
Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent




Abstract
:1. Introduction
2. What Is the Ethacrynic Acid?
3. Effects of ECA on Cancer Hallmarks
3.1. ECA Suppresses Proliferation and Growth of Tumor
3.2. ECA Induces Cell Death
3.3. Effects of ECA on Replicative Immortality
3.4. ECA Increases the Efficiency of Tumor Suppressors
3.5. Effects of ECA on Genome Instability and Mutation
3.6. ECA and Deregulating Cellular Energetics
3.7. ECA Impedes Metastasis and Invasion
3.8. ECA Hinders Angiogenesis
3.9. ECA and Immune Evasion
3.10. Effects of ECA on Tumor-Associated Inflammation
3.11. Effects of ECA on Tumor-Related Neural Input
4. Mechanism of Ethacrynic Acid on Cancer
4.1. Blocking of the Ion Channels
4.2. Inhibition of the Wnt Signaling Pathway
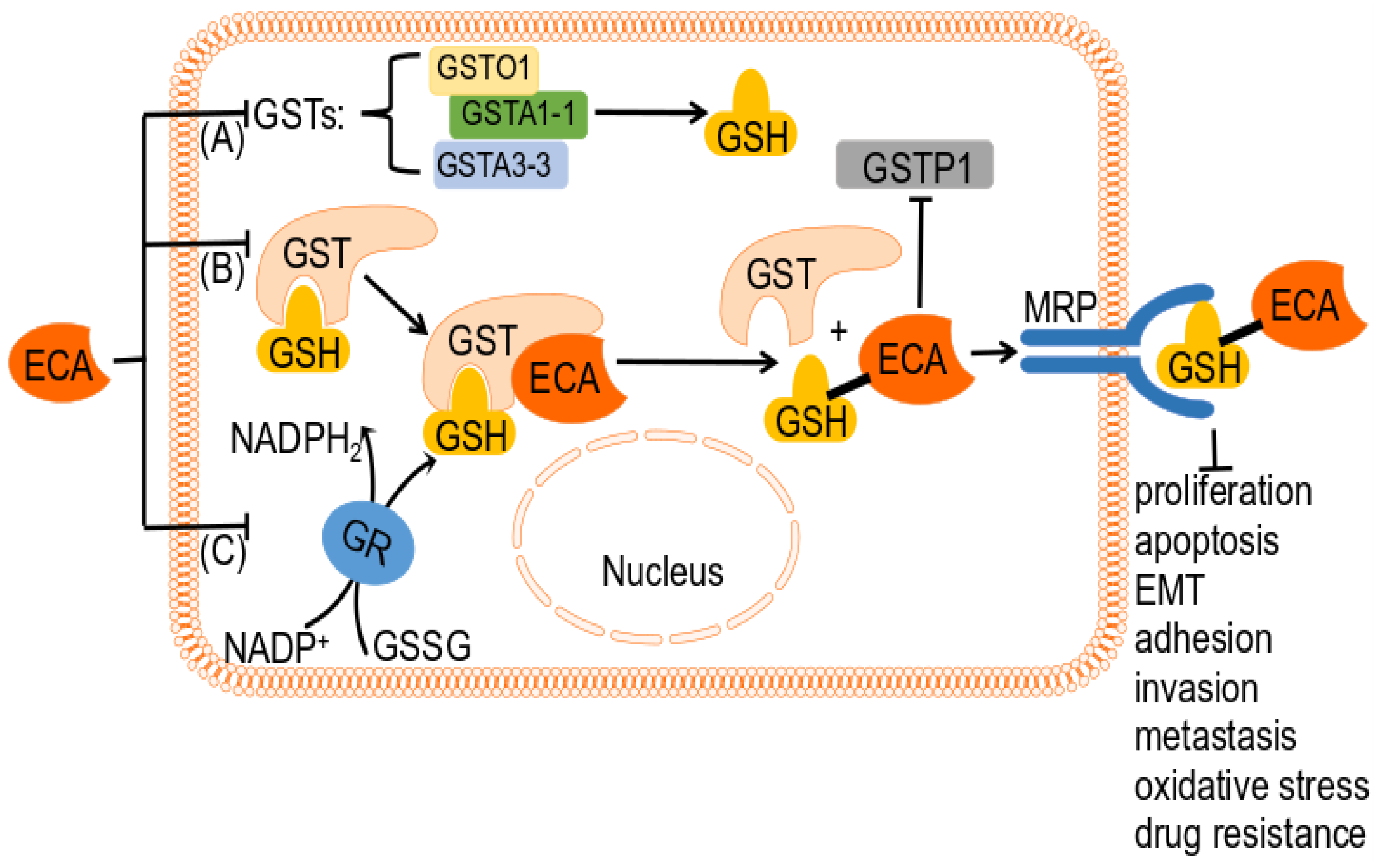
4.3. ECA as a GSH-Related Enzyme Inhibitor
4.4. Inhibition of the NF-κB Signaling Pathway
4.5. MAPK Signaling Pathway Inhibition
4.6. Inhibition of STAT3 and HIF-1 Signaling Pathways
4.7. NOTCH Signaling Pathway Inhibition and Induction of Oxidative Stress Pathway
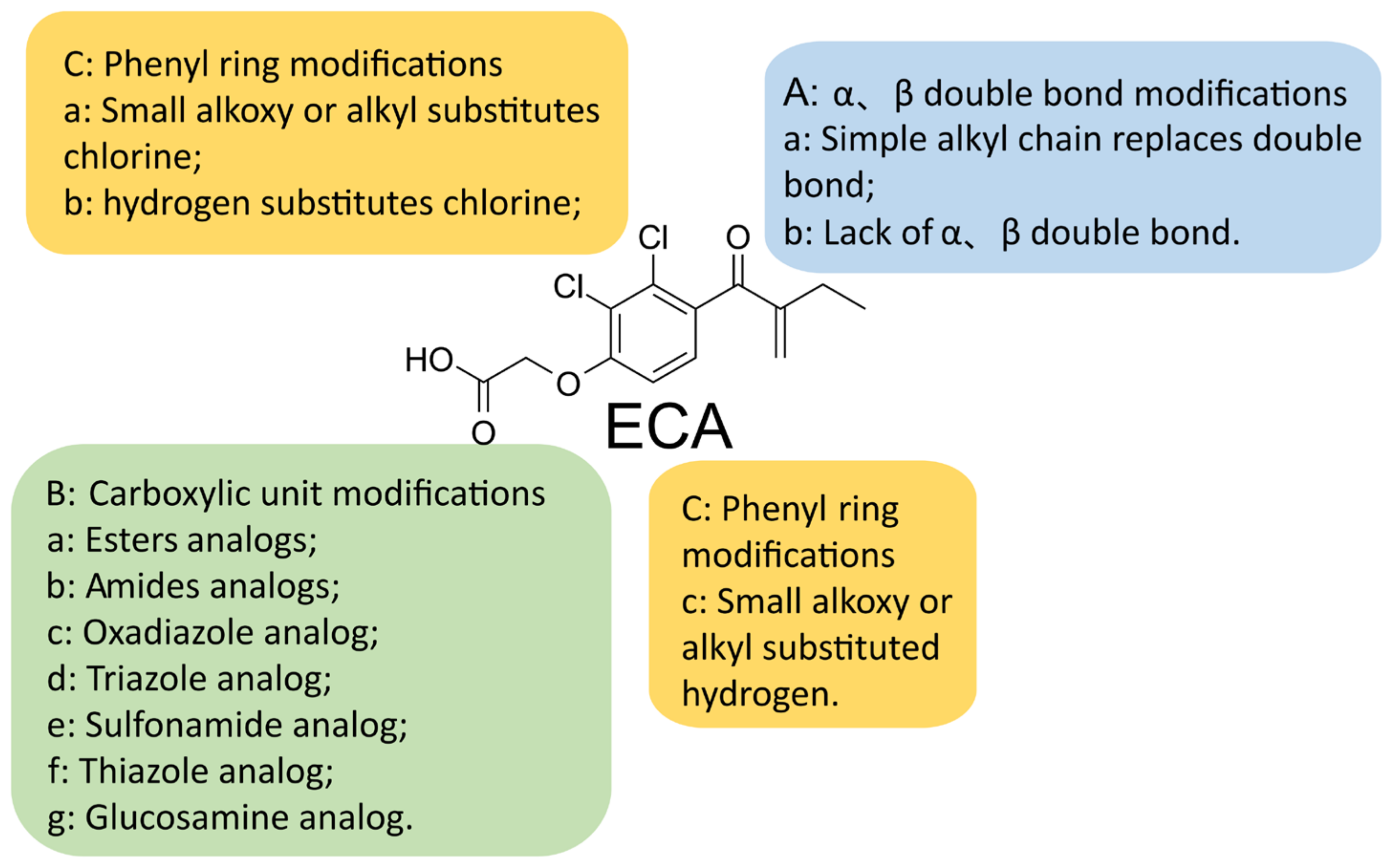
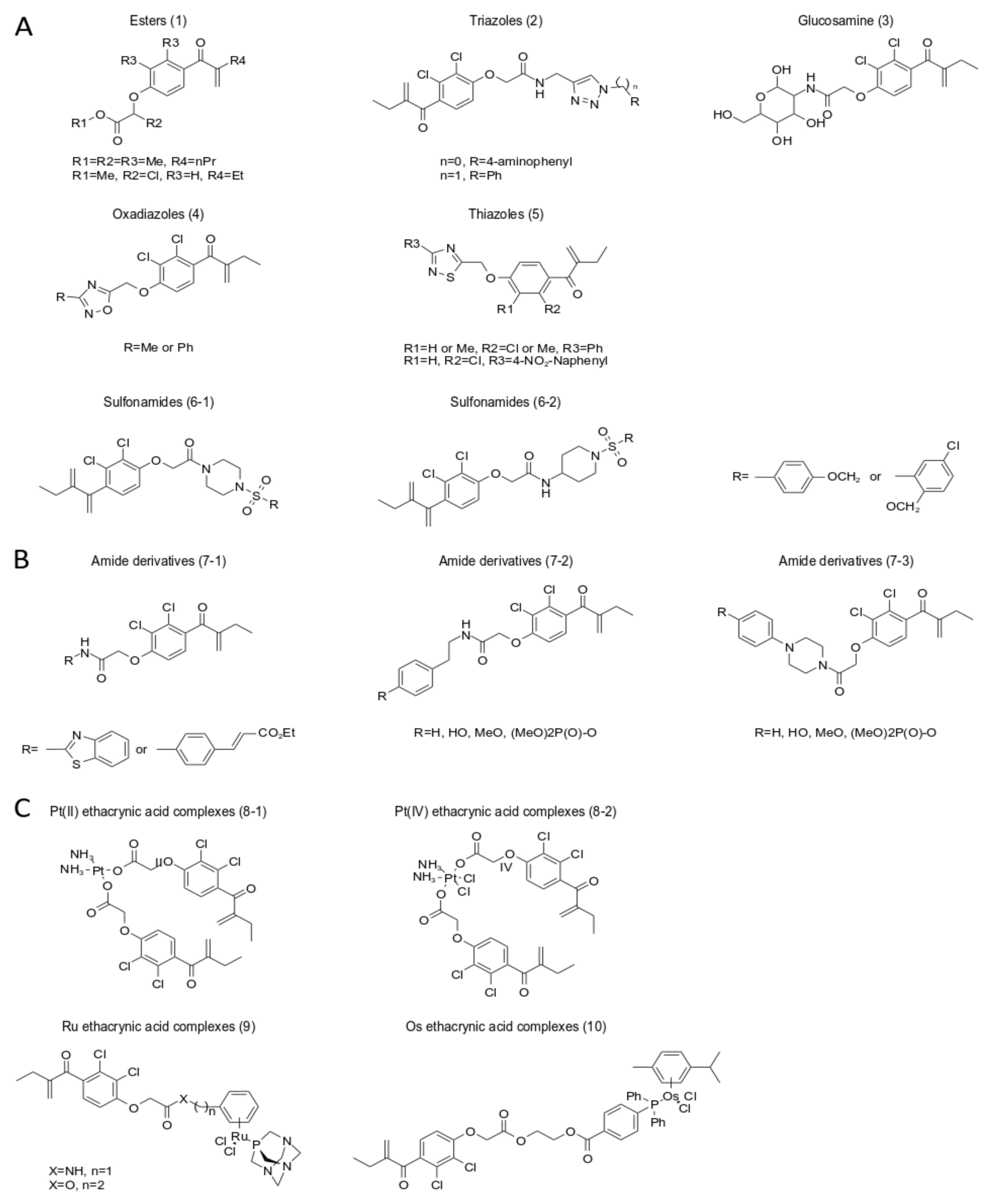
5. A New Analog of Ethacrynic Acid
6. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Stein, C.J.; Colditz, G.A. Modifiable risk factors for cancer. Br. J. Cancer 2004, 90, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Cho, J.; Lee, K. Tumour regression via integrative regulation of neurological, inflammatory, and hypoxic tumour microenvironment. Biomol. Ther. 2020, 28, 119. [Google Scholar] [CrossRef]
- Chen, W.; Sun, K.; Zheng, R.; Zeng, H.; Zhang, S.; Xia, C.; Yang, Z.; Li, H.; Zou, X.; He, J. Cancer incidence and mortality in China, 2014. Chin. J. Cancer Res. 2018, 30, 1. [Google Scholar] [CrossRef]
- Luengo-Fernandez, R.; Leal, J.; Gray, A.; Sullivan, R. Economic burden of cancer across the European Union: A population-based cost analysis. Lancet Oncol. 2013, 14, 1165–1174. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Lund, J.; Kepka, D.; Mariotto, A. Economic burden of cancer in the United States: Estimates, projections, and future research. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Won, M.; Koo, S.; Li, H.; Sessler, J.L.; Lee, J.Y.; Sharma, A.; Kim, J.S. An ethacrynic acid-brominated BODIPY photosensitizer (EA-BPS) construct enhances the lethality of reactive oxygen species in hypoxic tumor-targeted photodynamic therapy. Angew. Chem. Int. Ed. 2021, 60, 3196–3204. [Google Scholar] [CrossRef]
- Yenigül, M.; Akçok, İ.; Gencer Akçok, E.B. Ethacrynic acid and cinnamic acid combination exhibits selective anticancer effects on K562 chronic myeloid leukemia cells. Mol. Biol. Rep. 2022, 49, 7521–7530. [Google Scholar] [CrossRef]
- Yu, L.; Kim, H.J.; Park, M.K.; Byun, H.J.; Kim, E.J.; Kim, B.; Nguyen, M.T.; Kim, J.H.; Kang, G.J.; Lee, H. Ethacrynic acid, a loop diuretic, suppresses epithelial-mesenchymal transition of A549 lung cancer cells via blocking of NDP-induced WNT signaling. Biochem. Pharmacol. 2021, 183, 114339. [Google Scholar] [CrossRef]
- Beyer, K.H.; Baer, J.E.; Michaelson, J.K.; Russo, H.F. Renotropic characteristics of ethacrynic acid: A phenoxyacetic saluretic-diuretic agent. J. Pharmacol. Exp. Ther. 1965, 147, 1–22. [Google Scholar]
- Goldberg, M.; McCurdy, D.K.; Foltz, E.L.; Bluemle, L.W. Effects of ethacrynic acid (a new saluretic agent) on renal diluting and concentrating mechanisms: Evidence for site of action in the loop of Henle. J. Clin. Investig. 1964, 43, 201–216. [Google Scholar] [CrossRef]
- Molnar, J.; Somberg, J.C. The clinical pharmacology of ethacrynic acid. Am. J. Ther. 2009, 16, 86–92. [Google Scholar] [CrossRef]
- Schröder, G.; Sannerstedt, R.; Werkö, L. Clinical Experiences with Ethacrynic Acid, a New Non-Thiazide Saluretic Agent (MK-595). Acta Med. Scand. 1964, 175, 781–786. [Google Scholar] [CrossRef]
- Wang, R.; Li, C.; Song, D.; Zhao, G.; Zhao, L.; Jing, Y. Ethacrynic acid butyl-ester induces apoptosis in leukemia cells through a hydrogen peroxide–mediated pathway independent of glutathione S-transferase P1-1 inhibition. Cancer Res. 2007, 67, 7856–7864. [Google Scholar] [CrossRef] [Green Version]
- Lacreta, F.P.; Brennan, J.M.; Nash, S.L.; Comis, R.L.; Tew, K.D.; O’Dwyer, P.J. Pharmakokinetics and bioavailability study of ethacrynic acid as a modulator of drug resistance in patients with cancer. J. Pharmacol. Exp. Ther. 1994, 270, 1186–1191. [Google Scholar]
- Dormans, T.P.; Pickkers, P.; Russel, F.G.; Smits, P. Vascular effects of loop diuretics. Cardiovasc. Res. 1996, 32, 988–997. [Google Scholar] [CrossRef]
- Somberg, J.C.; Molnar, J. The pleiotropic effects of ethacrynic acid. Am. J. Ther. 2009, 16, 102–104. [Google Scholar] [CrossRef]
- Datey, K.; Deshmukh, S.; Dalvi, C.; Purandare, N. Hepatocellular damage with ethacrynic acid. Br. Med. J. 1967, 3, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Liu, H.; Qi, W.; Jiang, H.; Li, Y.; Wu, X.; Sun, H.; Gross, K.; Salvi, R. Ototoxic effects and mechanisms of loop diuretics. J. Otol. 2016, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Prazma, J.; Thomas, W.; Fischer, N.; Preslar, M.J. Ototoxicity of the ethacrynic acid. Arch. Otolaryngol. 1972, 95, 448–456. [Google Scholar] [CrossRef]
- Ding, D.; Jiang, H.; Salvi, R.J. Mechanisms of rapid sensory hair-cell death following co-administration of gentamicin and ethacrynic acid. Hear. Res. 2010, 259, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Jiang, H.; Wang, P.; Salvi, R. Cell death after co-administration of cisplatin and ethacrynic acid. Hear. Res. 2007, 226, 129–139. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Emekwue, N.P.C.; Weiher, H.; Schmidt-Wolf, I.G. Ethacrynic Acid and Ciclopiroxolamine in Various Cancer Cells. Int. J. Hematol. Ther. 2018, 4, 16–24. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yutaka, S.; Shinya, F.; Yasuhiko, F.; Toshio, K. Antiproliferative effects of glutathione S-transferase inhibitors on the K562 cell line. Biochem. Pharmacol. 1990, 39, 1263–1266. [Google Scholar] [CrossRef]
- Liu, B.; Huang, X.; Hu, Y.; Chen, T.; Peng, B.; Gao, N.; Jin, Z.; Jia, T.; Zhang, N.; Wang, Z. Ethacrynic acid improves the antitumor effects of irreversible epidermal growth factor receptor tyrosine kinase inhibitors in breast cancer. Oncotarget 2016, 7, 58038. [Google Scholar] [CrossRef] [Green Version]
- Wall, I.; Schmidt-Wolf, I.G. Effect of Wnt inhibitors in pancreatic cancer. Anticancer Res. 2014, 34, 5375–5380. [Google Scholar]
- Lee, Y.-J.; Song, H.; Yoon, Y.J.; Park, S.-J.; Kim, S.-Y.; Han, D.C.; Kwon, B.-M. Ethacrynic acid inhibits STAT3 activity through the modulation of SHP2 and PTP1B tyrosine phosphatases in DU145 prostate carcinoma cells. Biochem. Pharmacol. 2020, 175, 113920. [Google Scholar] [CrossRef]
- Manupati, K.; Debnath, S.; Goswami, K.; Bhoj, P.S.; Chandak, H.S.; Bahekar, S.P.; Das, A. Glutathione S-transferase omega 1 inhibition activates JNK-mediated apoptotic response in breast cancer stem cells. FEBS J. 2019, 286, 2167–2192. [Google Scholar] [CrossRef]
- Khil, M.S.; Kim, S.H.; Pinto, J.T.; Kim, J.H. Ethacrynic acid: A novel radiation enhancer in human carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 1996, 34, 375–380. [Google Scholar] [CrossRef]
- Rayn, K.N.; Hale, G.R.; Grave, G.P.; Agarwal, P.K. New therapies in nonmuscle invasive bladder cancer treatment. Indian J. Urol. 2018, 34, 11–19. [Google Scholar] [CrossRef]
- Schmidt, M.; Kim, Y.; Gast, S.M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G. Increased in vivo efficacy of lenalidomide and thalidomide by addition of ethacrynic acid. In Vivo 2011, 25, 325–333. [Google Scholar]
- Aizawa, S.; Ookawa, K.; Kudo, T.; Asano, J.; Hayakari, M.; Tsuchida, S. Characterization of cell death induced by ethacrynic acid in a human colon cancer cell line DLD-1 and suppression by N-acetyl-L-cysteine. Cancer Sci. 2003, 94, 886–893. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, C.; Cui, B.; Pang, Y.; Liang, R.; Luo, X. Ethacrynic Acid Enhances the Antitumor Effects of Afatinib in EGFR/T790M-Mutated NSCLC by Inhibiting WNT/Beta-Catenin Pathway Activation. Dis. Markers 2021, 2021, 1–17. [Google Scholar] [CrossRef]
- Lu, D.; Liu, J.X.; Endo, T.; Zhou, H.; Yao, S.; Willert, K.; Schmidt-Wolf, I.G.; Kipps, T.J.; Carson, D.A. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/β-catenin pathway. PLoS ONE 2009, 4, e8294. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, X.; Zhu, Y.; Song, T.; Chen, X.; Lei, X.; Wang, C. Chemoproteomic profiling reveals ethacrynic acid targets adenine nucleotide translocases to impair mitochondrial function. Mol. Pharm. 2018, 15, 2413–2422. [Google Scholar] [CrossRef]
- Yang, Q.; Xiao, H.; Cai, J.; Xie, Z.; Wang, Z.; Jing, X. Nanoparticle mediated delivery of a GST inhibitor ethacrynic acid for sensitizing platinum based chemotherapy. RSC Adv. 2014, 4, 61124–61132. [Google Scholar] [CrossRef]
- Al-Dali, A.M.; Weiher, H.; Schmidt-Wolf, I.G. Utilizing ethacrynic acid and ciclopirox olamine in liver cancer. Oncol. Lett. 2018, 16, 6854–6860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeel, L.C.; Schmeel, F.C.; Kim, Y.; Endo, T.; Lu, D.; Schmidt-Wolf, I.G. Targeting the Wnt/beta-catenin pathway in multiple myeloma. Anticancer. Res. 2013, 33, 4719–4726. [Google Scholar] [PubMed]
- Von Schulz-Hausmann, S.A.; Schmeel, L.C.; Schmeel, F.C.; Schmidt-Wolf, I.G. Targeting the Wnt/beta-catenin pathway in renal cell carcinoma. Anticancer Res. 2014, 34, 4101–4108. [Google Scholar] [PubMed]
- Kim, Y.; Gast, S.M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G. In vivo efficacy of the diuretic agent ethacrynic acid against multiple myeloma. Leuk. Res. 2012, 36, 598–600. [Google Scholar] [CrossRef]
- Muhammad, Y.; Kerr, S.G. Effects of alpha, beta-unsaturated cinnamyl-like aromatic compounds on the viability of a model human cancer (MCF-7) cell line. FASEB J. 2013, 27, lb637. [Google Scholar] [CrossRef]
- Niu, B.; Zhou, Y.; Liao, K.; Wen, T.; Lao, S.; Quan, G.; Xin, P.; Wu, C. “Pincer movement”: Reversing cisplatin resistance based on simultaneous glutathione depletion and glutathione S-transferases inhibition by redox-responsive degradable organosilica hybrid nanoparticles. Acta Pharmaceutica Sin. B. 2022, 12, 2074–2088. [Google Scholar]
- Huang, L.; Xie, L.; Qiu, Y.; Hu, P.; Ye, X. Ethacrynic acid promotes apoptosis in lung cancer A549 cells when combined with cisplatin chemotherapy. J. Third Mil. Med. Univ. 2017, 1720–1727. [Google Scholar]
- Kakizaki, I.; Ookawa, K.; Ishikawa, T.; Hayakari, M.; Aoyagi, T.; Tsuchida, S. Induction of apoptosis and cell cycle arrest in mouse colon 26 cells by benastatin A. Jpn. J. Cancer Res. 2000, 91, 1161–1168. [Google Scholar] [CrossRef]
- Wang, R.; Liu, C.; Xia, L.; Zhao, G.; Gabrilove, J.; Waxman, S.; Jing, Y. Ethacrynic Acid and a Derivative Enhance Apoptosis in Arsenic Trioxide–Treated Myeloid Leukemia and Lymphoma Cells: The Role of Glutathione S-Transferase P1-1Apoptosis Induction by Arsenic Trioxide plus Ethacrynic Acid. Clin. Cancer Res. 2012, 18, 6690–6701. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, J.; Soldner, F.; Schulz, J.; Klockgether, T.; Kovar, K.; Wüllner, U. Differential effects of L-buthionine sulfoximine and ethacrynic acid on glutathione levels and mitochondrial function in PC12 cells. Neurosci. Lett. 1999, 264, 1–4. [Google Scholar] [CrossRef]
- Rizzardini, M.; Lupi, M.; Bernasconi, S.; Mangolini, A.; Cantoni, L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J. Neurol. Sci. 2003, 207, 51–58. [Google Scholar] [CrossRef]
- Buseman, C.; Wright, W.; Shay, J. Is telomerase a viable target in cancer? Mutat. Res./Fundam. Mol. Mech. Mutagen. 2012, 730, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xie, L.Y.; Allan, S.; Beach, D.; Hannon, G.J. Myc activates telomerase. Genes Dev. 1998, 12, 1769–1774. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ou, C.C.; Feldman, R.I.; Nicosia, S.V.; Kruk, P.A.; Cheng, J.Q. Aurora-A kinase regulates telomerase activity through c-Myc in human ovarian and breast epithelial cells. Cancer Res. 2004, 64, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Khattar, E.; Tergaonkar, V. Transcriptional regulation of telomerase reverse transcriptase (TERT) by MYC. Front. Cell Dev. Biol. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-De Matte, M.Y.; Moses-Soto, H.; Kruk, P.A. Calcium-mediated telomerase activity in ovarian epithelial cells. Arch. Biochem. Biophys. 2002, 399, 239–244. [Google Scholar] [CrossRef]
- Hong, B.; van den Heuvel, P.J.; V Prabhu, V.; Zhang, S.; S El-Deiry, W. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef]
- Somasundaram, K.; El-Deiry, W.S. Tumor suppressor p53: Regulation and function. Front. Biosci. Landmark 2000, 5, 424–437. [Google Scholar] [CrossRef] [Green Version]
- Ward, W.M.; Hoffman, J.D.; Loo, G. Genotoxic effect of ethacrynic acid and impact of antioxidants. Toxicol. Appl. Pharmacol. 2015, 286, 17–26. [Google Scholar] [CrossRef]
- Yusuf, M.A.; Srivenugopal, K.S. GSTP1 expression in human cancers decreases p53 protein levels and markedly attenuates p53-dependent responses to therapy-induced DNA damage. Cancer Res. 2011, 71, 218. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punganuru, S.R.; Madala, H.R.; Srivenugopal, K.S. Induction of mild oxidative stress as a strategy for reactivation of mutant p53 proteins: KSS72, a small molecule derived from ethacrynic acid restores the biological functions of R248W/Q mutant. Cancer Res. 2017, 77, 2570. [Google Scholar] [CrossRef]
- Maximchik, P.; Kulikov, A.; Zhivotovsky, B.; Gogvadze, V. Cellular energetics as a target for tumor cell elimination. Biochemistry 2016, 81, 65–79. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726. [Google Scholar] [CrossRef] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Gordon, E.E.; de Hartog, M. The relationship between cell membrane potassium ion transport and glycolysis: The effect of ethacrynic acid. J. Gen. Physiol. 1969, 54, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Kusakari, J.; Ise, I.; Comegys, T.; Thalmann, I.; Thalmann, R. Effect of ethacrynic acid, furosemide, and ouabain upon the endolymphatic potential and upon high energy phosphates of the stria vascularis. Laryngoscope 1978, 88, 12–37. [Google Scholar] [CrossRef]
- Soltys, B.J.; Gupta, R.S. Presence and cellular distribution of a 60-kDa protein related to mitochondrial hsp60 in Giardia lamblia. J. Parasitol. 1994, 80, 580–590. [Google Scholar] [CrossRef]
- Gordon, E.E. Site of ethacrynic acid action on Ehrlich ascites tumor cells. Biochem. Pharmacol. 1968, 17, 1237–1242. [Google Scholar] [CrossRef]
- Huang, J.; Philbert, M.A. Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res. 1996, 711, 184–192. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.J.; Kang, K.J.; Park, M.K.; Lee, H.J.; Kang, J.H.; Lee, E.J.; Kim, Y.R.; Kim, H.J.; Kim, Y.W.; Jung, K.C. Ethacrynic acid inhibits sphingosylphosphorylcholine-induced keratin 8 phosphorylation and reorganization via transglutaminase-2 inhibition. Biomol. Ther. 2013, 21, 338. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, F.-L.; Jia, W. Glutathione S-transferase ω 1 promotes the proliferation, migration and invasion, and inhibits the apoptosis of non-small cell lung cancer cells, via the JAK/STAT3 signaling pathway. Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Bryant, Z.E.; Janser, R.F.; Jabarkhail, M.; Candelaria-Lyons, M.S.; Romero, B.B.; Steelant, W.F.; Janser, I. Inhibitory effects of ethacrynic acid analogues lacking the α, β-unsaturated carbonyl unit and para-acylated phenols on human cancer cells. Bioorganic Med. Chem. Lett. 2011, 21, 912–915. [Google Scholar] [CrossRef] [Green Version]
- Janser, R.F.; Meka, R.K.; Bryant, Z.E.; Adogla, E.A.; Vogel, E.K.; Wharton, J.L.; Tilley, C.M.; Kaminski, C.N.; Ferrey, S.L.; Steelant, W.F. Ethacrynic acid analogues lacking the α, β-unsaturated carbonyl unit—Potential anti-metastatic drugs. Bioorganic Med. Chem. Lett. 2010, 20, 1848–1850. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, M.; Wang, L.; Jiao, B. HIFs, angiogenesis, and cancer. J. Cell. Biochem. 2013, 114, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.-R.; Han, K.-C.; Park, H.; Yang, E.G. Menadione and ethacrynic acid inhibit the hypoxia-inducible factor (HIF) pathway by disrupting HIF-1α interaction with p300. Biochem. Biophys. Res. Commun. 2013, 434, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Vanhecke, E.; Saule, P.; Mougel, A.; Page, A.; Romon, R.; Nurcombe, V.; Le Bourhis, X.; Hondermarck, H. Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res. 2008, 68, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Romon, R.; Adriaenssens, E.; Lagadec, C.; Germain, E.; Hondermarck, H.; Le Bourhis, X. Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Mol. Cancer 2010, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How cancers escape immune destruction and mechanisms of action for the new significantly active immune therapies: Helping nonimmunologists decipher recent advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.S. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 35, pp. S185–S198. [Google Scholar]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wei, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Han, Y.; Englert, J.A.; Delude, R.L.; Fink, M.P. Ethacrynic acid inhibits multiple steps in the NF-κB signaling pathway. Shock 2005, 23, 45–53. [Google Scholar] [CrossRef]
- Wang, M.; Gorasiya, S.; Antoine, D.J.; Sitapara, R.A.; Wu, W.; Sharma, L.; Yang, H.; Ashby, C.R., Jr.; Vasudevan, D.; Zur, M. The compromise of macrophage functions by hyperoxia is attenuated by ethacrynic acid via inhibition of NF-κB–mediated release of high-mobility group box-1. Am. J. Respir. Cell Mol. Biol. 2015, 52, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Thornton, A.M.; Kinney, M.C.; Ma, C.A.; Spinner, J.J.; Fuss, I.J.; Shevach, E.M.; Jain, A. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor β (TGF-β) signaling and the development of regulatory T cells. J. Biol. Chem. 2011, 286, 40520–40530. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhu, H.; Fu, Y.; Shen, W.; Miao, K.; Hong, M.; Xu, W.; Fan, L.; Young, K.H.; Liu, P. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget 2016, 7, 21631. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Killeen, M.E.; Englert, J.A.; Stolz, D.B.; Song, M.; Han, Y.; Delude, R.L.; Kellum, J.A.; Fink, M.P. The phase 2 enzyme inducers ethacrynic acid, DL-sulforaphane, and oltipraz inhibit lipopolysaccharide-induced high-mobility group box 1 secretion by RAW 264.7 cells. J. Pharmacol. Exp. Ther. 2006, 316, 1070–1079. [Google Scholar] [CrossRef]
- Harada, T.; Fink, M.; Cruz, R.J., Jr. Ethacrynic acid decreases expression of proinflammatory intestinal wall cytokines and ameliorates gastrointestinal stasis in murine postoperative ileus. Clinics 2018, 73, e332. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, Q.; Lu, Z.; Wang, L.; Ding, L.; Xia, L.; Zhang, H.; Wang, M.; Chen, Y.; Li, G. Role of the nervous system in cancers: A review. Cell Death Discov. 2021, 7, 76. [Google Scholar] [CrossRef]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascal, D. Tumor progression: The neuronal input. Ann. Transl. Med. 2018, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Entschladen, F.; Drell, T.L.; Lang, K.; Joseph, J.; Zaenker, K.S. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004, 5, 254–258. [Google Scholar] [CrossRef]
- Shiroya, T.; Fukunaga, R.; Akashi, K.; Shimada, N.; Takagi, Y.; Nishino, T.; Hara, M.; Inagaki, C. An ATP-driven Cl− pump in the brain. J. Biol. Chem. 1989, 264, 17416–17421. [Google Scholar] [CrossRef]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Hirose, T.; Fukai, Y.; Zeng, X.-T.; Yasukura, T.; Ohnishi, S.; Uriu, T.; Inagaki, C. Ethacrynic acid-induced convulsions and brain noradrenaline in mice. Eur. J. Pharmacol. 1990, 179, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Hirose, T.; Inagaki, C. Ethacrynic acid-induced glutamate release from mouse brain synaptosomes. Brain Res. 1991, 543, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, J.; Omori, K.; Okugawa, G.; Fujiseki, Y.; Heizmann, C.; Inagaki, C. Long-lasting c-fos and NGF mRNA expressions and loss of perikaryal parvalbumin immunoreactivity in the development of epileptogenesis after ethacrynic acid-induced seizure. Brain Res. 1999, 834, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Knipper, M.; Leung, L.; Zhao, D.; Rylett, R. Short-term modulation of glutamatergic synapses in adult rat hippocampus by NGF. Neuroreport 1994, 5, 2433–2436. [Google Scholar] [CrossRef]
- Ding, Y.; Hao, K.; Li, Z.; Ma, R.; Zhou, Y.; Zhou, Z.; Wei, M.; Liao, Y.; Dai, Y.; Yang, Y. C-Fos separation from Lamin A/C by GDF15 promotes colon cancer invasion and metastasis in inflammatory microenvironment. J. Cell. Physiol. 2020, 235, 4407–4421. [Google Scholar] [CrossRef]
- Muhammad, N.; Bhattacharya, S.; Steele, R.; Phillips, N.; Ray, R.B. Involvement of c-Fos in the Promotion of Cancer Stem-like Cell Properties in Head and Neck Squamous Cell Carcinomac-Fos in the Enhancement of Cancer Stem-like Properties. Clin. Cancer Res. 2017, 23, 3120–3128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, D.; McQuade, J.S.; Behbehani, M.; Tsien, J.Z.; Xu, M. C-fos regulates neuronal excitability and survival. Nat. Genet. 2002, 30, 416–420. [Google Scholar] [CrossRef]
- Fiske, J.L.; Fomin, V.P.; Brown, M.L.; Duncan, R.L.; Sikes, R.A. Voltage-sensitive ion channels and cancer. Cancer Metastasis Rev. 2006, 25, 493–500. [Google Scholar] [CrossRef]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef] [Green Version]
- Schönherr, R. Clinical relevance of ion channels for diagnosis and therapy of cancer. J. Membr. Biol. 2005, 205, 175–184. [Google Scholar] [CrossRef]
- Shiozaki, A.; Miyazaki, H.; Niisato, N.; Nakahari, T.; Iwasaki, Y.; Itoi, H.; Ueda, Y.; Yamagishi, H.; Marunaka, Y. Furosemide, a blocker of Na+/K+/2Cl− cotransporter, diminishes proliferation of poorly differentiated human gastric cancer cells by affecting G0/G1 state. J. Physiol. Sci. 2006, 56, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-T.; Nagaba, Y.; Cross, H.S.; Wrba, F.; Zhang, L.; Guggino, S.E. The mRNA of L-type calcium channel elevated in colon cancer: Protein distribution in normal and cancerous colon. Am. J. Pathol. 2000, 157, 1549–1562. [Google Scholar] [CrossRef]
- Panet, R.; Marcus, M.; Atlan, H. Overexpression of the Na+/K+/Cl− cotransporter gene induces cell proliferation and phenotypic transformation in mouse fibroblasts. J. Cell. Physiol. 2000, 182, 109–118. [Google Scholar] [CrossRef]
- Katnik, C.; Cuevas, J. Loop Diuretics Inhibit Ischemia-Induced Intracellular Ca2+ Overload in Neurons via the Inhibition of Voltage-Gated Ca2+ and Na+ Channels. Front. Pharmacol. 2021, 12, 732922. [Google Scholar] [CrossRef]
- Zhao, X.-X.; Chen, W.-W.; Chen, Y.-Y.; Liu, M.-S.; Li, M.-Y.; Cao, L.; Liu, Q.-H. Ethacrynic acid inhibits airway smooth muscle contraction in mice. Sheng Li Xue Bao 2019, 71, 863–873. [Google Scholar]
- Taketo, M.M. Shutting down Wnt signal–activated cancer. Nat. Genet. 2004, 36, 320–322. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Nakayama, S.; Sng, N.; Carretero, J.; Welner, R.; Hayashi, Y.; Yamamoto, M.; Tan, A.J.; Yamaguchi, N.; Yasuda, H.; Li, D. β-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 2014, 74, 5891–5902. [Google Scholar] [CrossRef] [Green Version]
- Giles, R.H.; Van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta Rev. Cancer 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon: Functions and mechanisms of β-catenin-independent Wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.; Shaw, H.V.; Koval, A.; Katanaev, V.L. A second WNT for old drugs: Drug repositioning against WNT-dependent cancers. Cancers 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-H.; Hsieh, F.-L.; Zebisch, M.; Harlos, K.; Elegheert, J.; Jones, E.Y. Structure and functional properties of Norrin mimic Wnt for signalling with Frizzled4, Lrp5/6, and proteoglycan. Elife 2015, 4, e06554. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Harikumar, K.G.; Erice, C.; Chen, C.; Gu, X.; Wang, L.; Parker, N.; Cheng, Z.; Xu, W.; Williams, B.O. Structure and function of Norrin in assembly and activation of a Frizzled 4–Lrp5/6 complex. Genes Dev. 2013, 27, 2305–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389. [Google Scholar]
- Wang, Z.; Zhao, T.; Zhang, S.; Wang, J.; Chen, Y.; Zhao, H.; Yang, Y.; Shi, S.; Chen, Q.; Liu, K. The Wnt signaling pathway in tumorigenesis, pharmacological targets, and drug development for cancer therapy. Biomarker Res. 2021, 9, 1–16. [Google Scholar] [CrossRef]
- Lyou, Y.; Habowski, A.N.; Chen, G.T.; Waterman, M.L. Inhibition of nuclear Wnt signalling: Challenges of an elusive target for cancer therapy. Br. J. Pharmacol. 2017, 174, 4589–4599. [Google Scholar] [CrossRef] [Green Version]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Cazenave, L.A.; Moscow, J.A.; Myers, C.E.; Cowan, K.H. Glutathione S—Transferase and drug resistance. Drug Resist. Cancer Ther. 1989, 48, 171–187. [Google Scholar]
- Sau, A.; Tregno, F.P.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Agonigi, G.; Riedel, T.; Gay, M.P.; Biancalana, L.; Oñate, E.; Dyson, P.J.; Pampaloni, G.; Paunescu, E.; Esteruelas, M.A.; Marchetti, F. Arene osmium complexes with ethacrynic acid-modified ligands: Synthesis, characterization, and evaluation of intracellular glutathione S-transferase inhibition and antiproliferative activity. Organometallics 2016, 35, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Ruzza, P.; Rosato, A.; Rossi, C.R.; Floreani, M.; Quintieri, L. Glutathione transferases as targets for cancer therapy. Anti Cancer Agents Med. Chem. 2009, 9, 763–777. [Google Scholar] [CrossRef]
- Musdal, Y.; Hegazy, U.M.; Aksoy, Y.; Mannervik, B. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1-1. Chem. Biol. Interact. 2013, 205, 53–62. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; LaCreta, F.; Nash, S.; Tinsley, P.W.; Schilder, R.; Clapper, M.L.; Tew, K.D.; Panting, L.; Litwin, S.; Comis, R.L. Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic acid. Cancer Res. 1991, 51, 6059–6065. [Google Scholar]
- Hoffman, D.W.; Wiebkin, P.; Rybak, L.P. Inhibition of glutathione-related enzymes and cytotoxicity of ethacrynic acid and cyclosporine. Biochem. Pharmacol. 1995, 49, 411–415. [Google Scholar] [CrossRef]
- Shen, H.; Ranganathan, S.; Kuzmich, S.; Tew, K.D. Influence of ethacrynic acid on glutathione S-transferase π transcript and protein half-lives in human colon cancer cells. Biochem. Pharmacol. 1995, 50, 1233–1238. [Google Scholar] [CrossRef]
- Luo, W.; Kinsey, M.; Schiffman, J.D.; Lessnick, S.L. Glutathione S-transferases in pediatric cancer. Front. Oncol. 2011, 1, 39. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Findlay, V.L.; Tew, K.D. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005, 401, 287–307. [Google Scholar]
- Caffrey, P.B.; Zhu, M.; Zhang, Y.; Chinen, N.; Frenkel, G.D. Rapid development of glutathione-S-transferase-dependent drug resistance in vitro and its prevention by ethacrynic acid. Cancer Lett. 1999, 136, 47–52. [Google Scholar] [CrossRef]
- Smith, M.T.; Evans, C.G.; Doane-Setzer, P.; Castro, V.M.; Tahir, M.K.; Mannervik, B. Denitrosation of 1, 3-bis (2-chloroethyl)-1-nitrosourea by class mu glutathione transferases and its role in cellular resistance in rat brain tumor cells. Cancer Res. 1989, 49, 2621–2625. [Google Scholar]
- Rhodes, T.; Twentyman, P. A study of ethacrynic acid as a potential modifier of melphalan and cisplatin sensitivity in human lung cancer parental and drug-resistant cell lines. Br. J. Cancer 1992, 65, 684–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploemen, J.; Van Schanke, A.; Van Ommen, B.; Van Bladeren, P. Reversible conjugation of ethacrynic acid with glutathione and human glutathione S-transferase P1-1. Cancer Res. 1994, 54, 915–919. [Google Scholar] [PubMed]
- Zaman, G.J.; Cnubben, N.H.; van Bladeren, P.J.; Evers, R.; Borst, P. Transport of the glutathione conjugate of ethacrynic acid by the human multidrug resistance protein MRP. FEBS Lett. 1996, 391, 126–130. [Google Scholar] [CrossRef] [Green Version]
- van Iersel, M.L.; van Lipzig, M.M.; Rietjens, I.M.; Vervoort, J.; van Bladeren, P.J. GSTP1-1 stereospecifically catalyzes glutathione conjugation of ethacrynic acid. FEBS Lett. 1998, 441, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G. Interactions of glutathione S-transferase-π with ethacrynic acid and its glutathione conjugate. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1993, 1164, 173–178. [Google Scholar] [CrossRef]
- Ploemen, J.H.; van Ommen, B.; van Bladeren, P.J. Inhibition of rat and human glutathione S-transferase isoenzymes by ethacrynic acid and its glutathione conjugate. Biochem. Pharmacol. 1990, 40, 1631–1635. [Google Scholar] [CrossRef]
- Oakley, A.J.; Rossjohn, J.; Lo Bello, M.; Caccuri, A.M.; Federici, G.; Parker, M.W. The three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic acid and its glutathione conjugate. Biochemistry 1997, 36, 576–585. [Google Scholar] [CrossRef]
- Cameron, A.D.; Sinning, I.; L’Hermite, G.; Olin, B.; Board, P.G.; Mannervik, B.; Jones, T.A. Structural analysis of human alpha-class glutathione transferase A1-1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugate. Structure 1995, 3, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.-D.; Yang, Z.-P.; Tang, Q.-L.; Peng, T.; Zhang, Z.-B.; Zhou, S.-G.; Wang, G.-X.; He, B.; Zang, L.-Q. Expression and function of GSTA1 in lung cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 8631–8635. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, F.; Wang, C.; Wang, C.; Tang, Y.; Jiang, Z. Glutathione S-transferase A1 mediates nicotine-induced lung cancer cell metastasis by promoting epithelial-mesenchymal transition. Exp. Ther. Med. 2017, 14, 1783–1788. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Patrick, B.; Li, J.; Sharma, R.; Jeyabal, P.V.; Reddy, P.M.; Awasthi, S.; Awasthi, Y.C. Glutathione S-transferases as antioxidant enzymes: Small cell lung cancer (H69) cells transfected with hGSTA1 resist doxorubicin-induced apoptosis. Arch. Biochem. Biophys. 2006, 452, 165–173. [Google Scholar] [CrossRef]
- Srivastava, R.; Sharma, R.; Mishra, S. Human Glutathione S-Transferase A1-1 binding with naturally occurring ligands: Assessment by docking simulations. Int. J. Pharm. 2013, 1, 457–464. [Google Scholar]
- Kumar, M.; Martin, A.; Nirgude, S.; Chaudhary, B.; Mondal, S.; Sarkar, A. Quinacrine inhibits GSTA1 activity and induces apoptosis through G1/S arrest and generation of ROS in human non-small cell lung cancer cell lines. Oncotarget 2020, 11, 1603. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Peng, B.; Shao, Q.; Qian, W.; Zhang, J.-Y. Identification of glutathione S-transferase omega 1 (GSTO1) protein as a novel tumor-associated antigen and its autoantibody in human esophageal squamous cell carcinoma. Tumor Biol. 2014, 35, 10871–10877. [Google Scholar] [CrossRef]
- Yan, X.-D.; Pan, L.-Y.; Yuan, Y.; Lang, J.-h.; Mao, N. Identification of platinum-resistance associated proteins through proteomic analysis of human ovarian cancer cells and their platinum-resistant sublines. J. Proteome Res. 2007, 6, 772–780. [Google Scholar] [CrossRef]
- Piaggi, S.; Raggi, C.; Corti, A.; Pitzalis, E.; Mascherpa, M.C.; Saviozzi, M.; Pompella, A.; Casini, A.F. Glutathione transferase omega 1-1 (GSTO1-1) plays an anti-apoptotic role in cell resistance to cisplatin toxicity. Carcinogenesis 2010, 31, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Waxman, D.J. Role of cellular glutathione and glutathione S-transferase in the expression of alkylating agent cytotoxicity in human breast cancer cells. Biochem. Pharmacol. 1994, 47, 1079–1087. [Google Scholar] [CrossRef]
- Drozd, E.; Gruber, B.; Marczewska, J.; Drozd, J.; Anuszewska, E. Intracellular glutathione level and efflux in human melanoma and cervical cancer cells differing in doxorubicin resistance. Adv. Hyg. Exp. Med. 2016, 70, 319–328. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: Glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J. Biol. Chem. 2006, 281, 29542–29557. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Stokes, W.; Chater, E.; Roy, R.; De Bruin, E.; Hu, Y.; Liu, Z.; Smit, E.F.; Heynen, G.J.; Downward, J. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell Discov. 2016, 2, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tew, K.D.; Gaté, L. Glutathione S-transferases as emerging therapeutic targets. Expert Opin. Ther. Targets 2001, 5, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Nagourney, R.A.; Messenger, J.C.; Kern, D.H.; Weisenthal, L.M. Enhancement of anthracycline and alkylator cytotoxicity by ethacrynic acid in primary cultures of human tissues. Cancer Chemother. Pharmacol. 1990, 26, 318–322. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Kruh, G.D.; Tew, K.D. The influence of coordinate overexpression of glutathione phase II detoxification gene products on drug resistance. J. Pharmacol. Exp. Ther. 2000, 294, 480–487. [Google Scholar] [PubMed]
- Grant, C.E.; Valdimarsson, G.; Hipfner, D.R.; Almquist, K.C.; Cole, S.P.; Deeley, R.G. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res. 1994, 54, 357–361. [Google Scholar]
- Ciaccio, P.J.; Shen, H.; Kruh, G.D.; Tew, K.D. Effects of chronic ethacrynic acid exposure on glutathione conjugation and MRP expression in human colon tumor cells. Biochem. Biophys. Res. Commun. 1996, 222, 111–115. [Google Scholar] [CrossRef]
- Omata, Y.; Saito, Y.; Fujita, K.; Ogawa, Y.; Nishio, K.; Yoshida, Y.; Niki, E. Induction of adaptive response and enhancement of PC12 cell tolerance by lipopolysaccharide primarily through the upregulation of glutathione S-transferase A3 via Nrf2 activation. Free Radic. Biol. Med. 2008, 45, 1437–1445. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.-S.; Choi, D.-H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.-Y. Transglutaminase 2 induces nuclear factor-κB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K.; Lee, C.H. Role of sphingosylphosphorylcholine in tumor and tumor microenvironment. Cancers 2019, 11, 1696. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.I.; McGregor, L.M.; Jain, T.; Liu, D.R. Discovery of a covalent kinase inhibitor from a DNA-encoded small-molecule library× protein library selection. J. Am. Chem. Soc. 2017, 139, 10192–10195. [Google Scholar] [CrossRef]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in cancer immunotherapy. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Tamagnone, L.; Zacchigna, S.; Rehman, M. Taming the Notch transcriptional regulator for cancer therapy. Molecules 2018, 23, 431. [Google Scholar] [CrossRef] [Green Version]
- Hynes, N.E.; Stoelzle, T. Key signalling nodes in mammary gland development and cancer: Myc. Breast Cancer Res. 2009, 11, 1–9. [Google Scholar] [CrossRef]
- Lee, K.G.Z.; Babak, M.V.; Weiss, A.; Dyson, P.J.; Nowak-Sliwinska, P.; Montagner, D.; Ang, W.H. Development of an efficient dual-action GST-inhibiting anticancer platinum (IV) prodrug. ChemMedChem 2018, 13, 1210–1217. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Furfaro, A.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.; Pronzato, M.; Nitti, M. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxidative Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.P.; Hayashi, T.; Cottam, H.B.; Jin, G.; Yao, S.; Wu, C.C.; Rosenbach, M.D.; Corr, M.; Schwab, R.B.; Carson, D.A. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 7479–7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caminade, A.-M. Phosphorus dendrimers as nanotools against cancers. Molecules 2020, 25, 3333. [Google Scholar] [CrossRef] [PubMed]
- Mignani, S.; El Brahmi, N.; El Kazzouli, S.; Eloy, L.; Courilleau, D.; Caron, J.; Bousmina, M.M.; Caminade, A.-M.; Cresteil, T.; Majoral, J.-P. A novel class of ethacrynic acid derivatives as promising drug-like potent generation of anticancer agents with established mechanism of action. Eur. J. Med. Chem. 2016, 122, 656–673. [Google Scholar] [CrossRef]
- Dong, J.; Yang, D.; Zhao, G. Encouraging Effects of Ethacrynic Acid Derivatives Possessing a Privileged α, β-Unsaturated Carbonyl Structure Scaffold. Med. Chem. 2018, 8, 185–191. [Google Scholar] [CrossRef]
- Dude, M.-A.; Kaeppler, U.; Herb, M.; Schiller, M.; Schulz, F.; Vedder, B.; Heppner, S.; Pradel, G.; Gut, J.; Rosenthal, P.J. Synthesis and evaluation of non-peptidic cysteine protease inhibitors of P. falciparum derived from etacrynic acid. Molecules 2008, 14, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Chen, J.-H.; Dong, X.; Tang, M.-T.; Gao, L.-Y.; Zhao, G.-S.; Yu, L.-G.; Guo, X.-L. 6r, a novel oxadiazole analogue of ethacrynic acid, exhibits antitumor activity both in vitro and in vivo by induction of cell apoptosis and S-phase arrest. Biomed. Pharmacother. 2013, 67, 58–65. [Google Scholar] [CrossRef]
- El Brahmi, N.; Mignani, S.M.; Caron, J.; El Kazzouli, S.; Bousmina, M.M.; Caminade, A.-M.; Cresteil, T.; Majoral, J.-P. Investigations on dendrimer space reveal solid and liquid tumor growth-inhibition by original phosphorus-based dendrimers and the corresponding monomers and dendrons with ethacrynic acid motifs. Nanoscale 2015, 7, 3915–3922. [Google Scholar] [CrossRef]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Ghammaz, H.; El Fahime, E.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Improvement of the Chemical Reactivity of Michael Acceptor of Ethacrynic Acid Correlates with Antiproliferative Activities. Molecules 2023, 28, 910. [Google Scholar] [CrossRef]
- Chiang, L.-W.; Pei, K.; Chen, S.-W.; Huang, H.-L.; Lin, K.-J.; Yen, T.-C.; Yu, C.-S. Combining a solution-phase derived library with in-situ cellular bioassay: Prompt screening of amide-forming minilibraries using MTT assay. Chem. Pharm. Bull. 2009, 57, 714–718. [Google Scholar] [CrossRef] [Green Version]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Synthesis and biological evaluation of ethacrynic acid derivatives bearing sulfonamides as potent anti-cancer agents. Bioorganic Med. Chem. Lett. 2020, 30, 127426. [Google Scholar] [CrossRef]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Synthesis and evaluation of a novel class of ethacrynic acid derivatives containing triazoles as potent anticancer agents. Bioorganic Chem. 2021, 115, 105293. [Google Scholar] [CrossRef]
- Li, T.; Liu, G.; Li, H.; Yang, X.; Jing, Y.; Zhao, G. The synthesis of ethacrynic acid thiazole derivatives as glutathione S-transferase pi inhibitors. Bioorganic Med. Chem. 2012, 20, 2316–2322. [Google Scholar] [CrossRef]
- Liu, G.; Wang, R.; Wang, Y.; Li, P.; Zhao, G.; Zhao, L.; Jing, Y. Ethacrynic Acid Oxadiazole Analogs Induce Apoptosis in Malignant Hematologic Cells through Downregulation of Mcl-1 and c-FLIP, Which Was Attenuated by GSTP1-1Apoptosis Induction by Ethacrynic Acid Oxadiazole Analogs. Mol. Cancer Ther. 2013, 12, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Mostofa, A.; Madala, H.R.; Basak, D.; Srivenugopal, K.S. Potent anti-proliferative actions of a non-diuretic glucosamine derivative of ethacrynic acid. Bioorganic Med. Chem. Lett. 2016, 26, 2829–2833. [Google Scholar] [CrossRef]
- Su, Y.-H.; Chiang, L.-W.; Jeng, K.-C.; Huang, H.-L.; Chen, J.-T.; Lin, W.-J.; Huang, C.-W.; Yu, C.-S. Solution-phase parallel synthesis and screening of anti-tumor activities from fenbufen and ethacrynic acid libraries. Bioorganic Med. Chem. Lett. 2011, 21, 1320–1324. [Google Scholar] [CrossRef]
- Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y. Novel oxadiazole analogues derived from ethacrynic acid: Design, synthesis, and structure− activity relationships in inhibiting the activity of glutathione S-transferase P1-1 and cancer cell proliferation. J. Med. Chem. 2010, 53, 1015–1022. [Google Scholar] [CrossRef]
- Zhao, G.; Yu, T.; Wang, R.; Wang, X.; Jing, Y. Synthesis and structure–activity relationship of ethacrynic acid analogues on glutathione-s-transferase P1-1 activity inhibition. Bioorganic Med. Chem. 2005, 13, 4056–4062. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, X. Advance in antitumor agents targeting glutathione-S-transferase. Curr. Med. Chem. 2006, 13, 1461–1471. [Google Scholar] [CrossRef]
- Ploemen, J.H.; Bogaards, J.J.; Veldink, G.A.; van Ommen, B.; Jansen, D.H.; van Bladeren, P.J. Isoenzyme selective irreversible inhibition of rat and human glutathione S-transferases by ethacrynic acid and two brominated derivatives. Biochem. Pharmacol. 1993, 45, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, C.; Jin, S.; Liu, J.; Xue, X.; Eltahan, A.S.; Sun, J.; Tan, J.; Dong, J.; Liang, X.-J. Overcoming resistance to cisplatin by inhibition of glutathione S-transferases (GSTs) with ethacraplatin micelles in vitro and in vivo. Biomaterials 2017, 144, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Madala, H.R.; Punganuru, S.R.; Kalkunte, S. Synthesis of a novel non-diuretic, brain-penetrating, ethacrynic acid analog and demonstration of its potent efficacy in orthotopic glioblastoma (GBM) models. FASEB J. 2017, 31, 178-1. [Google Scholar]
- Daguer, J.P.; Zambaldo, C.; Abegg, D.; Barluenga, S.; Tallant, C.; Müller, S.; Adibekian, A.; Winssinger, N. Identification of covalent bromodomain binders through DNA display of small molecules. Angew. Chem. Int. Ed. 2015, 54, 6057–6061. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Cancer | Cell Lines | The IC50 of ECA (µmol/L)/h * | References |
|---|---|---|---|
| Lung cancer | A549 | 87.03/48 h | [38] |
| A549 | 178/48 h | [39] | |
| H1975 | 99.54/48 h | [38] | |
| Pancreatic cancer | DanG | 67.8/not indicated | [31] |
| PancO2 (mouse) | 141.7/not indicated | [31] | |
| Malignant Melanoma | A375 | 57.26 ± 6.6/not indicated | [40] |
| SK-Mel-28 | 122/48 h | [39] | |
| B16 (mouse) | 201/48 h | [39] | |
| cervical cancer | HeLa | 127.2 ± 13.1/not indicated | [40] |
| breast cancer | MCF7 | 45.53/24 h | [30] |
| MCF7 | 63/48 h | [39] | |
| MCF7 | 340/48 h, 475/72 h | [41] | |
| MDA-MB-231 | 39.64/24 h | [30] | |
| MDA-MB-231 | 42.74 ± 0.93/48 h | [33] | |
| MDA-MB-468 | 39.04 ± 1.12/48 h | [33] | |
| 4T1 (mouse) | 25.23/24 h | [30] | |
| Prostate Cancer | LNCap | 46/48 h | [39] |
| PC3 | 67/48 h | [39] | |
| Colon Cancer | HCT116 | 58/48 h | [39] |
| SW480 | 68/48 h | [39] | |
| HT29 | 56/48 h | [39] | |
| Hepatocellular carcinoma | HepG2 | 223/48 h | [39] |
| HepG2 | 14.8/not indicated | [42] | |
| Hep3B | 6.4/not indicated | [42] | |
| Multiple myeloma | OPM-2 | 22/72 h | [36,43] |
| U266 | 90/48 h | [39] | |
| U266 | 60/72 h | [36,43] | |
| RPMI-8226 | 8/72 h | [36,43] | |
| KMS-18 | 7/not indicated | [36] | |
| Lymphoma | Raji | 33/not indicated | [36] |
| OCI-Ly8 LAM53 | 57/not indicated | [36] | |
| SU-DHL 4 | 58/not indicated | [36] | |
| RAMOS | 174/48 h | [39] | |
| Plasmocytoma | MPC-11 (mouse) | 50/72 h | [36,43] |
| Kidney cancer | A498 | 50/72 h | [44] |
| A704 | 150/72 h | [44] | |
| Caki-2 | 70/72 h | [44] |
| GSTs Classification | Members |
|---|---|
| alpha (A) | GSTA1-GSTA4 |
| kappa (K) | GSTK1 |
| mu (M) | GSTM1-GSTM5 |
| omega (O) | GSTO1 |
| pi (P) | GSTP1 |
| sigma (S) | GSTS1 |
| theta (T) | GSTT1, GSTT2 |
| zeta (Z) | GSTZ1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Lee, H.; Rho, S.B.; Park, M.K.; Lee, C.H. Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent. Int. J. Mol. Sci. 2023, 24, 6712. https://doi.org/10.3390/ijms24076712
Yu L, Lee H, Rho SB, Park MK, Lee CH. Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent. International Journal of Molecular Sciences. 2023; 24(7):6712. https://doi.org/10.3390/ijms24076712
Chicago/Turabian StyleYu, Lu, Ho Lee, Seung Bae Rho, Mi Kyung Park, and Chang Hoon Lee. 2023. "Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent" International Journal of Molecular Sciences 24, no. 7: 6712. https://doi.org/10.3390/ijms24076712
APA StyleYu, L., Lee, H., Rho, S. B., Park, M. K., & Lee, C. H. (2023). Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent. International Journal of Molecular Sciences, 24(7), 6712. https://doi.org/10.3390/ijms24076712