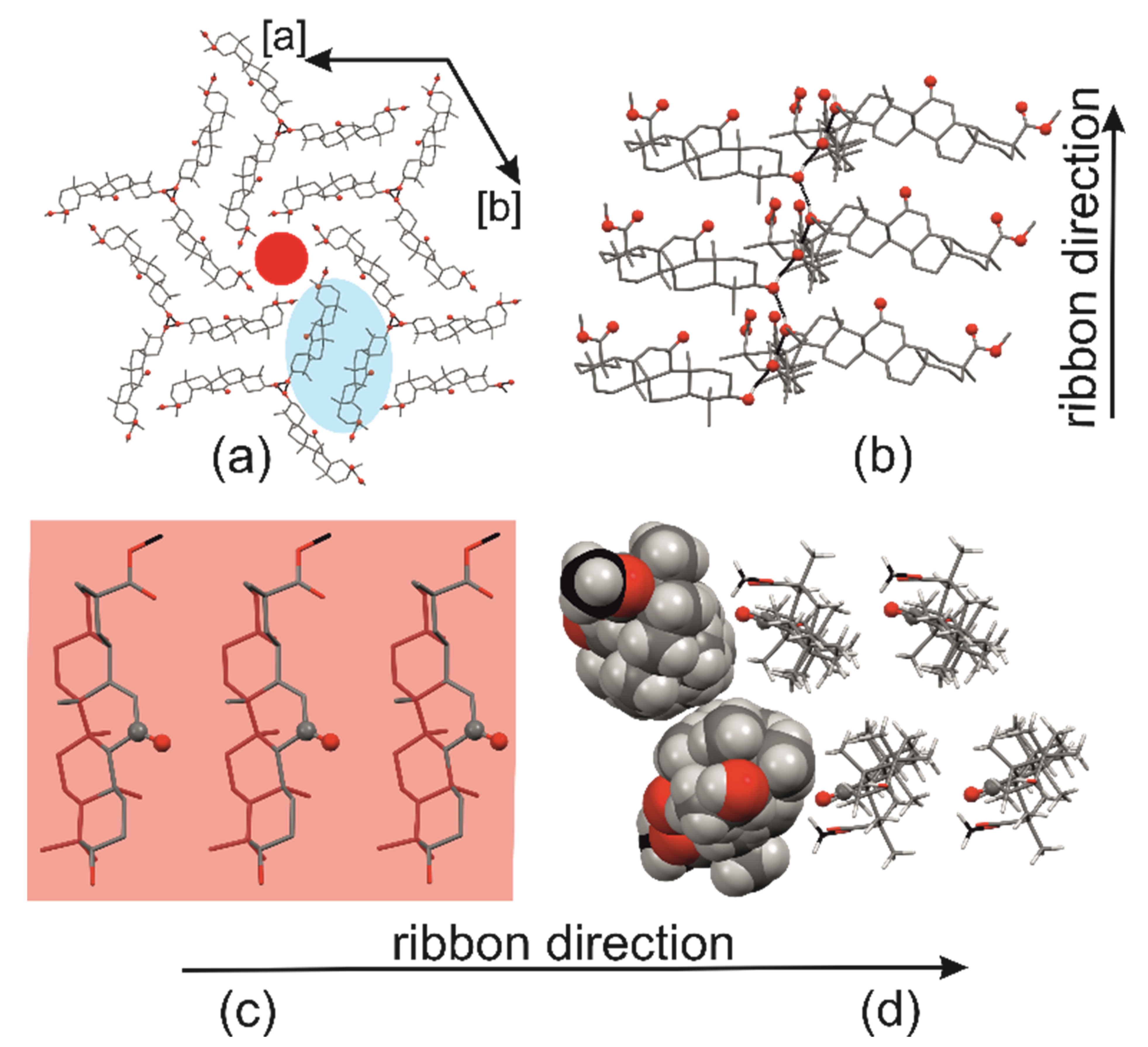
2.3. Crystal Structures with Self-Assembly Schemes Similar to R3-but-OA
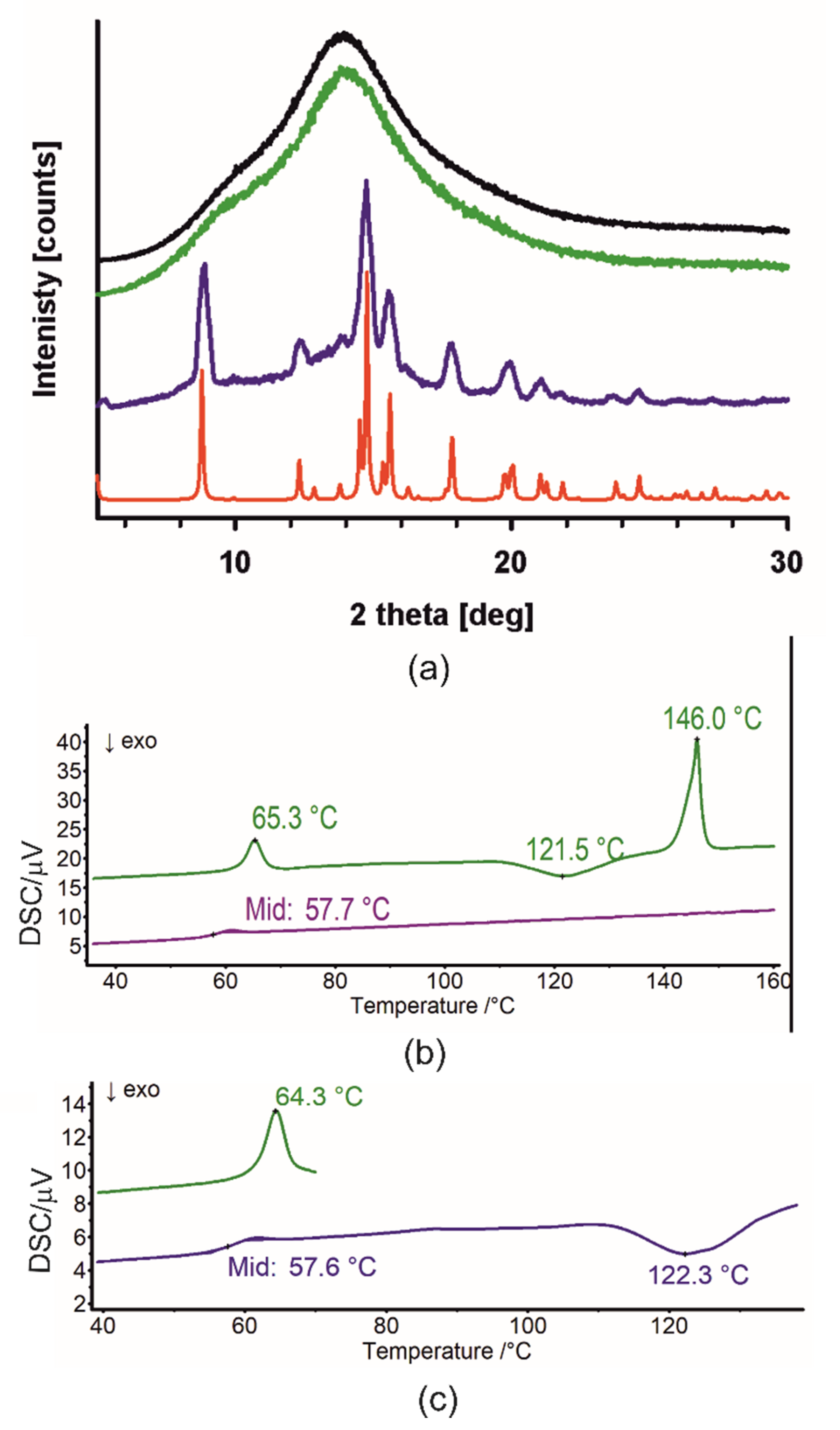
2.3.1. GE Methyl Ester Solvate [35]
2.3.2. Crystal Structure and Thermal Analysis of Me-11-oxo-OA
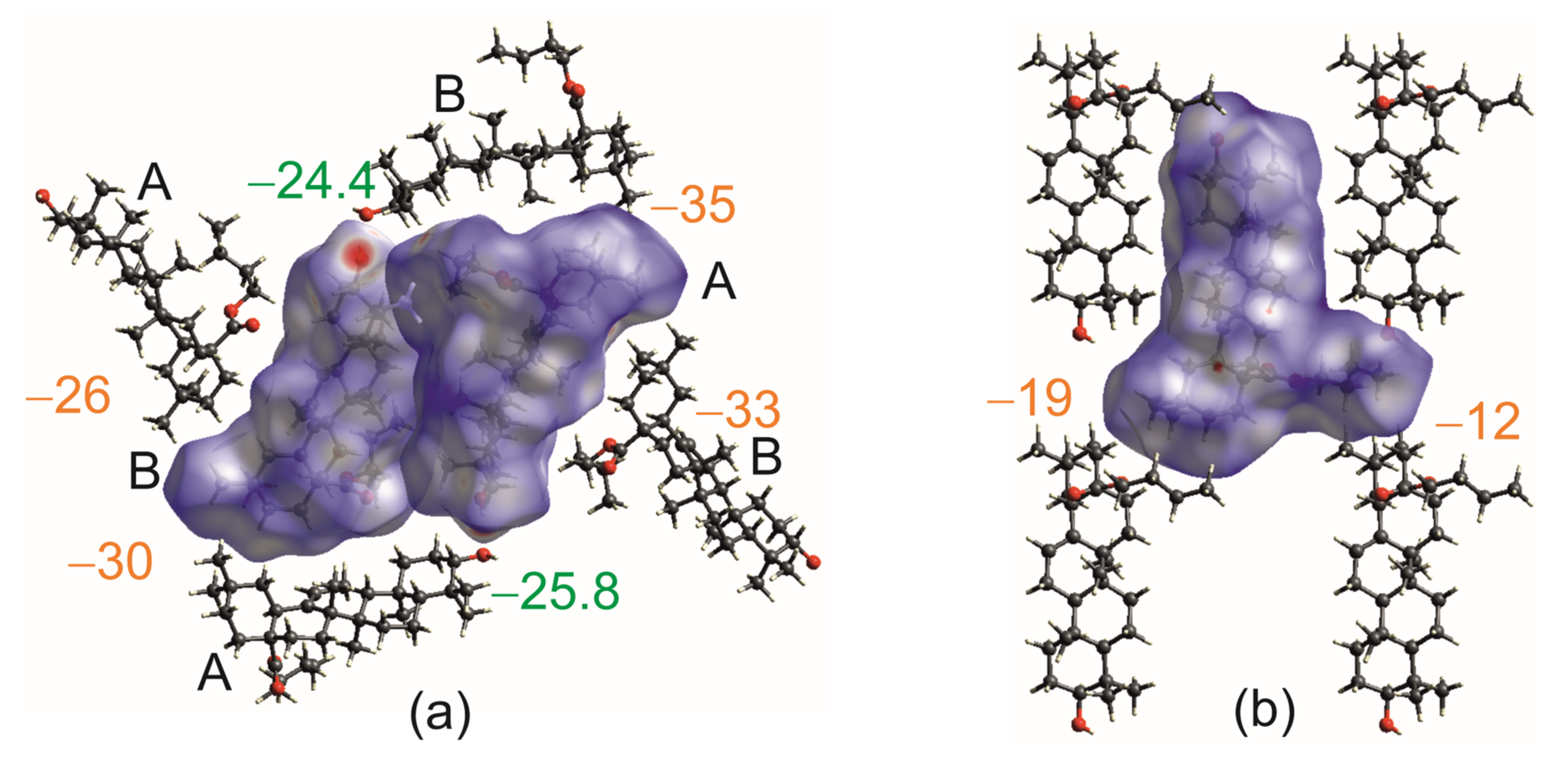
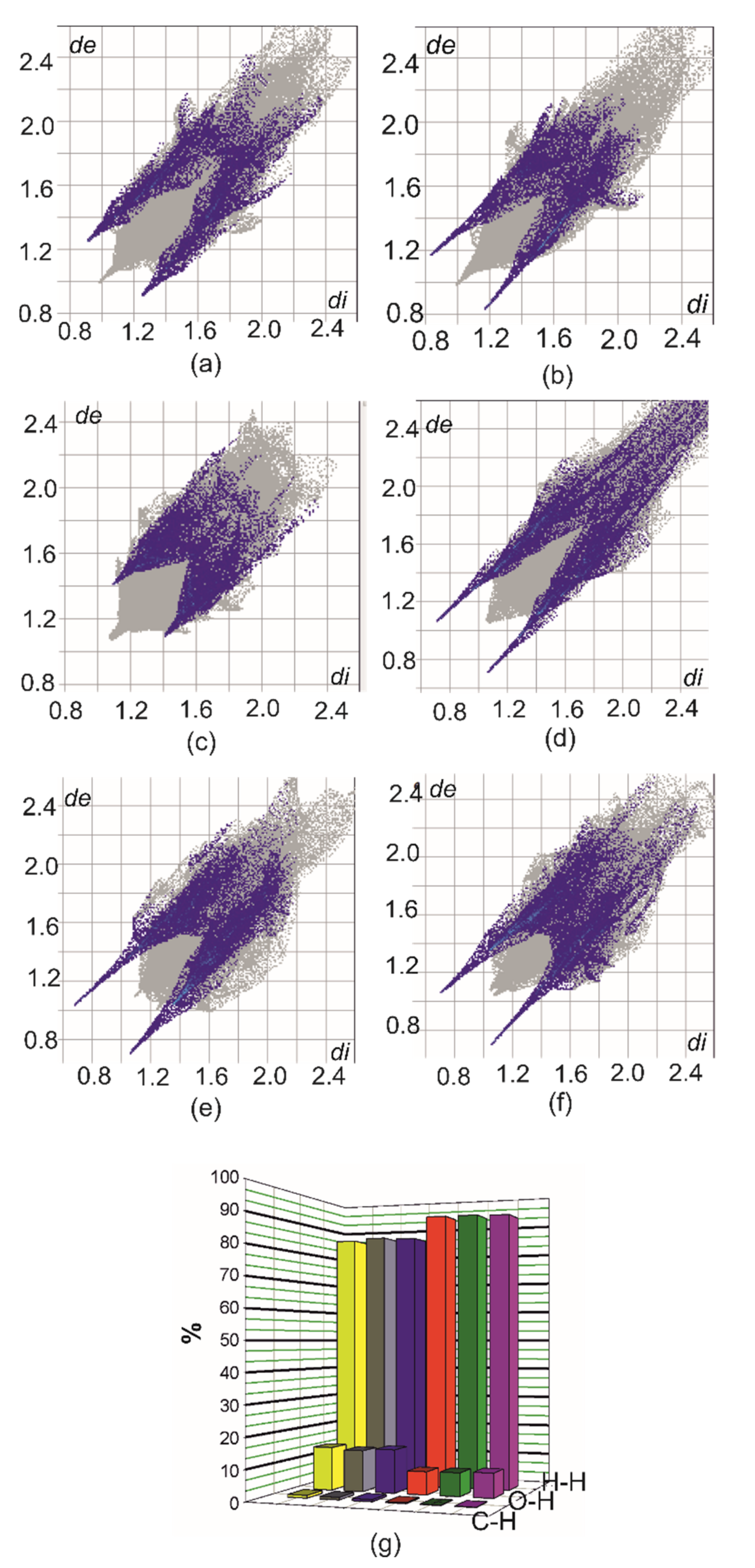
2.4. Hirshfeld Surface Analysis [49]
3. Materials and Methods
3.1. Synthesis and Crystallization
3.1.1. Synthesis
3.1.2. Crystallization
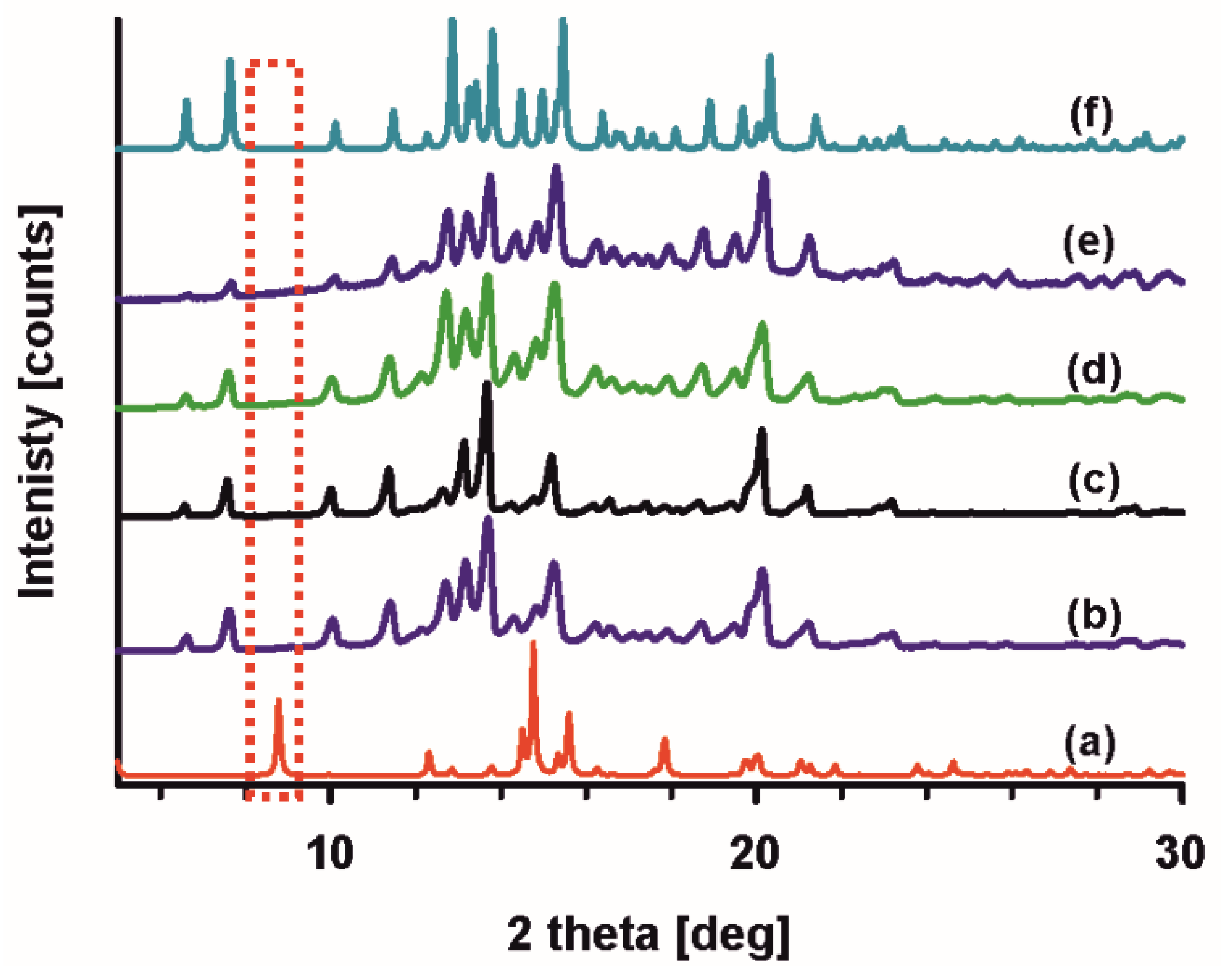
3.1.3. Screening for Polymorphs
3.1.4. Slurry Experiments
3.2. Characterization Techniques
3.2.1. X-ray Diffraction
3.2.2. Powder Diffraction Experiments
3.2.3. Differential Scanning Calorimetry (DSC)
3.2.4. Thermogravimetry (TGA)
3.2.5. Temperature Measurement by the Boetius Method
3.3. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2010; Volume 9780199236. [Google Scholar]
- Kras, W.; Carletta, A.; Montis, R.; Sullivan, R.A.; Cruz-Cabeza, A. Switching polymorph stabilities with impurities provides a thermodynamic route to benzamide form III. Commun. Chem. 2021, 438, 38. [Google Scholar] [CrossRef]
- Sacchi, P.; Reutzel-Edens, S.M.; Cruz-Cabeza, A.J. The unexpected discovery of the ninth polymorph of tolfenamic acid. CrystEngComm 2021, 23, 3636–3647. [Google Scholar] [CrossRef]
- Salazar-Rojas, D.; Kaufman, T.S.; Maggio, R.M. A comprehensive approach toward concomitant triclabendazole polymorphism in pharmaceutical products. J. Drug Deliv. Sci. Technol. 2021, 62, 102386. [Google Scholar] [CrossRef]
- Higashi, K.; Ueda, K.; Moribe, K. Recent progress of structural study of polymorphic pharmaceutical drugs. Adv. Drug. Deliv. Rev. 2017, 117, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Polymorphism in pharmaceutical solids. J. Control. Release 2001, 71, 354–355. [Google Scholar] [CrossRef]
- Chistyakov, D.; Sergeec, G. The Polymorphism of Drugs: New Approaches to the Synthesis of Nanostructured Polymorphs. Pharmaceutics 2020, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.H.; Young, V., Jr.; Grant, D.J. Polymorph screening: Influence of solvents on the rate of solvent-mediated polymorphic transformation. J. Pharm. Sci. 2001, 90, 1878–1890. [Google Scholar] [CrossRef]
- Langer, D.; Wicher, B.; Tykarska, E. Single-crystal-to-single-crystal phase transition of 18β-glycyrrhetinic acid isopropyl ester. Acta Cryst. 2022, B78, 450–458. [Google Scholar] [CrossRef]
- Brittain, H.G. Effects of mechanical processing on phase composition. J. Pharm. Sci. 2002, 91, 1573–1580. [Google Scholar] [CrossRef]
- Gong, N.; Wang, X.; Wang, Y.; Yang, S.; Song, J.; Lu, Y.; Du, G. Control over Polymorph Formation of Polydatin in Binary Solvent System and Structural Characterization. J. Pharm. Biomed. Anal. 2020, 190, 113260. [Google Scholar] [CrossRef]
- Lin, J.; Shi, P.; Wang, Y.; Wang, L.; Ma, Y.; Liu, F.; Wu, S.; Gong, J. Template design based on molecular and crystal structure similarity to regulate conformational polymorphism nucleation: The case of α,ω-alkanedicarboxylic acids. IUCrJ 2021, 8, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Sudha, C.; Srinivasan, K. Nucleation control and separation of paracetamol polymorphs through swift cooling crystallization process. J. Cryst. Growth 2014, 401, 248–251. [Google Scholar] [CrossRef]
- Svärd, M.; Rasmuson, Å.C. m -Hydroxybenzoic Acid: Quantifying Thermodynamic Stability and Influence of Solvent on the Nucleation of a Polymorphic System. Cryst. Growth Des. 2013, 13, 1140–1152. [Google Scholar] [CrossRef] [Green Version]
- McCrone, W.C. Polymorphism. In Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M.M., Weissberger, M., Eds.; Wiley Interscience: New York, NY, USA, 1965; Volume 2, pp. 725–767. [Google Scholar]
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Verhoff, M.; Seitz, S.; Paul, M.; Noha, M.S.; Jauch, J.; Schuster, D.; Werz, O. Tetra- and Pentacyclic Triterpene Acids from the Ancient Anti-inflammatory Remedy Frankincense as Inhibitors of Microsomal Prostaglandin E2 Synthase-1. J. Nat. Prod. 2014, 77, 1445–1451. [Google Scholar] [CrossRef]
- Fiore, C.; Eisenhut, M.; Ragazzi, E.; Zanchin, G.; Armanini, D. A history of the therapeutic use of liquorice in Europe. J. Ethnopharmacol. 2005, 99, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Muffler, K.; Leipold, D.; Scheller, M.-C.; Haas, C.; Steingroewer, J.; Bley, T.; Neuhaus, H.E.; Mirata, M.A.; Schrader, J.; Ulber, R. Biotransformation of triterpenes. Process Biochem. 2011, 46, 1–15. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene Biosynthesis in Plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Hussain, H.; Green, R.I.; Shamraiz, U.; Saleem, M.; Badshah, A.; Abbas, G.; Ur Rehman, N.; Irshad, M. Therapeutic potential of glycyrrhetinic acids: A patent review (2010–2017). Expert Opin. Ther. Pat. 2018, 5, 383–398. [Google Scholar] [CrossRef]
- Ayeleso, B.T.; Matumba, M.G.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk-Cwynar, B.; Zaprutko, L.; Marciniak, J.; Lewandowski, G.; Szulc, M.; Kaminska, E.; Wachowiak, N.; Mikolajczak, P.Ł. The analgesic and anti-inflammatory effect of new oleanolic acid acyloxyimino derivative. Eur. J. Pharm. Sci. 2012, 47, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Ismail, M.A.H.; El-Mekkawy, S.; Ismail, N.S.M.; Hanna, A.G. Synthesis and biological activity of new 18β-glycyrrhetinic acid derivatives. Arab. J. Chem. 2016, 9, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Sen, A. Prophylactic and therapeutic roles of oleanolic acid and its derivatives in several diseases. World J. Clin. Cases 2020, 8, 1767–1792. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Mei, L.; Wang, M.; Huang, Q.; Huand, R. Anti-inflammatory and Pro-apoptotic Effects of 18beta-Glycyrrhetinic Acid In Vitro and In Vivo Models of Rheumatoid Arthritis. Front. Pharmacol. 2021, 12, 681525. [Google Scholar] [CrossRef]
- Quan, W.; Kong, S.; Ouyang, Q.; Tao, J.; Lu, S.; Huang, Y.; Li, S.; Luo, H. Use of 18β-glycyrrhetinic acid nanocrystals to enhance anti-inflammatory activity by improving topical delivery. Colloids Surf. B Biointerfaces 2021, 205, 111791. [Google Scholar] [CrossRef]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Antiviral Activities of Oleanolic Acid and Its Analogues. Molecules 2018, 23, 2300. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-J.; Geng, C.-A.; Ma, Y.-B.; Huang, X.-Y.; Luo, J.; Chen, H.; Zhang, X.-M.; Chen, J.J. Synthesis, biological evaluation and structure–activity relationships of glycyrrhetinic acid derivatives as novel anti-hepatitis B virus agents. Bioorg. Med. Chem. Lett. 2012, 22, 3473–3479. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk-Cwynar, B.; Ruszkowski, P.; Jarosz, T.; Krukiewicz, K. Enhancing anticancer activity through the combination of bioreducing agents and triterpenes. Future Med. Chem. 2018, 10, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Huai, X.-D.; Zheng, Q.-X.; Wang, W.; Li, H.-J.; Huai, Q.-Y. Design and preparation of derivatives of oleanolic and glycyrrhetinic acids with cytotoxic properties. Drug Des. Devel. Ther. 2018, 12, 1321–1336. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yang, W.; Fan, Y.; Dehaen, W.; Li, Y.; Li, H.; Wang, W.; Zheng, Q.; Huai, Q. Design and synthesis of the novel oleanolic acid-cinnamic acid ester derivatives and glycyrrhetinic acid-cinnamic acid ester derivatives with cytotoxic properties. Bioorg. Chem. 2019, 88, 102951. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Wang, C.; Xu, W.-T.; Zhang, Y.; Zhang, T.; Xue, H.; Li, Y.-N.; Fu, Z.-R.; Wang, Y.; Jin, C.h.-H. 18β-Glycyrrhetinic Acid Has Anti-Cancer Effects via Inducing Apoptosis and G2/M Cell Cycle Arrest, and Inhibiting Migration of A549 Lung Cancer Cells. Onco Targets Ther. 2021, 14, 5131–5144. [Google Scholar] [CrossRef]
- Langer, D.; Wicher, B.; Bendzinska-Berus, W.; Bednarczyk-Cwynar, B.; Tykarska, E. Insights into isostructural and non-isostructural crystals of esters of oleanolic acid and its 11-oxo derivatives. Acta Cryst. 2022, B78, 606–617. [Google Scholar] [CrossRef]
- Beseda, I.; Czollner, L.; Shah, P.S.; Khunt, R.; Gaware, R.; Kosma, P.; Stanetty, C.; del Ruiz-Ruiz, M.C.; Amer, H.; Mereiter, K.; et al. Synthesis of glycyrrhetinic acid derivatives for the treatment of metabolic diseases. Bioorg. Med. Chem. 2010, 18, 433–454. [Google Scholar] [CrossRef] [PubMed]
- Campsteyn, H.; Dupont, L.; Lamotte, J.O.; Dideberg, O.; Vermeire, M. Crystal and molecular structure of glycyrrhetinic acid acetone monohydrate. Acta Crystallogr. 1977, B33, 3443–3448. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, T.; Liu, H.; Zheng, Y.; Li, N.; Zhao, Q.; Chen, Y.; Liu, B. Synthesis, toxicities and cell proliferation inhibition of CO-releasing molecules containing cobalt. Transit. Met. Chem. 2015, 40, 413–426. [Google Scholar] [CrossRef]
- Langer, D.; Wicher, B.; Szczolko, W.; Gdaniec, M.; Tykarska, E. Self-assembly modes of glycyrrhetinic acid esters in view of the crystal packing of related triterpene molecules. Acta Cryst. 2016, B72, 584–592. [Google Scholar] [CrossRef]
- Tykarska, E.; Gdaniec, M. Solid-state supramolecular architecture of carbenoxolone-comparative studies with glycyrrhetinic and glycyrrhizic acids. Acta Cryst. 2015, B71, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Rev. A.02); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Burger, A.; Ramberger, R. On the polymorphism of pharmaceuticals and other molecular crystals. I.; Theory of thermodynamic rules. Microchim. Acta 1979, 72, 259–271. [Google Scholar] [CrossRef]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [Green Version]
- Gavezzotti, A. Calculation of lattice energies of organic crystals: The PIXEL integration method in comparison with more traditional methods. Z. Krist. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. Psi4 1.4: Open-Source Software for High-Throughput Quantum Chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.; Bowling, P.E.; Herbert, J.M. Systematic evaluation of counterpoise correction in density functional theory. J. Chem. Theory Comput. 2022, 18, 6742–6756. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Rigaku. CrysAlis PRO Software; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2015. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Reeves, M.G.; Wood, P.A.; Parsons, S. MrPIXEL: Automated execution of Pixel calculations via the Mercury interface. J. Appl. Cryst. 2020, 53, 1154–1162. [Google Scholar] [CrossRef]
- Bond, A.D. processPIXEL: A program to generate energy-vector models from Gavezzotti’s PIXEL calculations. J. Appl. Cryst. 2014, 47, 1777–1780. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F., III. Large-scale symmetry-adapted perturbation theory computations via density fitting and laplace transformation techniques: Investigating the fundamental forces of DNA-intercalator interactions. J. Chem. Phys. 2011, 135, 174107. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on basis sets beautiful: Seasonal plantings of diffuse basis functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Schriber, J.B.; Sirianni, D.A.; Smith, D.G.; Burns, L.A.; Sitkoff, D.; Cheney, D.L.; Sherrill, C.D. Optimized damping parameters for empirical dispersion corrections to symmetry-adapted perturbation theory. J. Chem. Phys. 2021, 154, 234107. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Parrish, R.M.; Parker, T.M.; Sherrill, C.D. Chemical Assignment of Symmetry-Adapted Perturbation Theory Interaction Energy Components: The Functional-Group SAPT Partition. J. Chem. Theory Comput. 2014, 10, 4417–4431. [Google Scholar] [CrossRef]
- Kruse, H.; Banáš, P.; Šponer, J. Investigations of stacked DNA base-pair steps: Highly accurate stacking interaction energies, energy decomposition, and many-body stacking effects. J. Chem. Theory Comput. 2018, 15, 95–115. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-practice DFT protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| R3-but-OA | ||||
| O3A—H3A1···O3A i | 0.84 | 2.30 | 3.115 (4) | 164 |
| O3B—H3B1···O3B ii | 0.84 | 2.06 | 2.878 (4) | 163 |
| Me-11-oxo-OA | ||||
| O3A—H3OA···O1S | 0.84 | 1.86 | 2.696 (4) | 173 |
| O3B—H3OB···O3A iii | 0.84 | 1.90 | 2.708 (4) | 160 |
| O1S—H1S···O3B | 0.84 | 1.90 | 2.735 (4) | 174 |
| Me-GE * | ||||
| O1-H39···O1 iv | 0.84 | 1.93 | 2.745 | 162 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langer, D.; Wicher, B.; Dutkiewicz, Z.; Bendzinska-Berus, W.; Bednarczyk-Cwynar, B.; Tykarska, E. Polymorphism of Butyl Ester of Oleanolic Acid—The Dominance of Dispersive Interactions over Electrostatic. Int. J. Mol. Sci. 2023, 24, 6572. https://doi.org/10.3390/ijms24076572
Langer D, Wicher B, Dutkiewicz Z, Bendzinska-Berus W, Bednarczyk-Cwynar B, Tykarska E. Polymorphism of Butyl Ester of Oleanolic Acid—The Dominance of Dispersive Interactions over Electrostatic. International Journal of Molecular Sciences. 2023; 24(7):6572. https://doi.org/10.3390/ijms24076572
Chicago/Turabian StyleLanger, Dominik, Barbara Wicher, Zbigniew Dutkiewicz, Wioletta Bendzinska-Berus, Barbara Bednarczyk-Cwynar, and Ewa Tykarska. 2023. "Polymorphism of Butyl Ester of Oleanolic Acid—The Dominance of Dispersive Interactions over Electrostatic" International Journal of Molecular Sciences 24, no. 7: 6572. https://doi.org/10.3390/ijms24076572
APA StyleLanger, D., Wicher, B., Dutkiewicz, Z., Bendzinska-Berus, W., Bednarczyk-Cwynar, B., & Tykarska, E. (2023). Polymorphism of Butyl Ester of Oleanolic Acid—The Dominance of Dispersive Interactions over Electrostatic. International Journal of Molecular Sciences, 24(7), 6572. https://doi.org/10.3390/ijms24076572