
Crystal Chemistry of the Copper Oxalate Biomineral Moolooite: The First Single-Crystal X-ray Diffraction Studies and Thermal Behavior
 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Infrared Spectroscopy
2.2. Single-Crystal X-ray Diffraction Analysis: Data Processing Details
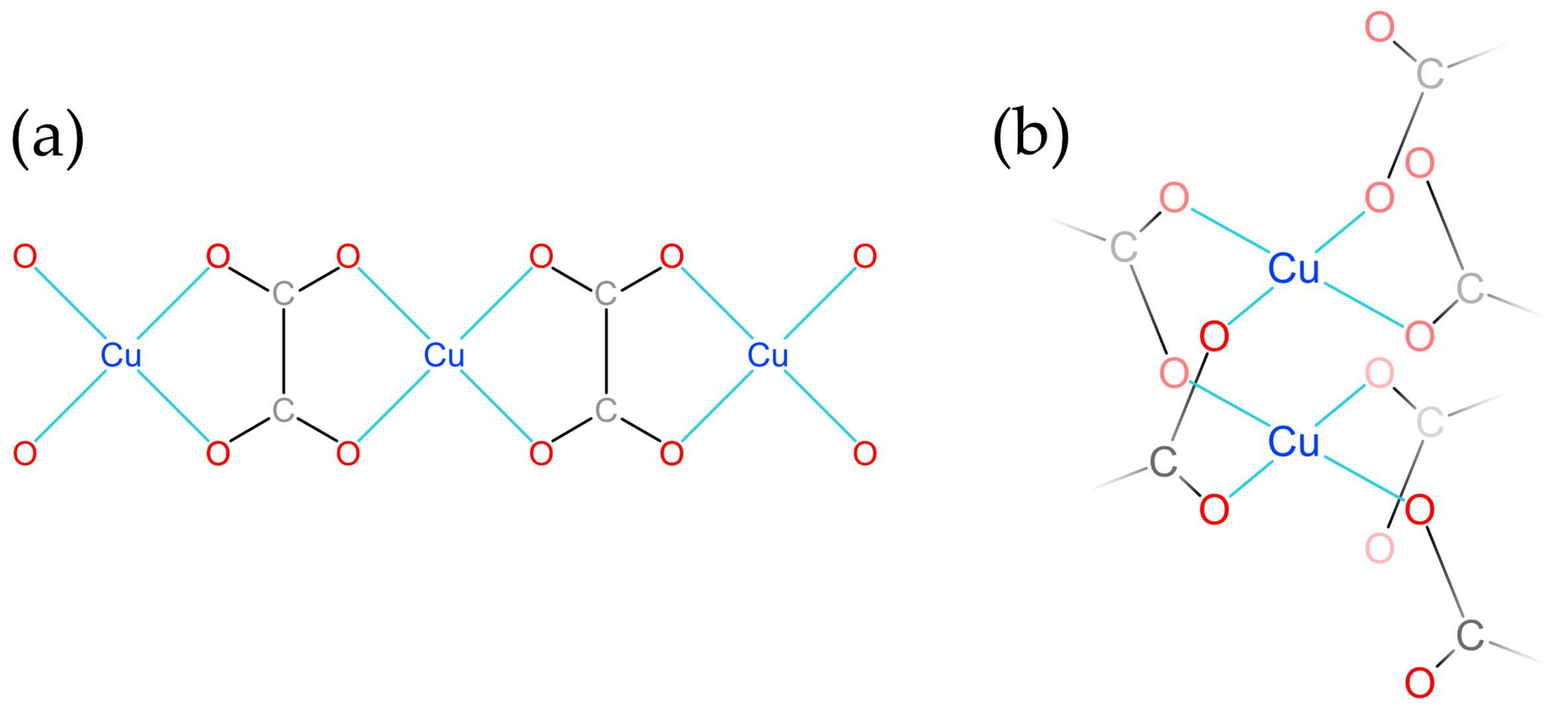
2.3. Crystal Structure Description
2.4. Structural Disorder Modeling Based on the Diffuse Scattering Simulation
- The crystal structure in the OP cell is not suitable for calculations due to its disordered nature. As it was mentioned above, the orthorhombic cell defines the translation vector along c to be equal to ~2.5 Å, which is only half of the shortest distance to maintain an order within the Cu–(C2O4)–Cu chain. Thus, calculations should be conducted on the basis of the monoclinic unit cell with full translation.
- The process of a diffuse scattering simulation demands the availability of a model demonstrating clear and visible disorder with split sites. The availability of the MC4e model satisfies these requirements with one important exception: the Wykoff site of Cu is 4e, which means that the structure contains a single symmetrically nonequivalent Cu site, and thus, the hypothetical ordered model cannot be obtained. On the other hand, the MC2 model shows a clear splitting of the Cu and C positions into two sets, which makes it possible to conduct calculations based on two hypothetically ordered models: the MC2a contains Cu atoms at the vertices and the center of the unit cell (the 2a Wyckoff sites; see Figure 7a), whereas the Cu atoms in the MC2b are arranged at the center of the c edge (0,0,1/2) and in the center of the ab face (1/2,1/2,0) of the unit cell (the 2b Wyckoff sites; see Figure 7b).
- Several features complicating calculations should be taken into account. First of all, both MC2a and MC2b are still based upon diffraction data obtained from the crystal formed by two superimposed “polytypes”, which is expressed in nonconventional bond lengths (see Section 2.3). Secondly, the diffraction intensity of diffuse scattering is too low to establish reasonable intensity vs. hkl correlations (the period of intensity variation). Additionally, the presence of twin domains, as described above, can possibly affect the observed diffuse scattering. Thus, the goal of the calculations is to confirm the origin of the disorder and to simulate the general diffraction pattern.
- The Cu atoms of both models are located at the 2a and 2b Wyckoff sites with sublattices, satisfying the following reflections conditions: h + k + l = 2n and h + l = 2n (on the h0l plane);
- The C atoms are located at 4e Wyckoff sites and have only one meaningful condition: h + l = 2n (on the h0l plane);
- The O atoms are also located at 4e Wyckoff sites with the same condition: h + l = 2n (on the h0l plane).

3. Discussion
3.1. Thermal Behavior
3.2. H2O Content in the Structure of Moolooite
4. Materials and Methods
4.1. Synthesis
4.2. Single-Crystal X-ray Diffraction
4.3. High-Temperature Powder X-ray Diffraction Studies
4.4. Infrared Spectroscopy
4.5. Thermogravimetry Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baran, E.J. Review: Natural oxalates and their analogous synthetic complexes. J. Coord. Chem. 2014, 67, 3734–3768. [Google Scholar] [CrossRef]
- Frank-Kamenetskaya, O.V.; Zelenskaya, M.S.; Izatulina, A.R.; Vereshagin, O.S.; Vlasov, D.Y.; Himelbrant, D.E.; Pankin, D.V. Copper oxalate formation by lichens and fungi. Sci. Rep. 2021, 11, 24239. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetskaya, O.V.; Ivanyuk, G.Y.; Zelenskaya, M.S.; Izatulina, A.R.; Kalashnikov, A.O.; Vlasov, D.Y.; Polyanskaya, E.I. Calcium Oxalates in Lichens on Surface of Apatite-Nepheline Ore (Kola Peninsula, Russia). Minerals 2019, 9, 656. [Google Scholar] [CrossRef] [Green Version]
- Gadd, G.M.; Bahri-Esfahani, J.; Li, Q.; Rhee, Y.J.; Wei, Z.; Fomina, M.; Liang, X. Oxalate production by fungi: Significance in geomycology, biodeterioration and bioremediation. Fungal Biol. Rev. 2014, 28, 36–55. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Pekov, I.V. Moolooite Cu(C2O4)·H2O from the Sarbai deposit—Is the first find in the CIS (The Commonwealth of Independent States). In Materials of the Ural Summer Mineralogical School—96; The Zavaritsky Institute of Geology and Geochemistry: Ekaterinburg, Russia, 1996; pp. 183–185. (In Russian) [Google Scholar]
- Syers, J.K.; Birnie, A.C.; Mitchell, B.B. The calcium oxalate content of some lichens growing on limestone. Lichenologist 1967, 3, 409–414. [Google Scholar] [CrossRef]
- Clarke, R.M.; Williams, I.R. Moolooite, a naturally occurring hydrated copper oxalate from Western Australia. Mineral. Mag. 1986, 50, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Atencio, D.; Coutinho, J.M.V.; Graeser, S.; Matioli, P.A.; Menezes Filho, L.A.D. Lindbergite, a new manganese oxalate dihydrate from Boca Rica mine, Galiléia, Minas Gerais, Brazil, and Parsettens, Oberhalbstein, Switzerland. Amer. Mineral. 2004, 89, 1087–1091. [Google Scholar] [CrossRef]
- Vlasov, D.Y.; Zelenskaya, M.S.; Izatulina, A.R.; Janson, S.Y.; Frank-Kamenetskaya, O.V. Oxalate Crystallization under the Action of Brown Rot Fungi. Crystals 2023, 13, 432. [Google Scholar] [CrossRef]
- Purvis, O.W. The occurrence of copper oxalate in lichens growing on copper sulphide-bearing rocks in Scandinavia. Lichenologist 1984, 16, 197–204. [Google Scholar] [CrossRef]
- Chisholm, J.E.; Jones, G.C.; Purvis, O.W. Hydrated copper oxalate, moolooite in lichens. Mineral. Mag. 1987, 51, 766–803. [Google Scholar] [CrossRef]
- Purvis, O.W.; Pawlik-Skowronska, B.; Cressey, G.; Jones, G.C.; Kearsley, A.; Spratt, J. Mineral phases and elemental composition of the copper hyperaccumulator lichen Lecanora polytropa. Mineral. Mag. 2008, 72, 607–616. [Google Scholar] [CrossRef]
- Vereshchagin, O.S.; Frank-Kamenetskaya, O.V.; Vlasov, D.Y.; Zelenskaya, M.S.; Rodina, O.A.; Chernyshova, I.A.; Himelbrant, D.E.; Stepanchikova, I.S.; Britvin, S.N. Microbial biomineralization under extreme conditions: Case study of basaltic rocks, Tolbachik Volcano, Kamchatka, Russia. Catena 2023, 226, 107048. [Google Scholar] [CrossRef]
- Ren, W.-X.; Li, P.-J.; Geng, Y.; Li, X.-J. Biological leaching of heavy metals from a contaminated soil by Aspergillus niger. J. Hazard. Mater. 2009, 167, 164–169. [Google Scholar] [CrossRef]
- Glukhova, L.B.; Frank, Y.A.; Danilova, E.V.; Avakyan, M.R.; Tuovinen, O.H.; Karnachuk, O.V. Isolation, characterization, and metal response of novel, acid tolerant Penicillium spp. from extremely metal-rich waters at a mining site in Transbaikal (Siberia, Russia). Microb. Ecol. 2018, 76, 911–924. [Google Scholar] [CrossRef] [Green Version]
- Fomina, M.; Hillier, S.; Charnock, J.M.; Melville, K.; Alexander, I.J.; Gadd, G.M. Role of oxalic acid overexcretion in transformations of toxic metal minerals by Beauveria caledonica. Appl. Environ. Microbiol. 2005, 71, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Tsekova, K.; Todorova, D.; Ganeva, S. Removal of heavy metals from industrial wastewater by free and immobilized cells of Aspergillus niger. Int. Biodeterior. Biodegr. 2010, 64, 447–451. [Google Scholar] [CrossRef]
- Frank-Kamenetskaya, O.V.; Zelenskaya, M.S.; Izatulina, A.R.; Gurzhiy, V.V.; Rusakov, A.V.; Vlasov, D.Y. Oxalate formation by Aspergillus niger on minerals of manganese ores. Amer. Mineral. 2022, 107, 100–109. [Google Scholar] [CrossRef]
- Zelenskaya, M.S.; Izatulina, A.R.; Frank-Kamenetskaya, O.V.; Vlasov, D.Y. Iron Oxalate Humboldtine Crystallization by Fungus Aspergillus niger. Crystals 2021, 11, 1591. [Google Scholar] [CrossRef]
- Donkova, B.; Mehandjev, D. Thermal-magnetic investigation of the decomposition of copper oxalate-a precursor for catalysts. J. Mater. Sci. 2005, 40, 3881–3886. [Google Scholar] [CrossRef]
- Behnoudnia, F.; Dehghani, H. Copper(II) oxalate nanospheres and its usage in preparation of Cu(OH)2, Cu2O and CuO nanostructures: Synthesis and growth mechanism. Polyhedron 2013, 56, 102–108. [Google Scholar] [CrossRef]
- Aimable, A.; Puentes, T.; Bowen, P. Synthesis of porous and nanostructured particles of CuO via a copper oxalate route. Powder Technol. 2011, 208, 467–471. [Google Scholar] [CrossRef]
- Wu, J.; Huang, K. Precipitation of flaky moolooite and its thermal decomposition. Int. J. Miner. Metall. Mater. 2016, 23, 976–980. [Google Scholar] [CrossRef]
- Singh, G.; Kapoor, I.P.S.; Dubey, R.; Srivastava, P. Preparation, characterization and catalytic effects of copper oxalate nanocrystals. J. Alloys Comp. 2012, 513, 499–505. [Google Scholar] [CrossRef]
- Dubicki, L.; Harris, C.M.; Kokot, E.; Martin, L. Magnetic Studies with Copper(II) Salts. VII. The Structure of Copper(II) α,ϖ-Dicarboxylates and Their Amine Derivatives. Inorg. Chem. 1966, 5, 93–100. [Google Scholar] [CrossRef]
- Schmittler, H. Zum Strukturprinzip des fehlǵeordneten Kupfer(II)-Oxalats CuC2O4∙nH2O. Monatsber. Dtsch. Akad. Wiss. Berlin 1968, 10, 581–604. [Google Scholar]
- Fichtner-Schmittler, H. On Some Features of X-Ray Powder Patterns of OD Structures. Krist. Tech. 1979, 14, 1079–1088. [Google Scholar] [CrossRef]
- Michalowicz, A.; Girerd, J.J.; Goulon, J. EXAFS Determination of the Copper Oxalate Structure. Relation between Structure and Magnetic Properties. Inorg. Chem. 1979, 18, 3004–3010. [Google Scholar] [CrossRef]
- Fichtner-Schmittler, H. Comments on the Structure of Copper (II) oxalate: Discussion of X-ray Powder Diffraction and EXAFS Results as a Basic for the Interpretation of Magnetic Properties. Cryst. Res. Technol. 1984, 19, 1225–1230. [Google Scholar] [CrossRef]
- Gleizes, A.; Maury, F.; Galy, J. Crystal Structure and Magnetism of Sodium Bis(oxalato)cuprate(II) Dihydrate, Na2Cu(C2O4)2∙2H2O. A Deductive Proposal for the Structure of Copper Oxalate, CuC2O4∙xH2O (0 ≤ x ≤ 1). Inorg. Chem. 1980, 19, 2074–2078. [Google Scholar] [CrossRef]
- Kondrashev, Y.D.; Bogdanov, V.S.; Golubev, S.N.; Pron, G.F. Crystal Structure of the Ordered Phase of Zinc Oxalate and the Structure of Anhydrous Fe2+, Co2+, Ni2+, Cu2+, and Zn2+ oxalates. J. Struct. Chem. 1985, 26, 74–77. [Google Scholar] [CrossRef]
- Jongen, N.; Bowen, P.; Lemaître, J.; Valmalette, J.-C.; Hofmann, H. Precipitation of Self-Organized Copper Oxalate Polycrystalline Particles in the Presence of Hydroxypropylmethylcellulose (HPMC): Control of Morphology. J. Colloid Interface Sci. 2000, 226, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.N.; Lebech, B.; Andersen, N.H.; Grivel, J.-C. The crystal structure of paramagnetic copper(II) oxalate (CuC2O4): Formation and thermal decomposition of randomly stacked anisotropic nano-sized crystallites. Dalton Trans. 2014, 43, 16754–16768. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, H.; Clarke, R.M.; Kimpton, J.A. Synchrotron radiation diffraction study of the mineral moolooite, and synthetic copper oxalates. Powder Diffr. 2019, 34, 21–34. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Version 1.171.41.94a; Rigaku Oxford Diffraction: Oxford, UK, 2021.
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital degeneracy. Proc. R. Soc. A 1937, 161, 220–236. [Google Scholar]
- Hathaway, B.J. A New Look at the Stereochemistry and Electronic Properties of Complexes of the Copper(II) Ion. Struct. Bond. 1984, 57, 56–118. [Google Scholar]
- Halcrow, M.A. Jahn-Teller distortions in transitional metal compounds, and their importance in functional molecular and inorganic materials. Chem. Soc. Rev. 2013, 42, 1784–1795. [Google Scholar] [CrossRef] [Green Version]
- Burns, P.C.; Hawthorne, F.C. Static and Dynamic Jahn-Teller effects in Cu2+ oxysalts minerals. Can. Mineral. 1996, 34, 1089–1105. [Google Scholar]
- Kornyakov, I.V. Synthesis and Crystal Chemistry of Novel Mineral-Related Divalent Copper Compound. PhD Thesis, Saint Petersburg State University, Saint Petersburg, Russia, 2021. [Google Scholar]
- Dewar, M.J.S.; Zheng, Y.-J. Structure of the Oxalate Ion. J. Mol. Struct. THEOCHEM 1990, 209, 157–162. [Google Scholar] [CrossRef]
- Naumov, D.Y.; Podberezskaya, N.V.; Boldyreva, E.V.; Virovets, A.V. Crystal-Chemical Analysis of the Structures of Oxalic Acid and its Salts Mx(C2O4)y∙nH2O (n = 0-3). J. Struct. Chem. 1996, 37, 480–503. [Google Scholar] [CrossRef]
- Izatulina, A.R.; Gurzhiy, V.V.; Krzhizhanovskaya, M.G.; Kuz’mina, M.A.; Leoni, M.; Frank-Kamenetskaya, O.V. Hydrated Calcium Oxalates: Crystal Structures, Thermal Stability, and Phase Evolution. Cryst. Growth Des. 2018, 18, 5465–5478. [Google Scholar] [CrossRef]
- Proffen, T.; Neder, R.B. DISCUS: A program for diffuse scattering and defect-structure simulation. J. Appl. Cryst. 1997, 30, 171–175. [Google Scholar] [CrossRef] [Green Version]
- Neder, R.B.; Proffen, T. Diffuse Scattering and Defect Structure Simulations—A Cook Book Using the Programs DISCUS, 1st ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Diffuse Program Collection. Available online: https://github.com/tproffen/DiffuseCode (accessed on 10 March 2023).
- Welberry, T.R.; Butler, B.D. Interpretation of Diffuse X-ray Scattering via Models of Disorder. J. Appl. Cryst. 1994, 27, 205–231. [Google Scholar] [CrossRef]
- Welberry, T.R. Diffuse Scattering and Models of Disorder, 1st ed.; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Hahn, T. (Ed.) The 230 space groups. In International Tables for Crystallography Volume A: Space-Group Symmetry, 5th ed.; Springer: Dordrecht, The Netherlands, 2002; Volume A, pp. 112–717. [Google Scholar]
- Calos, N.J.; Forrester, J.S.; Schaffer, G.B. A Crystallographic Contribution to the Mechanism of a Mechanically Induced Solid State Reaction. J. Solid State Chem. 1996, 122, 273–280. [Google Scholar] [CrossRef]
- Schmittler, H.; Physikalische Chemie der Akademie der Wissenschaften, Berlin, Germany. Personal communication, 1969.
- Dollimore, D. The Thermal Decomposition of Oxalates. A Review. Thermochim. Acta 1987, 117, 331–363. [Google Scholar] [CrossRef]
- Higashiyama, T.; Hasegawa, S. The Differential Thermal Analysis of Potassium Oxalate. Bull. Chem. Soc. Jpn. 1971, 44, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.K.; Gilligan, J.V.; Jones, S.A. Characterisation And Thermal Analysis Of Vanadium(II) Oxalate. J. Therm. Anal. 1996, 46, 1567–1576. [Google Scholar] [CrossRef]
- Frost, R.L.; Weier, M.L. Thermal treatment of weddellite—A Raman and infrared emission spectroscopic study. Thermochim. Acta 2003, 406, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.L.; Weier, M.L. Thermal treatment of whewellite—A thermal analysis and Raman spectroscopic study. Thermochim. Acta 2004, 409, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.L.; Erickson, K.; Weier, M.L. Thermal treatment of moolooite. A high resolution thermogravimetric and hot stage Raman spectroscopic study. J. Therm. Anal. Calorim. 2004, 77, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Glasser, L. Effective Volumes of Waters of Crystallization: Ionic Systems. Cryst. Growth Des. 2019, 19, 3397–3401. [Google Scholar] [CrossRef]
- Fraser, W. Diffractometers for modern X-ray crystallography: The XtaLAB Synergy X-ray diffractometer platform. Rigaku J. 2020, 36, 37–47. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Visualisation and characterisation of voids in crystalline materials. CrystEngComm 2011, 13, 1804–1813. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Topas 5.0. General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2014. [Google Scholar]
- Langreiter, T.; Kahlenberg, V. TEV—A Program for the Determination of the Thermal Expansion Tensor from Diffraction Data. Crystals 2015, 5, 143–153. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Orthorhombic Setting | Orthorhombic Setting with Twin Domain Accounted for | Monoclinic Setting (MC4e) | Monoclinic Setting (MC2) |
|---|---|---|---|---|
| Crystallographic Data | ||||
| Space Group | Pnnm | Pnnm | P21/n | P21/n |
| a (Å) | 5.601(2) | 5.607(2) | 6.147(3) | 6.111(4) |
| b (Å) | 5.415(3) | 5.428(2) | 5.433(2) | 5.467(3) |
| c (Å) | 2.5553(8) | 2.5579(7) | 5.1173(18) | 5.114(2) |
| β (o) | 90 | 90 | 114.51(5) | 114.68(7) |
| V (Å3) | 77.50(5) | 77.85(5) | 155.51(12) | 155.21(18) |
| Z | 1 | 1 | 2 | 2 |
| Data Collection Parameters | ||||
| Angle range 2θ (o) | 10.476–65.388 | 10.456–65.894 | 8.764–66.906 | 8.778–66.536 |
| Index range | −8 ≤ h ≤ 6, −8 ≤ k ≤ 6, −3 ≤ l ≤ 3 | −8 ≤ h ≤ 8, −8 ≤ k ≤ 6, −3 ≤ l ≤ 3 | −7 ≤ h ≤ 9, −8 ≤ k ≤ 7, −7 ≤ l ≤ 7 | −9 ≤ h ≤ 7, −8 ≤ k ≤ 7, −7 ≤ l ≤ 7 |
| Total reflections | 753 | 382 | 1583 | 1538 |
| Unique reflections | 169 | 382 | 515 | 512 |
| Reflections with F2 > 2σ(F2) | 129 | 316 | 333 | 336 |
| Rint, Rσ (%) | 4.66, 3.30 | N/A 1, 2.17 | 4.71, 5.47 | 6.83, 7.07 |
| Refinement Parameters | ||||
| R1 (F2 > 2σ(F2)), wR2 (F2 > 2σ(F2)) | 0.0726, 0.1854 | 0.0810, 0.2135 | 0.0958, 0.2519 | 0.1325, 0.3690 |
| R1 and wR2 (all data) | 0.0905, 0.2027 | 0.0994, 0.2580 | 0.1334, 0.2706 | 0.1757, 0.3923 |
| BASF | – | 0.66(1):0.33(1) | – | – |
| S | 1.135 | 1.207 | 1.270 | 1.229 |
| ρmax, ρmin (e− Å−3) | 1.59/−0.95 | 1.84/−1.30 | 1.50/−0.60 | 2.00/−1.48 |
| CSD 2 | 2249854 | 2249855 | 2249856 | 2249857 |
| Bond, Å | This Study 1 | Schmittler [26] | Christensen et al. [33] | O’Connor et al. [34] |
|---|---|---|---|---|
| Cu–Oeq | 1.835(6)–1.856(6) | 1.85(8) | 1.918(8), 2.081(7) | 1.827(3) |
| Cu–Oap | 2.512(5)–2.628(5) | 2.54(8) | Not reported | 2.535(5) |
| C-O | 1.378(13)–1.406(14) | 1.37(11) | 1.26(1), 1.27(1) | 1.314(9) |
| C–C | 1.54(3)–1.58(2) | 1.66(8) | 1.576(6) | 1.705(14) |
| T (°C) | α11 | α22 | α33 | <α11a | <α22b | <α33c |
|---|---|---|---|---|---|---|
| 30 | 5.6 | 47.5 | –9.9 | 31.5 | 0 | 172.9 |
| 100 | 19.7 | 48.9 | –4.6 | 28.6 | 0 | 175.7 |
| 150 | 29.8 | 50.0 | –0.8 | 27.6 | 0 | 176.7 |
| 200 | 39.8 | 51.0 | 2.9 | 27.0 | 0 | 177.3 |
| 240 | 47.8 | 51.9 | 5.9 | 26.6 | 0 | 177.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornyakov, I.V.; Gurzhiy, V.V.; Kuz’mina, M.A.; Krzhizhanovskaya, M.G.; Chukanov, N.V.; Chislov, M.V.; Korneev, A.V.; Izatulina, A.R. Crystal Chemistry of the Copper Oxalate Biomineral Moolooite: The First Single-Crystal X-ray Diffraction Studies and Thermal Behavior. Int. J. Mol. Sci. 2023, 24, 6786. https://doi.org/10.3390/ijms24076786
Kornyakov IV, Gurzhiy VV, Kuz’mina MA, Krzhizhanovskaya MG, Chukanov NV, Chislov MV, Korneev AV, Izatulina AR. Crystal Chemistry of the Copper Oxalate Biomineral Moolooite: The First Single-Crystal X-ray Diffraction Studies and Thermal Behavior. International Journal of Molecular Sciences. 2023; 24(7):6786. https://doi.org/10.3390/ijms24076786
Chicago/Turabian StyleKornyakov, Ilya V., Vladislav V. Gurzhiy, Mariya A. Kuz’mina, Maria G. Krzhizhanovskaya, Nikita V. Chukanov, Mikhail V. Chislov, Anatolii V. Korneev, and Alina R. Izatulina. 2023. "Crystal Chemistry of the Copper Oxalate Biomineral Moolooite: The First Single-Crystal X-ray Diffraction Studies and Thermal Behavior" International Journal of Molecular Sciences 24, no. 7: 6786. https://doi.org/10.3390/ijms24076786
APA StyleKornyakov, I. V., Gurzhiy, V. V., Kuz’mina, M. A., Krzhizhanovskaya, M. G., Chukanov, N. V., Chislov, M. V., Korneev, A. V., & Izatulina, A. R. (2023). Crystal Chemistry of the Copper Oxalate Biomineral Moolooite: The First Single-Crystal X-ray Diffraction Studies and Thermal Behavior. International Journal of Molecular Sciences, 24(7), 6786. https://doi.org/10.3390/ijms24076786