
Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI

 ,
,  ,
,
Abstract
:1. Introduction
2. Patient Report
3. Results
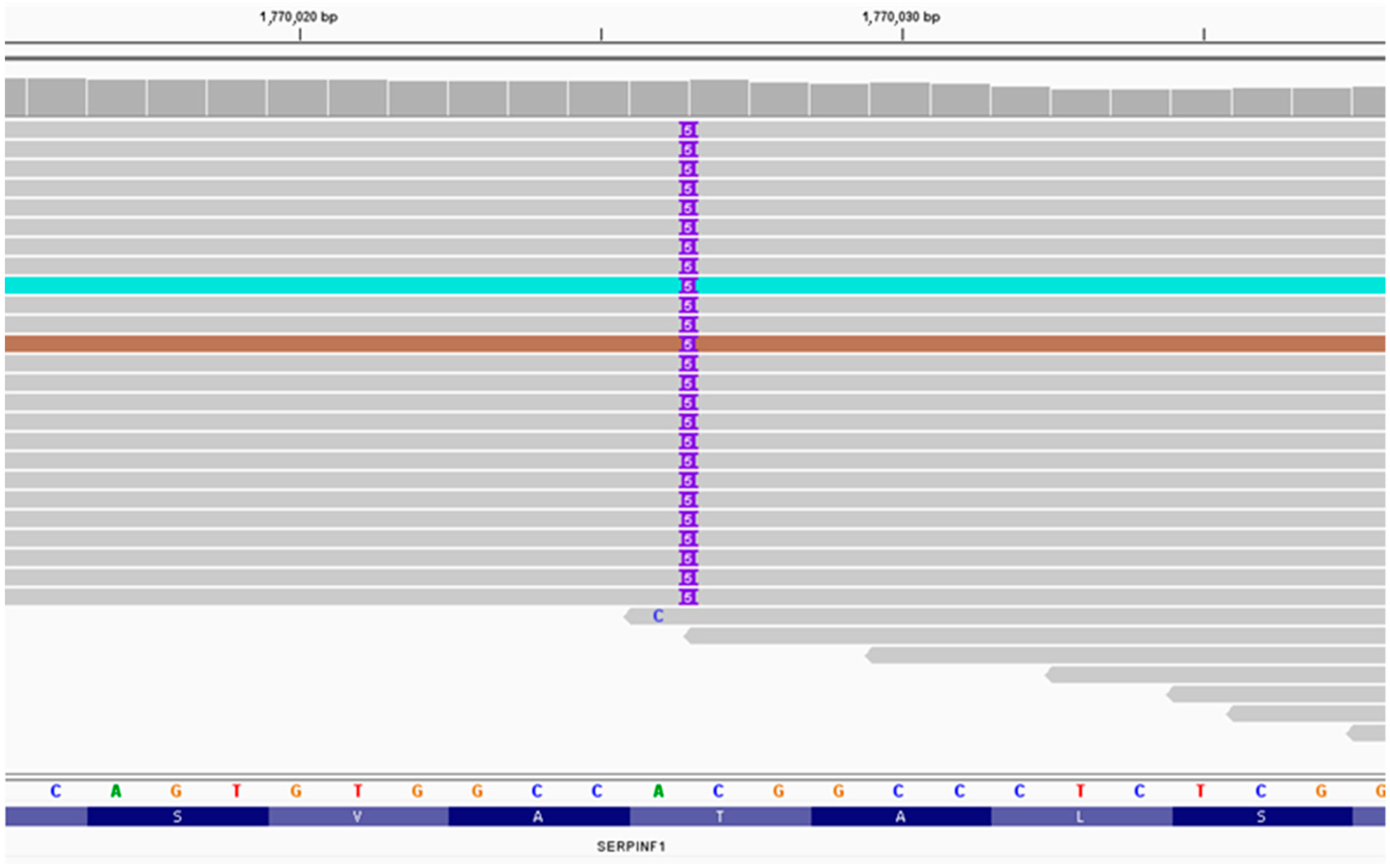
3.1. Massively Parallel Sequencing
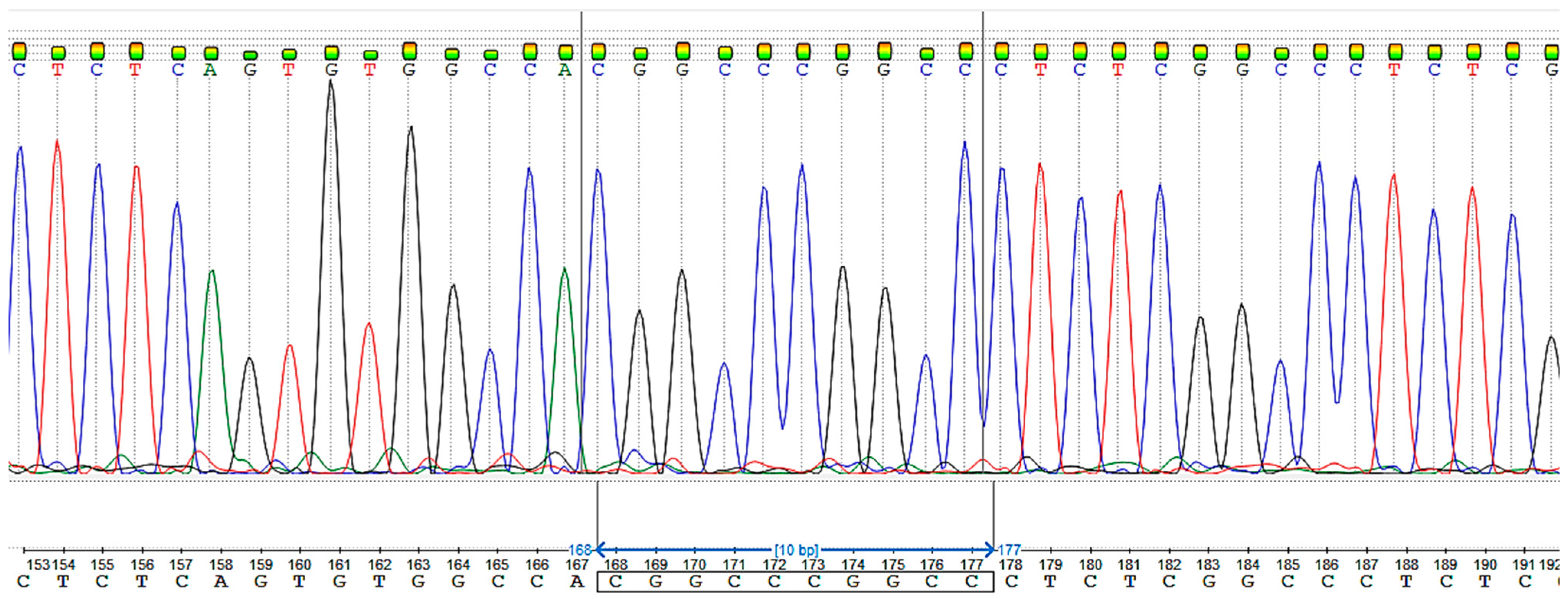
3.2. Sanger Sequencing
3.3. Likelihood Ratio (LR) for Relative Pairs Using Autosomal STRs
4. Discussion
5. Materials and Methods
5.1. Massively Parallel Sequencing and Sanger Sequencing
5.2. Calculating the Degree of Relationships of the Proband’s Parents
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, F.H.; Rauch, F.; Plotkin, H.; Ward, L.; Travers, R.; Roughley, P.; Lalic, L.; Glorieux, D.F.; Fassier, F.O.; Bishop, N.J. Type V Osteogenesis Imperfecta: A New Form of Brittle Bone Disease*. J. Bone Miner. Res. 1997, 12, 1650–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, J.C.; Do, A.N.D. Osteogenesis Imperfecta; MDText.com, Inc.: Dartmouth, MA, USA, 2020. [Google Scholar]
- Li, R.; Li, H.; Peng, D.; Hao, B.; Wang, Z.; Huang, E.; Wu, R.; Sun, H. Improved pairwise kinship analysis using massively parallel sequencing. Forensic Sci. Int. Genet. 2019, 38, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, F.H.; Ward, L.M.; Rauch, F.; Lalic, L.; Roughley, P.J.; Travers, R. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002, 17, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.; Semler, O.; Gilissen, C.; Li, Y.; Bolz, H.J.; Giunta, C.; Bergmann, C.; Rohrbach, M.; Koerber, F.; Zimmermann, K.; et al. Exome Sequencing Identifies Truncating Mutations in Human SERPINF1 in Autosomal-Recessive Osteogenesis Imperfecta. Am. J. Hum. Genet. 2011, 88, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, E.P.; Rauch, F.; Grafe, I.; Lietman, C.; Doll, J.A.; Dawson, B.; Bertin, T.; Napierala, D.; Morello, R.; Gibbs, R.; et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J. Bone Miner. Res. 2011, 26, 2798–2803. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Martin, J.A.; Valencia, M.; Pulido, V.; Martínez-Glez, V.; Rueda-Arenas, I.; Amr, K.; Farra, C.; Lapunzina, P.; Ruiz-Perez, V.L.; Temtamy, S.; et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. Part A 2013, 161, 1354–1369. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Ki, C.-S.; Sohn, Y.B.; Kim, S.J.; Maeng, S.H.; Jin, D.-K. Osteogenesis Imperfecta Type VI with Severe Bony Deformities Caused by Novel Compound Heterozygous Mutations in SERPINF1. J. Korean Med. Sci. 2013, 28, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturi, G.; Gandini, A.; Monti, E.; Carbonare, L.D.; Corradi, M.; Vincenzi, M.; Valenti, M.T.; Valli, M.; Pelilli, E.; Boner, A.; et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type i collagen. J. Bone Miner. Res. 2012, 27, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, N.; Tombran-Tink, J.; Niyibizi, C. Pigment Epithelium-Derived Factor Enhances Differentiation and Mineral Deposition of Human Mesenchymal Stem Cells. Stem Cells 2013, 31, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Semler, O.; Netzer, C.; Hoyer-Kuhn, H.; Becker, J.; Eysel, P.; Schoenau, E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J. Musculoskelet. Neuronal Interact. 2012, 12, 183–188. [Google Scholar] [PubMed]
- Stepanov, V.A.; Kolesnikov, N.A.; Valikhova, L.V.; Zarubin, A.A.; Khitrinskaya, I.Y.; Kharkov, V.N. Structure and origin of Tuvan gene poolaccording to autosome SNP and Y-chromosome haplogroups. Vavilov J. Genet. Breed. 2023, 27, 36. [Google Scholar] [CrossRef] [PubMed]
- Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes 2020, 11, 833. [Google Scholar] [CrossRef] [PubMed]
- Klimov, L.Y.; Vdovina, T.M.; Pechenkina, V.A.; Zhelezniakova, T.V.; Zakharova, I.N.; Dolbnya, S.V.; Kuryaninova, V.A.; Kastarnov, A.V.; Shmalko, D.A.; Tsutsaev, R.O.; et al. Osteogenesis imperfecta: A literature review and a clinical case of a perinatal-lethal type of disease. In Meditsinskiy Sovet; Remedium Group Ltd.: Singapore, 2021; pp. 226–234. [Google Scholar] [CrossRef]
- Puzyrev, V.P.; Erdynieva, L.S.; Kucher, A.N.; Nazarenko, L.P. Genetiko-Epidemiologicheskoe Issledovanie Naseleniya Tuvy (Genetic Epidemiological Study of the Tuva Population); STT: Tomsk, Russia, 1999. [Google Scholar]
- Kling, D.; Tillmar, A.O.; Egeland, T. Familias 3—Extensions and new functionality. Forensic Sci. Int. Genet. 2014, 13, 121–127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Person 1 | Person 2 | Relationship | LR | IBS = 2 | IBS = 1 | IBS = 0 |
|---|---|---|---|---|---|---|
| TKT150 | TKT151 | Parent–child | 50,281,500 | 27.27% | 72.73% | 0% |
| TKT150 | TKT151 | Siblings | 4,092,930 | 27.27% | 72.73% | 0% |
| TKT72 | TKT73 | Siblings | 271,411 | 40.91% | 36.36% | 22.73% |
| TKT150 | TKT151 | Cousins | 2166.26 | 27.27% | 72.73% | 0% |
| TKT81 | TKT141 | Parent–child | 317.711 | 4.55% | 95.45% | 0% |
| TKT18 | TKT142 | Cousins | 2.26667 | 18.18% | 45.45% | 36.36% |
| TKT152 | TKT178 | Cousins | 2.26382 | 9.09% | 50% | 40.91% |
| D6262 | D6263 | Cousins | 2.26355 | 15% | 55% | 30% |
| TKT50 | TKT143 | Cousins | 2.25966 | 9.09% | 54.55% | 36.36% |
| TKT68 | TKT76 | Cousins | 2.2494 | 18.18% | 40.91% | 40.91% |
| TKT138 | TKT182 | Cousins | 2.24883 | 18.18% | 63.64% | 18.18% |
| TKT171 | TKT177 | Second cousins | 2.24573 | 4.55% | 54.55% | 40.91% |
| D6262 | D6263 | Second cousins | 1.45411 | 15% | 55% | 30% |
| D6262 | D6263 | Half-siblings | 1.1542 | 15% | 55% | 30% |
| D6262 | D6263 | Direct match | 2.7 × 10−16 | 15% | 55% | 30% |
| D6262 | D6263 | Parent–child | 0.0 | 15% | 55% | 30% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhalsanova, I.Z.; Postrigan, A.E.; Valiakhmetov, N.R.; Kolesnikov, N.A.; Zhigalina, D.I.; Zarubin, A.A.; Petrova, V.V.; Minaycheva, L.I.; Seitova, G.N.; Skryabin, N.A.; et al. Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI. Int. J. Mol. Sci. 2023, 24, 6672. https://doi.org/10.3390/ijms24076672
Zhalsanova IZ, Postrigan AE, Valiakhmetov NR, Kolesnikov NA, Zhigalina DI, Zarubin AA, Petrova VV, Minaycheva LI, Seitova GN, Skryabin NA, et al. Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI. International Journal of Molecular Sciences. 2023; 24(7):6672. https://doi.org/10.3390/ijms24076672
Chicago/Turabian StyleZhalsanova, Irina Zh., Anna Evgenievna Postrigan, Nail Raushanovich Valiakhmetov, Nikita Aleksandrovich Kolesnikov, Daria Ivanovna Zhigalina, Aleksei Andreevich Zarubin, Valeria Viktorovna Petrova, Larisa Ivanovna Minaycheva, Gulnara Narimanovna Seitova, Nikolay Alekseevich Skryabin, and et al. 2023. "Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI" International Journal of Molecular Sciences 24, no. 7: 6672. https://doi.org/10.3390/ijms24076672
APA StyleZhalsanova, I. Z., Postrigan, A. E., Valiakhmetov, N. R., Kolesnikov, N. A., Zhigalina, D. I., Zarubin, A. A., Petrova, V. V., Minaycheva, L. I., Seitova, G. N., Skryabin, N. A., & Stepanov, V. A. (2023). Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI. International Journal of Molecular Sciences, 24(7), 6672. https://doi.org/10.3390/ijms24076672