
Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma

Abstract
:1. Introduction
2. Markers of Stem Cells in Pancreatic Ductal Adenocarcinoma
2.1. CD44
2.2. Epithelial Cell Adhesion Molecule (Epithelial Specific Antigen)
2.3. CD133
2.4. CXCR4
2.5. CD24
2.6. ALDH1
2.7. C-Met
2.8. DCLK1
3. Cancer Stem Cells: Assays
4. Signalling Pathways in PDAC CSC
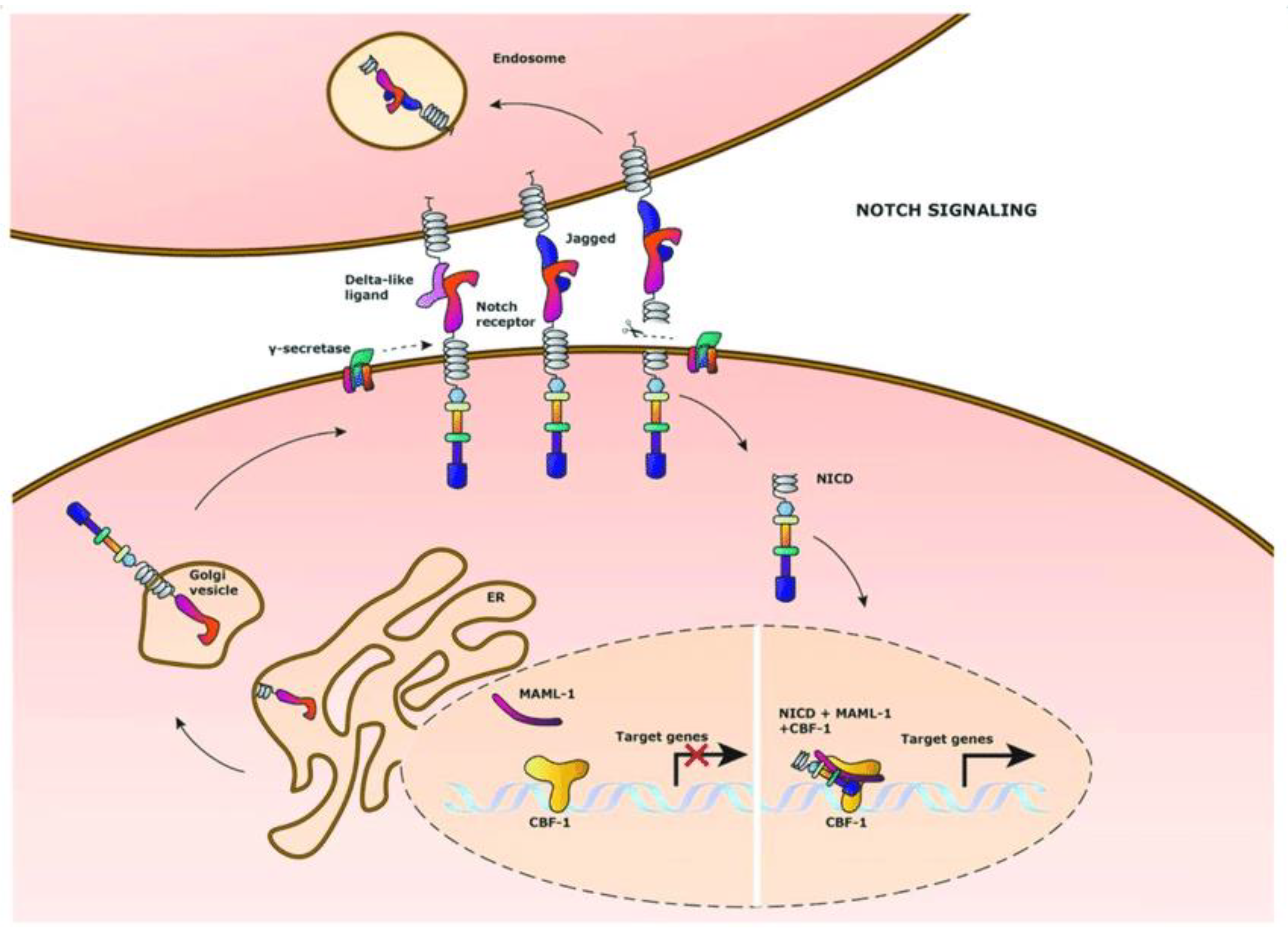
4.1. Notch Pathway
4.2. Hedgehog Signalling Pathway
4.3. Wnt Pathway
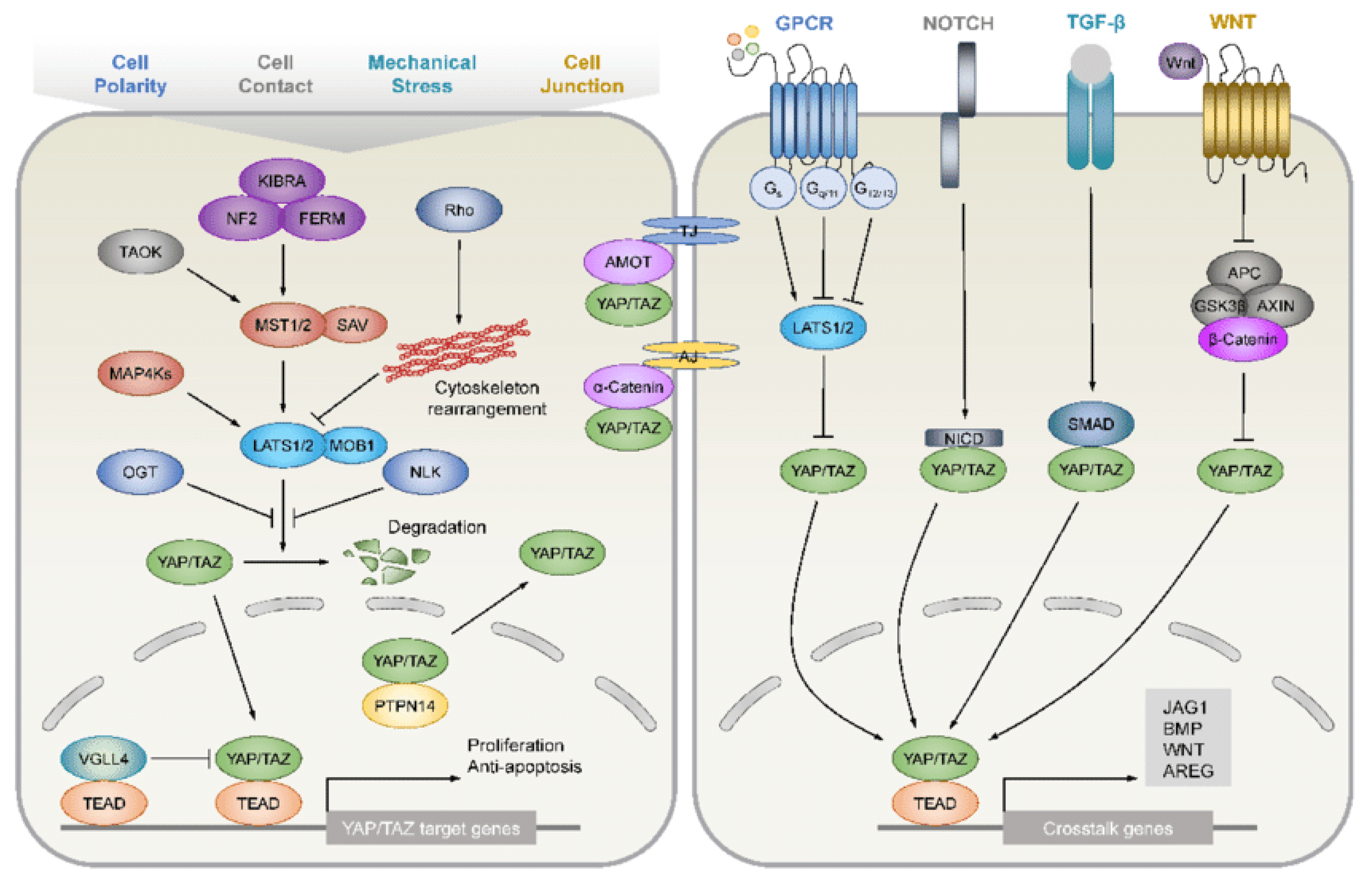
4.4. Hippo Pathway
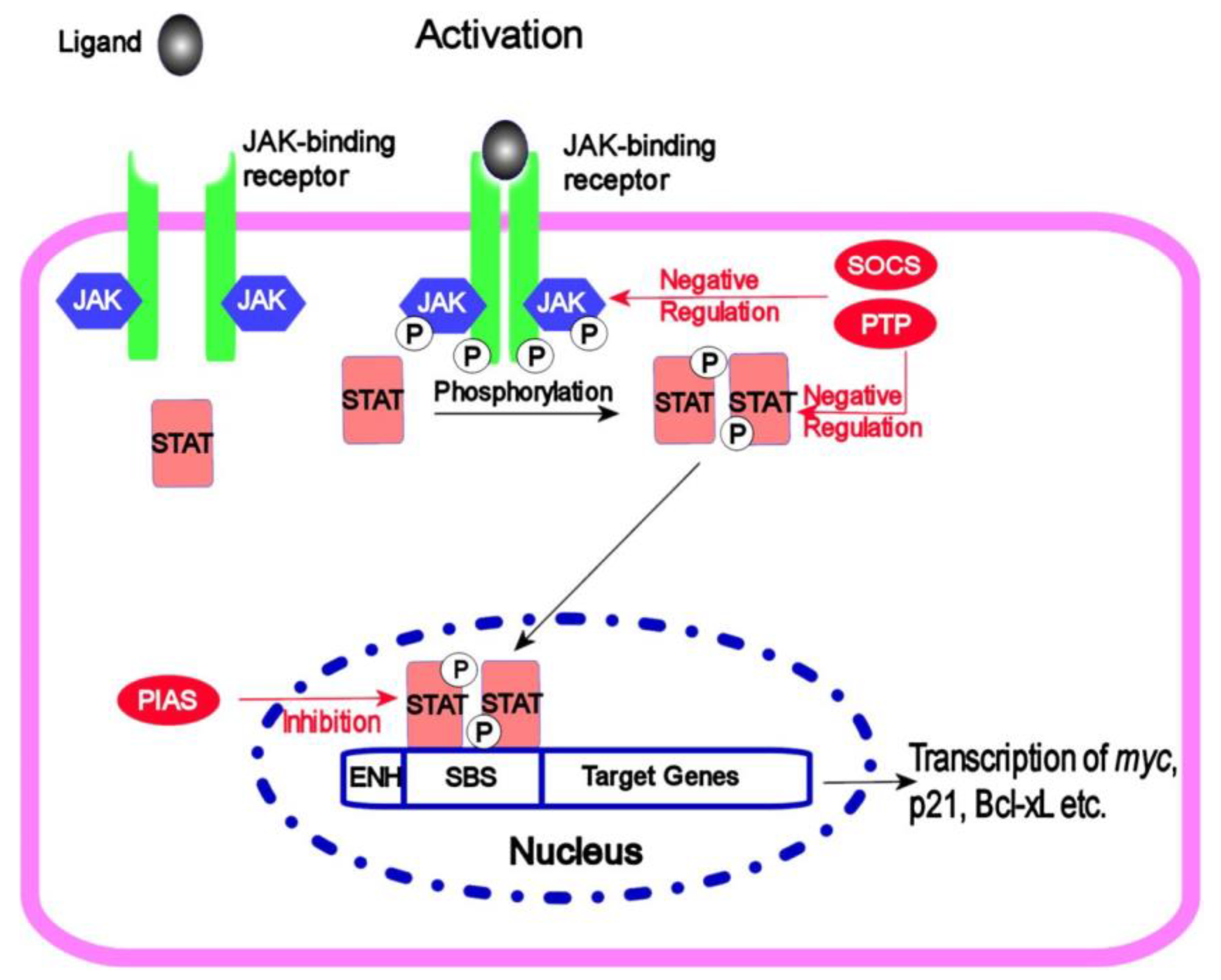
4.5. JAK/STAT Signalling
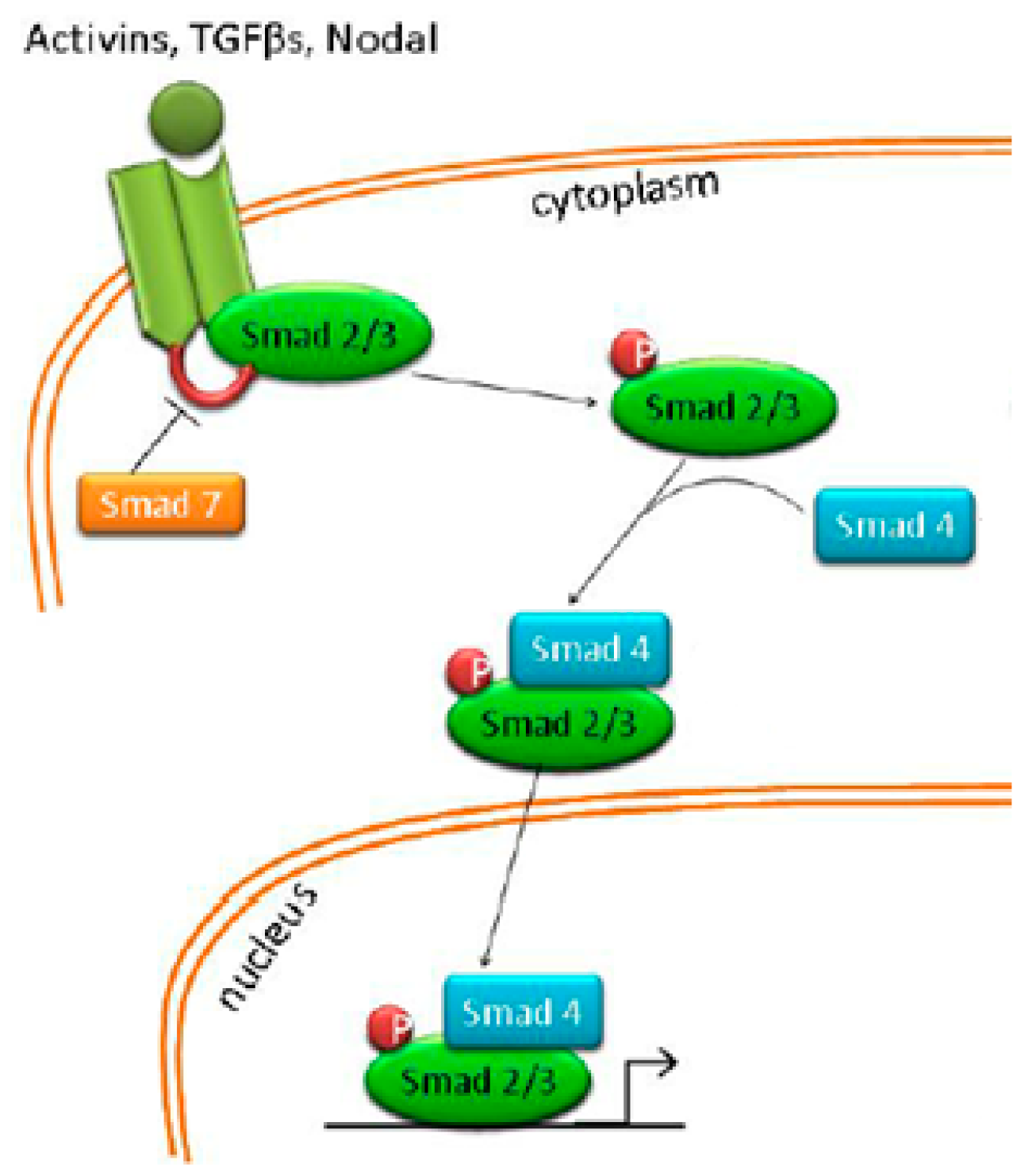
4.6. Nodal/Activin Pathway
4.7. K-Ras Mutation and Its Involvement in CSCs and Signalling Pathway Regulation
5. Metabolism of Stem Cells in Pancreatic Ductal Adenocarcinoma
6. Epithelial–Mesenchymal Transition and Its Association with Stem Cells in Pancreatic Ductal Adenocarcinoma
7. Epigenetic Events in Pancreatic Cancer Stem Cells
8. PDAC Cancer Stem Cells: Treatment Options
8.1. Salinomycin
8.2. Gramicidin A
8.3. Chloroquine
8.4. Aspirin
8.5. Disulfiram
8.6. EpCam/CD3 Bispecific T-Cell Engaging Antibody
8.7. Metformin
8.8. Decitabine and Vorinostat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Therapy Targets | Reference |
|---|---|---|
| Salinomycin | Wnt pathway (LARP6), CD133+ cells | [115,116,117] |
| Gramicidin A | Mitochondrial ultrastructure | [118] |
| Chloroquine | Hedgehog pathway (Smo) | [119] |
| Aspirin | NF-κB | [120] |
| Disulfiram | ALDH, NF-κB | [122] |
| MT110 | EpCAM+ cells | [123] |
| Metformin | NF-κB, MAPK/mTOR | [91,124] |
| Decitabine | miR-34a | [114] |
| Vorinostat | miR-34a, ZEB1 | [114] |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fossaert, V.; Mimmo, A.; Rhaiem, R.; Rached, L.J.; Brasseur, M.; Brugel, M.; Pegoraro, F.; Sanchez, S.; Bouché, O.; Kianmanesh, R.; et al. Neoadjuvant chemotherapy for borderline resectable and upfront resectable pancreatic cancer increasing overall survival and disease-free survival? Front. Oncol. 2022, 12, 980659. [Google Scholar] [CrossRef] [PubMed]
- Pedrazzoli, S. Surgical treatment of pancreatic cancer: Currently debated topics on morbidity, mortality, and lymphadenectomy. Surg. Oncol. 2022, 45, 101858. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510. [Google Scholar] [CrossRef]
- Shi, Y.-H.; Xu, Q.-C.; Zhu, Y.-Q.; Liu, Z.-D.; Zhao, G.-Y.; Liu, Q.; Wang, X.-Y.; Wang, J.-Q.; Xu, X.; Su, Q.; et al. Imatinib facilitates gemcitabine sensitivity by targeting epigenetically activated PDGFC signaling in pancreatic cancer. Mol. Ther. 2022, 31, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, B.; Xiang, L.; Deng, J.; Xu, B.; He, P.; Pu, W.; Wang, H.; Fan, Y.; Chen, H. Case Report: Anlotinib combined with PD-1 inhibitor and sequential GA regimen or FOLFIRINOX Chemotherapy in treatment of KRAS G12V mutated pancreatic ductal adenocarcinoma with liver metastasis: A case and literature review. Front. Immunol. 2022, 13, 1016647. [Google Scholar] [CrossRef]
- Schaal, J.L.; Bhattacharyya, J.; Brownstein, J.; Strickland, K.C.; Kelly, G.; Saha, S.; Milligan, J.; Banskota, S.; Li, X.; Liu, W.; et al. Brachytherapy via a depot of biopolymer-bound 131I synergizes with nanoparticle paclitaxel in therapy-resistant pancreatic tumours. Nat. Biomed. Eng. 2022, 6, 1148–1166. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, R.; Debnath, D.; Hartley, M.L.; Noel, M.S. The Role of Immunotherapy in Pancreatic Cancer. Curr. Oncol. 2022, 29, 6864–6892. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.P.; Sacks, G.D.; Rochefort, M.M.; Donahue, T.R.; Reber, H.A.; Tomlinson, J.S.; Dawson, W.; Eibl, G.; Hines, O.J. Long-term survival in patients with pancreatic ductal adenocarcinoma. Surgery 2016, 159, 1520–1527. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Cronin, K.A.; Scott, S.; Firth, A.U.; Sung, H.; Henley, S.J.; Sherman, R.L.; Siegel, R.L.; Anderson, R.N.; Kohler, B.A.; Benard, V.B.; et al. Annual report to the nation on the status of cancer, part 1: National cancer statistics. Cancer 2022, 128, 4251–4284. [Google Scholar] [CrossRef]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Taga, T. Cancer ego-system in glioma: An iron-replenishing niche network systemically self-organized by cancer stem cells. Inflamm. Regen. 2022, 42, 54. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Henzi, T.; Schwaller, B.; Pecze, L. Biological noise and positional effects influence cell stemness. J. Biol. Chem. 2018, 293, 5247–5258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Monteiro, M.J.; Liu, J.; Gu, W. Mechanisms of cancer stem cell senescence: Current understanding and future perspectives. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1185–1202. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sainz, B., Jr. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cell Tumor Stem Cell Biol. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Weygant, N.; Chandrakesan, P.; Houchen, C.W.; Peng, J.; Qu, D. Tuft and Cancer Stem Cell Marker DCLK1: A New Target to Enhance Anti-Tumor Immunity in the Tumor Microenvironment. Cancers 2020, 12, 3801. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Y.; Yuan, Z. Pancreatic Cancer Stem Cells. Am. J. Cancer Res. 2015, 5, 894–906. [Google Scholar]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briede, I.; Balodis, D.; Gardovskis, J.; Strumfa, I. Stemness, Inflammation and Epithelial-Mesenchymal Transition in Colorectal Carcinoma: The Intricate Network. Int. J. Mol. Sci. 2021, 22, 12891. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kure, S.; Matsuda, Y.; Hagio, M.; Ueda, J.; Naito, Z.; Ishiwata, T. Expression of cancer stem cell markers in pancreatic intraepithelial neoplasias and pancreatic ductal adenocarcinomas. Int. J. Oncol. 2012, 41, 1314–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Chao, Y.; Tung, H.; Wang, H.; Shan, Y. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askan, G.; Sahin, I.H.; Chou, J.F.; Yavas, A.; Capanu, M.; Iacobuzio-Donahue, C.A.; Basturk, O.; O’Reilly, E.M. Pancreatic cancer stem cells may define tumor stroma characteristics and recurrence patterns in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 385. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Piva, F.; Marinelli, O.; Maggi, F.; Bianchi, F.; Bittoni, A.; Berardi, R.; Giampieri, R.; et al. Expression profiling of circulating tumor cells in pancreatic ductal adenocarcinoma patients: Biomarkers predicting overall survival. Front. Oncol. 2019, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, Y.; Yang, F.; Liu, S.; Zhu, Z.; Lei, Z.; Guo, J. Functions of EpCAM in physiological processes and diseases (Review). Int. J. Mol. Med. 2018, 42, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Patil, K.; Khan, F.B.; Akthar, S.; Ahmad, A.; Uddin, S. The plasticity of pancreatic cancer stem cells: Implications in therapeutic resistance. Cancer Metastasis Rev. 2021, 40, 691–720. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-H.; Sun, R.; Zhou, X.-M.; Zhang, M.-Y.; Lu, J.-B.; Yang, Y.; Zeng, L.-S.; Yang, X.-Z.; Shi, L.; Xiao, R.-W.; et al. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.C.; Kuo, K.K.; Su, H.T.; Hsiao, P.J.; Chen, Y.W.; Wu, D.C.; Hung, W.C.; Cheng, K.H. Pancreatic tumor progression associated with CD133 overexpression: Involvement of increased TERT expression and epidermal growth factor receptor-dependent Akt activation. Pancreas 2016, 45, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Kamachi, H.; Mitsuhashi, T.; Tsuruga, Y.; Hatanaka, Y.; Kamiyama, T.; Matsuno, Y.; Taketomi, A. Immunohistochemical analysis of cancer stem cell markers in pancreatic adenocarcinoma patients after neoadjuvant chemoradiotherapy. BMC Cancer 2014, 14, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, K.; Rodriguez-Aznar, E.; Ferreira, M.S.V.; Frappart, P.O.; Dittrich, T.; Tiwary, K.; Meessen, S.; Lerma, L.; Daiss, N.; Schulte, L.A.; et al. Telomerase and pluripotency factors jointly regulate stemness in pancreatic cancer stem cells. Cancers 2021, 13, 3145. [Google Scholar] [CrossRef]
- Liou, G.-Y. CD133 as a regulator of cancer metastasis through the cancer stem cells. Int. J. Biochem. Cell Biol. 2018, 106, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Azad, B.B.; Nimmagadda, S. The Intricate Role of CXCR4 in Cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [Green Version]
- Ercan, G.; Karlitepe, A.; Ozpolat, B. Pancreatic Cancer Stem Cells and Therapeutic Approaches. Anticancer Res. 2017, 37, 2761–2775. [Google Scholar] [CrossRef] [Green Version]
- Aigner, S.; Sthoeger, Z.M.; Fogel, M.; Weber, E.; Zarn, J.; Ruppert, M.; Zeller, Y.; Vestweber, D.; Stahel, R.; Sammar, M.; et al. CD24, a Mucin-Type Glycoprotein, Is a Ligand for P-Selectin on Human Tumor Cells. Blood 1997, 89, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cremers, N.; Kroese, F.; Orend, G.; Chiquet-Ehrismann, R.; Uede, T.; Yagita, H.; Sleeman, J.P. CD24 Expression Causes the Acquisition of Multiple Cellular Properties Associated with Tumor Growth and Metastasis. Cancer Res. 2005, 65, 10783–10793. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.L.; Oshi, M.; Endo, I.; Takabe, K. Clinical relevance of stem cell surface markers CD133, CD24, and CD44 in colorectal cancer. Am. J. Cancer Res. 2021, 11, 5141–5154. [Google Scholar] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Chmielowiec, J.; Borowiak, M.; Morkel, M.; Stradal, T.; Munz, B.; Werner, S.; Wehland, J.; Birchmeier, C.; Birchmeier, W. c-Met is essential for wound healing in the skin. J. Cell Biol. 2007, 177, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Sesillo, F.S.; Sacco, A. Tumorsphere derivation and treatment from primary tumor cells isolated from mouse rhabdomyosacomas. J. Vis. Exp. 2019, 151, e59897. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef]
- Broeck, A.V.D.; Vankelecom, H.; Van Delm, W.; Gremeaux, L.; Wouters, J.; Allemeersch, J.; Govaere, O.; Roskams, T.; Topal, B. Human Pancreatic Cancer Contains a Side Population Expressing Cancer Stem Cell-Associated and Prognostic Genes. PLoS ONE 2013, 8, e73968. [Google Scholar] [CrossRef]
- Liu, W.-H.; Wang, X.; You, N.; Tao, K.-S.; Wang, T.; Tang, L.-J.; Dou, K.-F. Efficient Enrichment of Hepatic Cancer Stem-Like Cells from a Primary Rat HCC Model via a Density Gradient Centrifugation-Centered Method. PLoS ONE 2012, 7, e35720. [Google Scholar] [CrossRef] [PubMed]
- Miltenyi, S.; Müller, W.; Weichel, W.; Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 1990, 11, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Fatima, I.; Singh, A.; Dhawan, P. Pancreatic Cancer and Therapy: Role and Regulation of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 4765. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.L.; Kissil, J.L. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol. Med. 2013, 19, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, L.; Avila, J.L.; Demarest, R.M.; Troutman, S.; Allen, M.; Ratti, F.; Rustgi, A.K.; Stanger, B.Z.; Radtke, F.; Adsay, V.; et al. Notch1 Functions as a Tumor Suppressor in a Model of K-ras—Induced Pancreatic Ductal Adenocarcinoma. Cancer Res. 2010, 70, 4280–4286. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011, 31, 1105–1113. [Google Scholar] [PubMed]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling pathways in cancer-associated fibroblasts: Recent advances and future perspectives. Cancer Commun. 2022, 43, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Roo, J.; Staal, F. Cell Signaling Pathway Reporters in Adult Hematopoietic Stem Cells. Cells 2020, 9, 2264. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog Signaling Antagonist GDC-0449 (Vismodegib) Inhibits Pancreatic Cancer Stem Cell Characteristics: Molecular Mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.H.; Ghosh, B.; Alam Rizvi, M.; Ali, M.; Kaur, L.; Mondal, A.C. Neural crest cells development and neuroblastoma progression: Role of Wnt signaling. J. Cell. Physiol. 2022, 238, 306–328. [Google Scholar] [CrossRef]
- Min, J.K.; Park, H.-S.; Lee, Y.-B.; Kim, J.-G.; Kim, J.-I.; Park, J.-B. Cross-Talk between Wnt Signaling and Src Tyrosine Kinase. Biomedicines 2022, 10, 1112. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.L.; Ortolano, N.A.; Romero-Morales, A.I.; Gama, V. Wnt Signaling and Its Impact on Mitochondrial and Cell Cycle Dynamics in Pluripotent Stem Cells. Genes 2018, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, K.Y.; Dawson, D.W. WNT Ligand Dependencies in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 671022. [Google Scholar] [CrossRef] [PubMed]
- Ilmer, M.; Boiles, A.R.; Regel, I.; Yokoi, K.; Michalski, C.W.; Wistuba, I.I.; Rodriguez, J.; Alt, E.; Vykoukal, J. RSPO2 Enhances Canonical Wnt Signaling to Confer Stemness-Associated Traits to Susceptible Pancreatic Cancer Cells. Cancer Res. 2015, 75, 1883–1896. [Google Scholar] [CrossRef] [Green Version]
- Schindler, A.J.; Watanabe, A.; Howell, S. LGR5 and LGR6 in stem cell biology and ovarian cancer. Oncotarget 2018, 9, 1346–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, D.; Ohlsson, H.; Althini, C.; Bauden, M.; Zhou, Q.; Hu, D.; Andersson, R. The Hippo Signaling Pathway in Pancreatic Cancer. Anticancer Res. 2019, 39, 3317–3321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Aegerter, P.; Nipper, M.; Ramjit, L.; Liu, J.; Wang, P. Hippo Signaling Pathway in Pancreas Development. Front. Cell Dev. Biol. 2021, 9, 663906. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Jho, E.-H. The history and regulatory mechanism of the Hippo pathway. BMB Rep. 2018, 51, 106–118. [Google Scholar] [CrossRef]
- Ervin, E.H.; French, R.; Chang, C.H.; Pauklin, S. Inside the stemness engine: Mechanistic links between deregulated transcription factors and stemness in cancer. Semin. Cancer Biol. 2022, 87, 48–83. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef]
- Lee, J.; Yakubov, B.; Ivan, C.; Jones, D.R.; Caperell-Grant, A.; Fishel, M.; Cardenas, H.; Matei, D. Tissue Transglutaminase Activates Cancer-Associated Fibroblasts and Contributes to Gemcitabine Resistance in Pancreatic Cancer. Neoplasia 2016, 18, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue transglutaminase mediated tumor-stroma interaction promotes pancreatic cancer progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Furqan, M.; Mukhi, N.; Lee, B.; Liu, D. Dysregulation of JAK-STAT pathway in haematological malignancies and JAK inhibitors for clinical application. Biomark. Res. 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Marimuthu, S.; Bhardwaj, A.; Deshmukh, S.K.; Srivastava, S.K.; Singh, A.P.; McClellan, S.; Carter, J.E.; Singh, S. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016, 370, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Pauklin, S.; Vallier, L. Activin/Nodal signalling in stem cells. Development 2015, 142, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Papanayoutu, C.; Collignon, J. Activin/Nodal signalling before implantation: Setting the stage for embryo patterning. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soncin, F.; Ward, C.M. The Function of E-Cadherin in Stem Cell Pluripotency and Self-Renewal. Genes 2011, 2, 229–259. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Lorenzo, I.M.; Zagorac, S.; Alcala, S.; Rodriquez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-Canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS Activates Hedgehog Signaling Pathway in Pancreatic Cancer Cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, H.-H.; Gao, C.-Y.; Zhang, X.-X.; Jiang, J.-X.; Zhang, Y.; Fang, J.; Zhao, F.; Chen, Z.-G. Energy metabolism in cancer stem cells. World J. Stem Cells 2020, 12, 448–461. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; Cola, A.D.; Scavo, E.; Carollo, R.; D’ Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multi-drug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1α balance determines phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Hockenbery, D. MYC and Mitochondrial Biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef] [Green Version]
- Domenichini, A.; Edmands, J.S.; Adamska, A.; Begicevic, R.-R.; Paternoster, S.; Falasca, M. Pancreatic cancer tumorspheres are cancer stem-like cells with increased chemoresistance and reduced metabolic potential. Adv. Biol. Regul. 2019, 72, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Urrutia, R. Basics of TGF-β and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Tao, G.-Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.-Q. TGF-β in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; Herreros, A.G.D.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Li, Z.-X.; Liu, X.; Wang, R.; Li, L.-W.; Zhang, Q. The Wnt/β-catenin and PI3K/Akt signaling pathways promote EMT in gastric cancer by epigenetic regulation via H3 lysine 27 acetylation. Tumor Biol. 2017, 39, 1010428317712617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [Green Version]
- Lamceva, J.; Uljanovs, R.; Strumfa, I. The main theories on the pathogenesis of endometriosis. Int. J. Mol. Sci. 2023, 24, 4254. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, S.; García-Bermejo, L.; Sainz, J.B. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes 2018, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, S.; Alcala, S.; Bayon, G.F.; Kheir, T.B.; Schoenhals, M.; González-Neira, A.; Fraga, M.F.; Aicher, A.; Heeschen, C.; Sainz, B. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17-92 Cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.-R.; Hsu, M.-C.; Luo, C.-W.; Chen, L.-T.; Shan, Y.-S.; Hung, W.-C. The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget 2016, 7, 61136–61151. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Tateishi, K.; Fujiwara, H.; Ijichi, H.; Yamamoto, K.; Nakatsuka, T.; Kakiuchi, M.; Sano, M.; Kudo, Y.; Hayakawa, Y.; et al. Deletion of Histone Methyltransferase G9a Suppresses Mutant Kras-Driven Pancreatic Carcinogenesis. Cancer Genomics Proteomics 2020, 17, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef]
- Ma, Y.-S.; Yang, X.-L.; Liu, Y.-S.; Ding, H.; Wu, J.-J.; Shi, Y.; Jia, C.-Y.; Lu, G.-X.; Zhang, D.-D.; Wang, H.-M.; et al. Long non-coding RNA NORAD promotes pancreatic cancer stem cell proliferation and self-renewal by blocking microRNA-202-5p-mediated ANP32E inhibition. J. Transl. Med. 2021, 19, 400. [Google Scholar] [CrossRef] [PubMed]
- Bimonte, S.; Barbieri, A.; Leongito, M.; Palma, G.; del Vecchio, V.; Falco, M.; Palaia, R.; Albino, V.; Piccirillo, M.; Amore, A.; et al. The Role of miRNAs in the Regulation of Pancreatic Cancer Stem Cells. Stem Cells Int. 2016, 2016, 8352684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortoglou, M.; Miralles, F.; Arisan, E.D.; Dart, A.; Jurcevic, S.; Lange, S.; Uysal-Onganer, P. microRNA-21 Regulates Stemness in Pancreatic Ductal Adenocarcinoma Cells. Int. J. Mol. Sci. 2022, 23, 1275. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.-N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting Epigenetic Regulation of miR-34a for Treatment of Pancreatic Cancer by Inhibition of Pancreatic Cancer Stem Cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.-N.; Liang, Y.; Zhou, L.-J.; Chen, S.-P.; Chen, G.; Zhang, T.-P.; Kang, T.; Zhao, Y.-P. Combination of salinomycin and gemcitabine eliminates pancreatic cancer cells. Cancer Lett. 2011, 313, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Choi, M.Y.; Yu, J.; Castro, J.E.; Kipps, T.J.; Carson, D.A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13253–13257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.-Q.; Geng, J.; Sheng, W.-J.; Liu, X.-J.; Jiang, M.; Zhen, Y.-S. The ionophore antibiotic gramicidin A inhibits pancreatic cancer stem cells associated with CD47 down-regulation. Cancer Cell Int. 2019, 19, 145. [Google Scholar] [CrossRef]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine Targets Pancreatic Cancer Stem Cells via Inhibition of CXCR4 and Hedgehog Signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, L.; Fan, P.; Bauer, N.; Gladkich, J.; Ryschich, E.; Bazhin, A.V.; Giese, N.A.; Strobel, O.; Hackert, T.; et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget 2015, 6, 9999–10015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Wang, Y.; Zhang, X.; Zhang, N.; Liu, L.; Soukup, K.; Michelakos, T.; Hong, T.; DeLeo, A.; Cai, L.; et al. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017, 409, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-Cell Engaging Antibody MT110 Eliminates Primary Human Pancreatic Cancer Stem Cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin Inhibits Cell Proliferation, Migration and Invasion by Attenuating CSC Function Mediated by Deregulating miRNAs in Pancreatic Cancer Cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bubin, R.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7030. https://doi.org/10.3390/ijms24087030
Bubin R, Uljanovs R, Strumfa I. Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(8):7030. https://doi.org/10.3390/ijms24087030
Chicago/Turabian StyleBubin, Roman, Romans Uljanovs, and Ilze Strumfa. 2023. "Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 24, no. 8: 7030. https://doi.org/10.3390/ijms24087030
APA StyleBubin, R., Uljanovs, R., & Strumfa, I. (2023). Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 24(8), 7030. https://doi.org/10.3390/ijms24087030