
OBHS Drives Abnormal Glycometabolis Reprogramming via GLUT1 in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
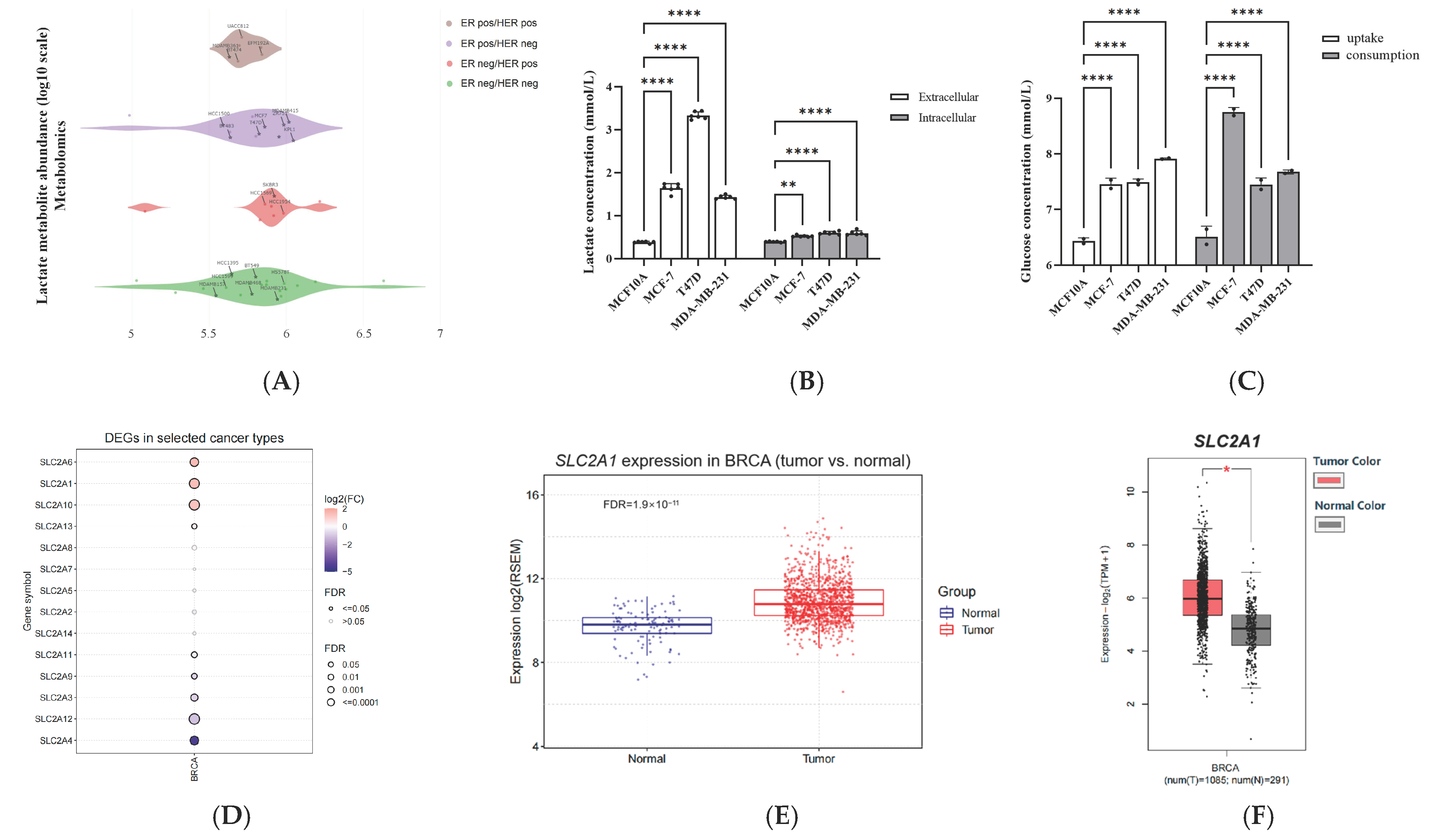
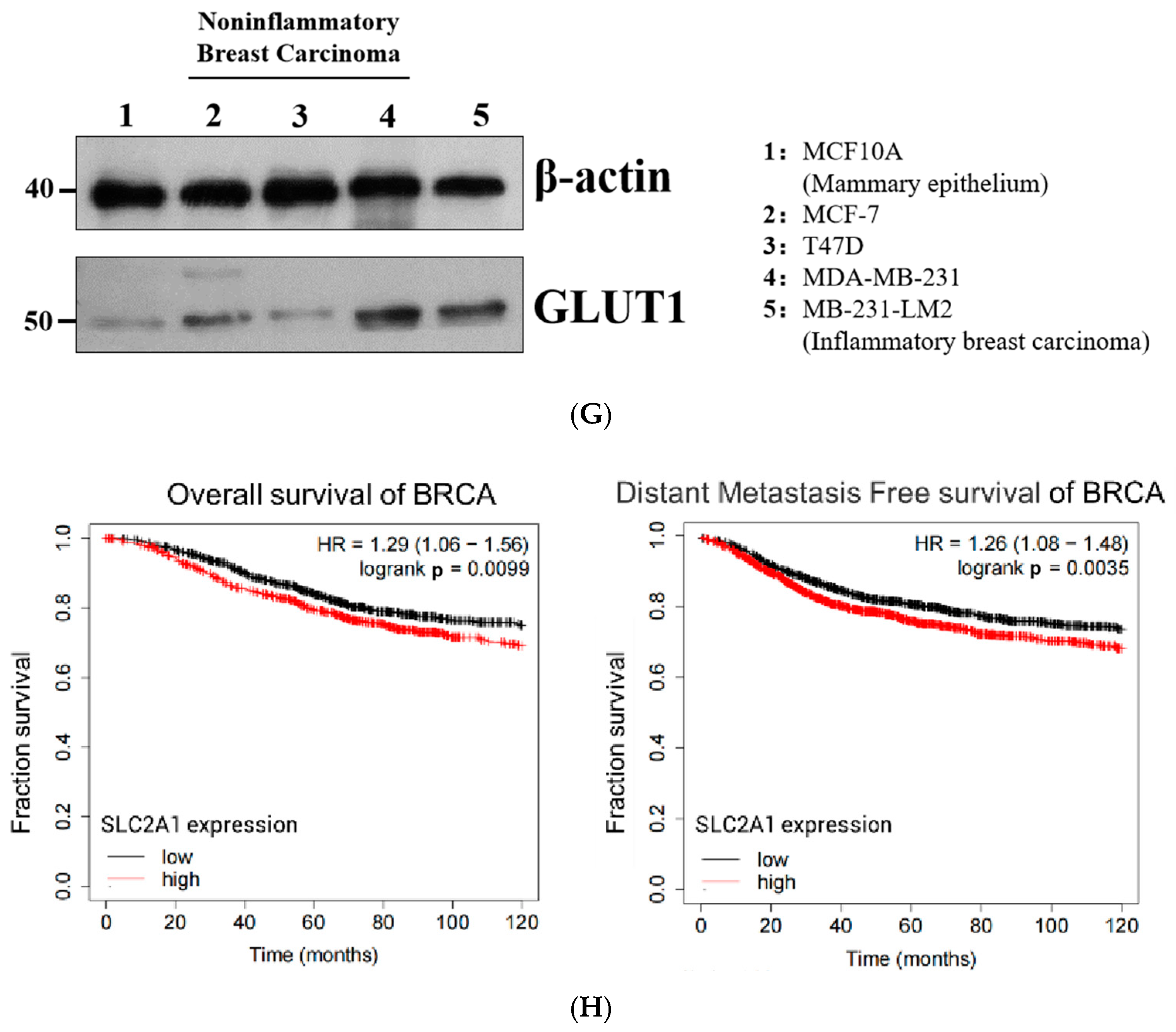
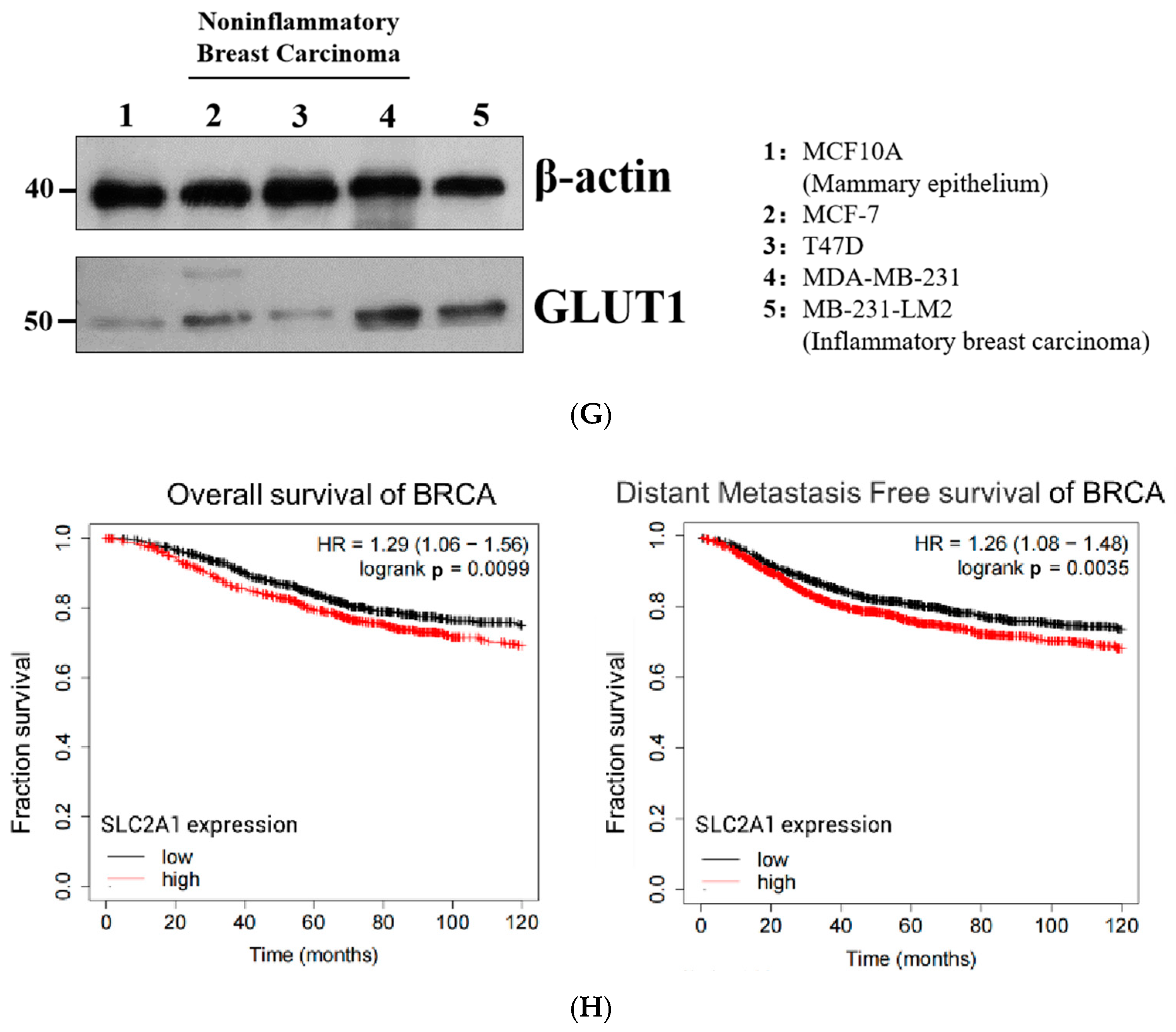
2.1. GLUT1 Expression Significantly Correlated with Poorer Survival Statistics among Patients with Breast Cancer in Clinical Settings
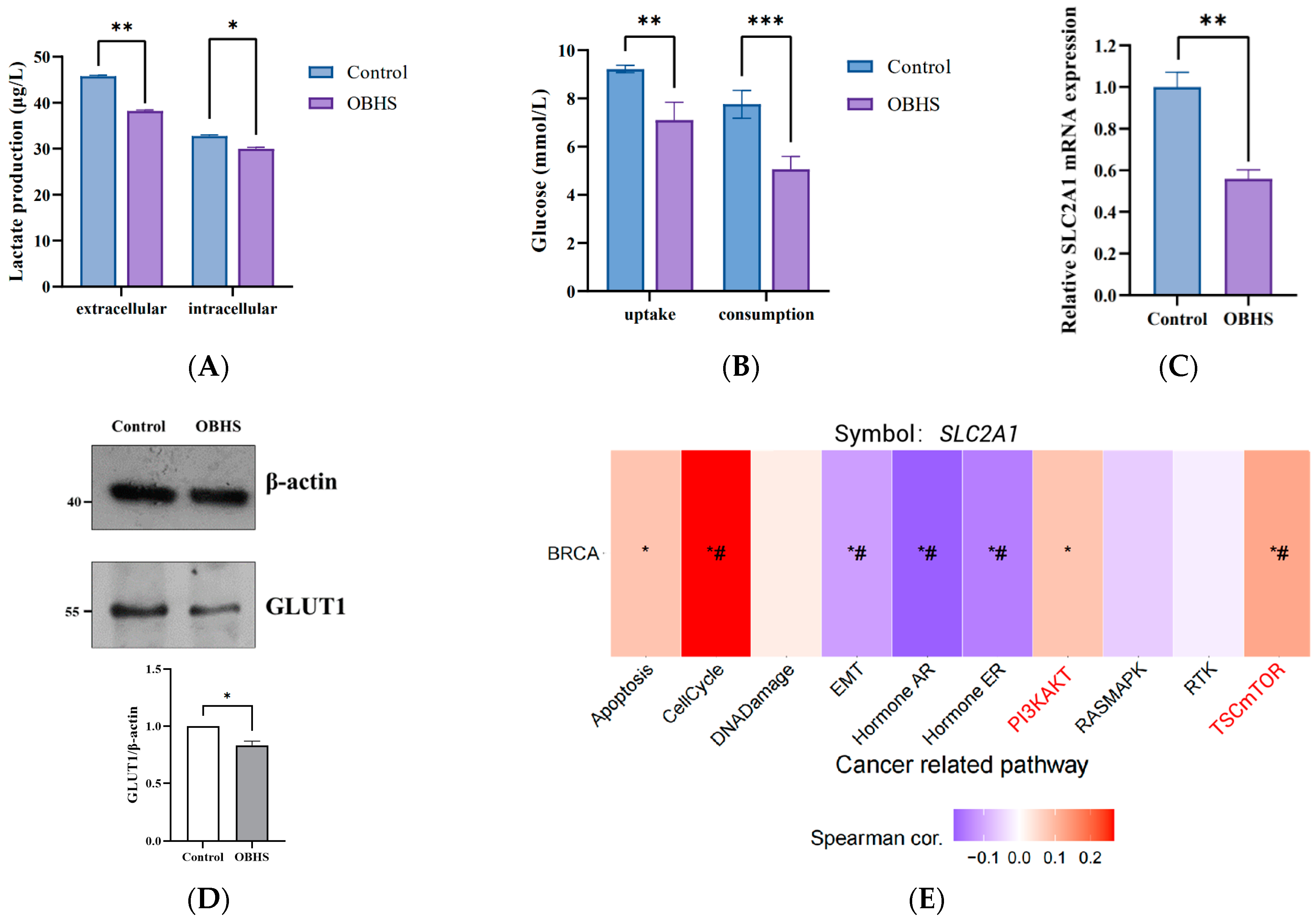
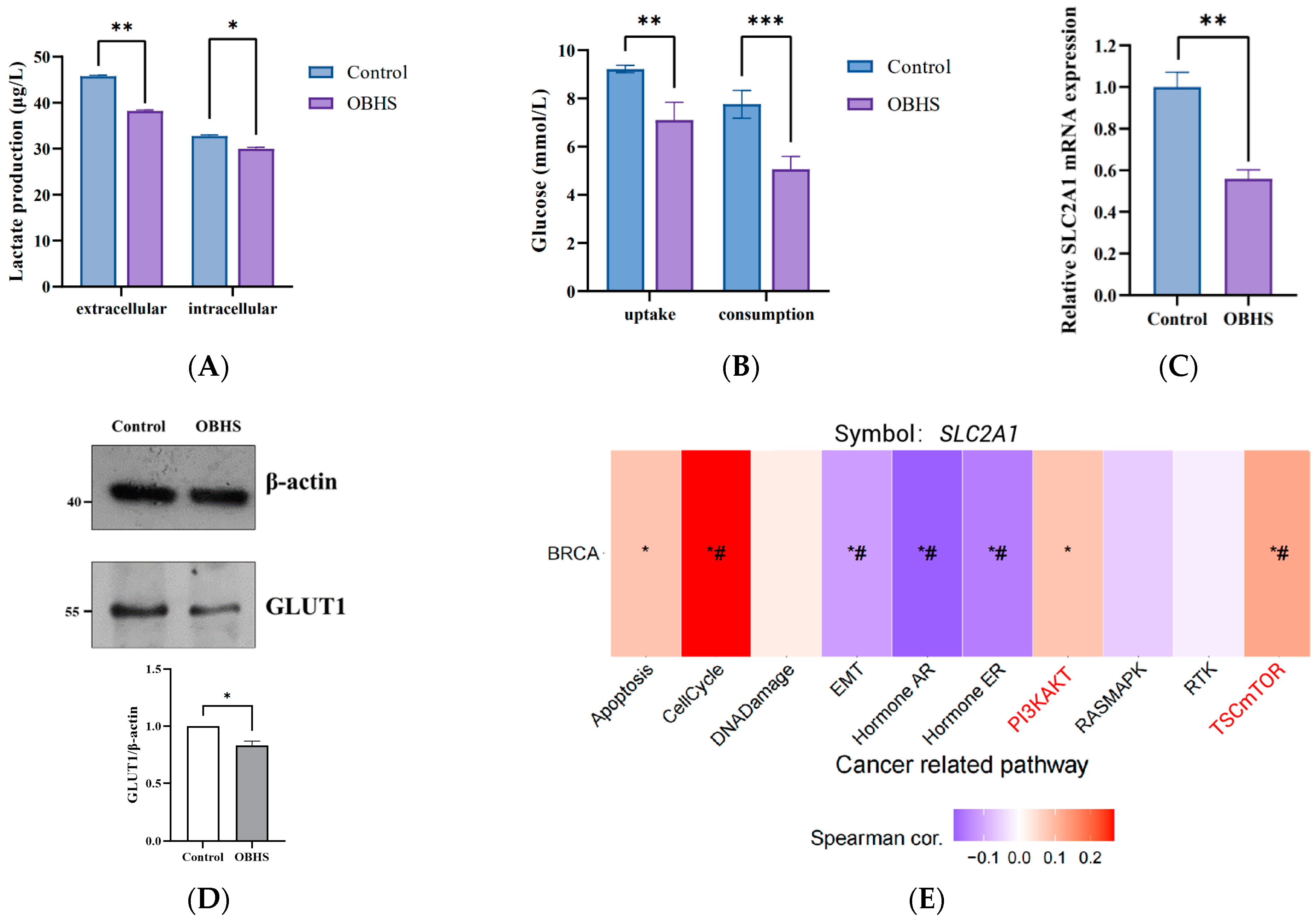
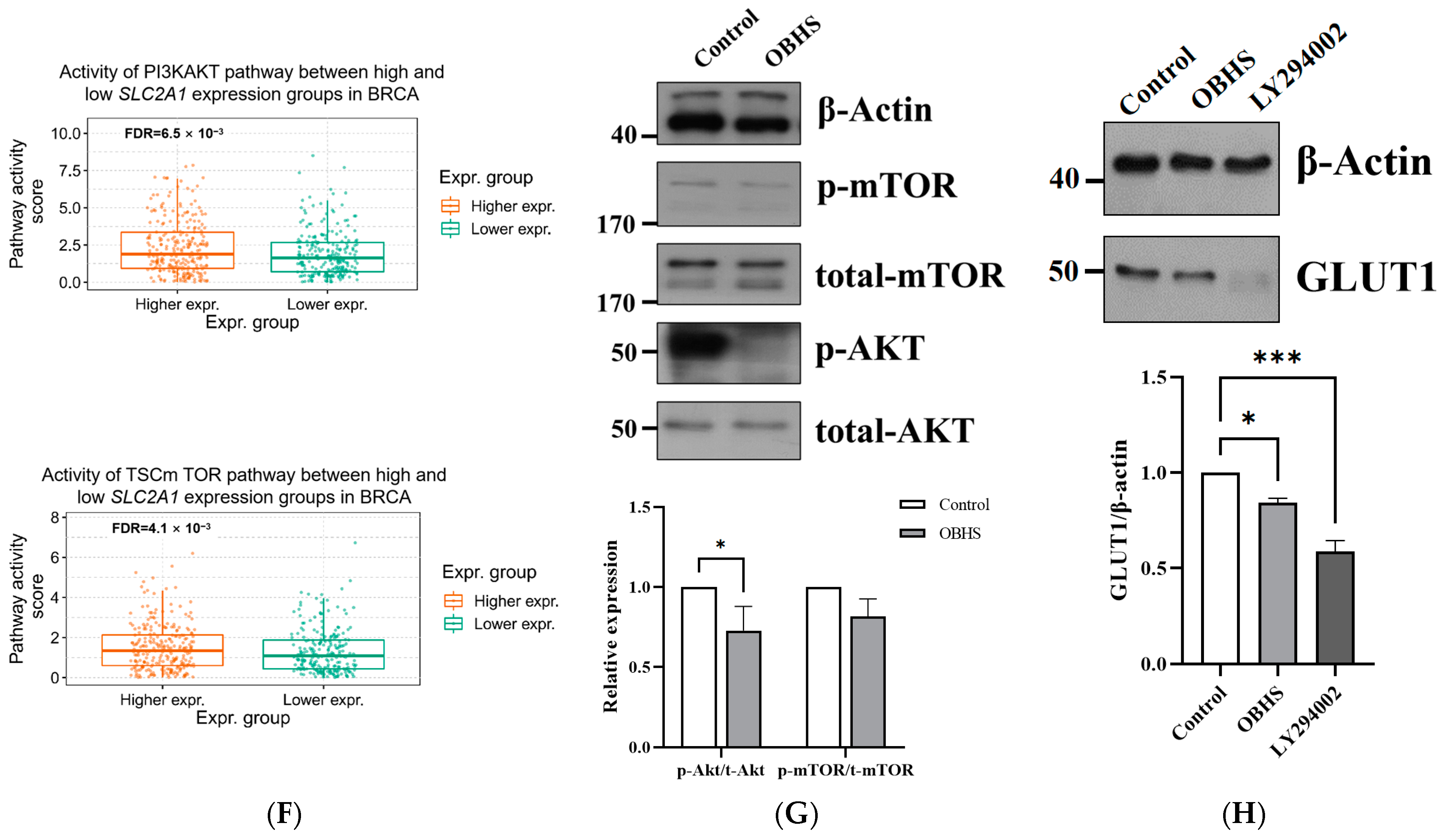
2.2. OBHS Decreased GLUT1 Expression Requires PI3K/Akt Signaling Pathway in MCF-7 Cells
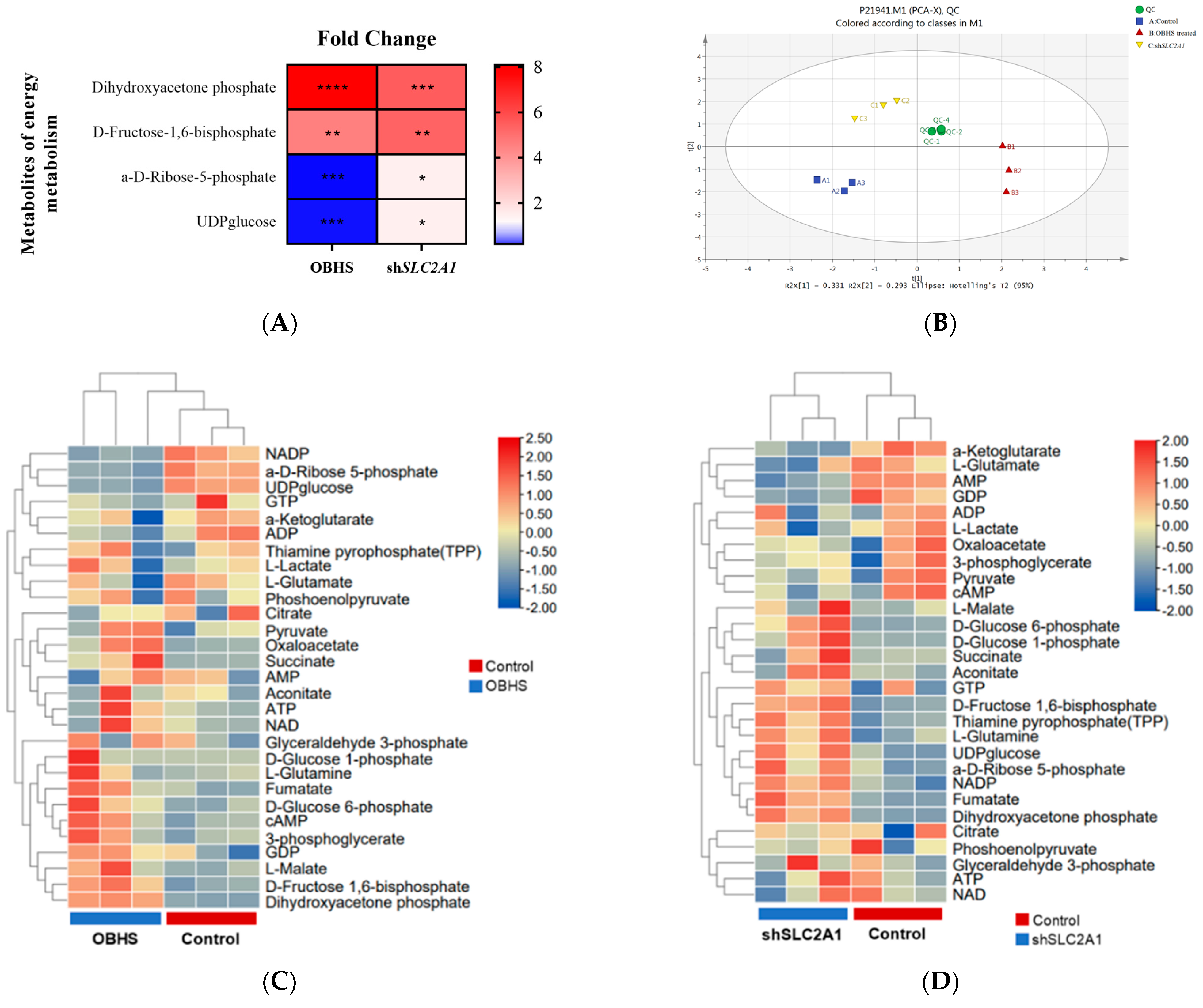
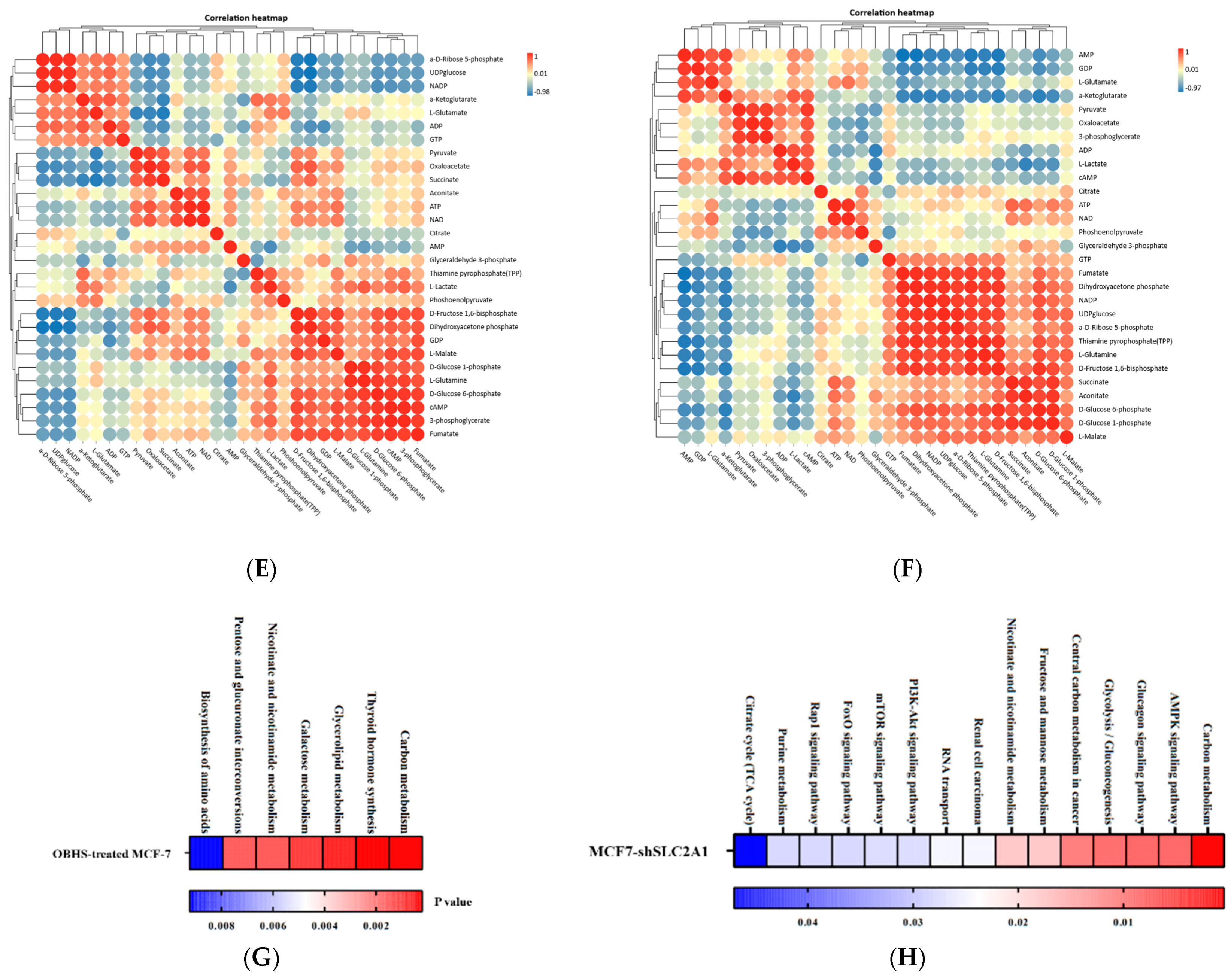
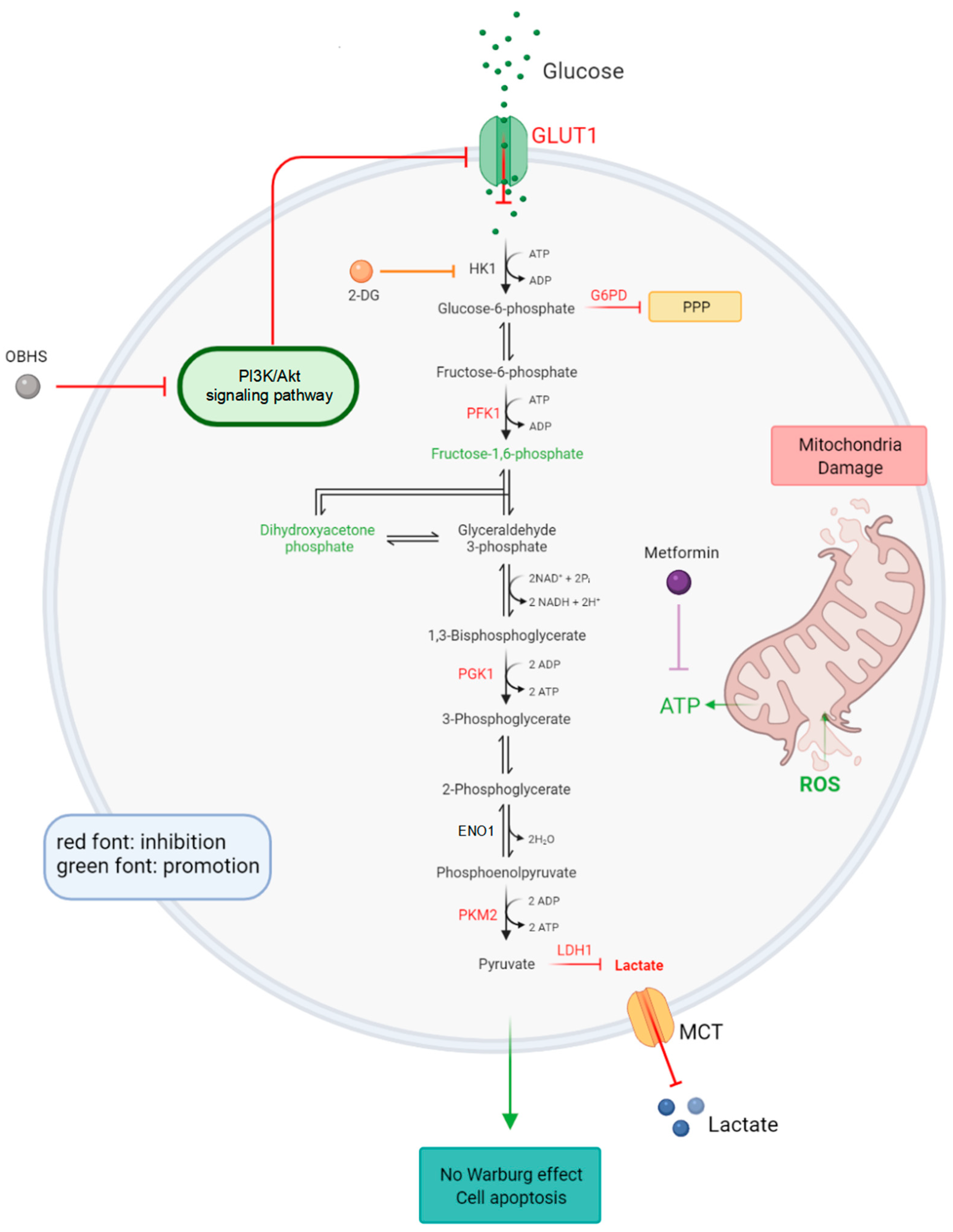
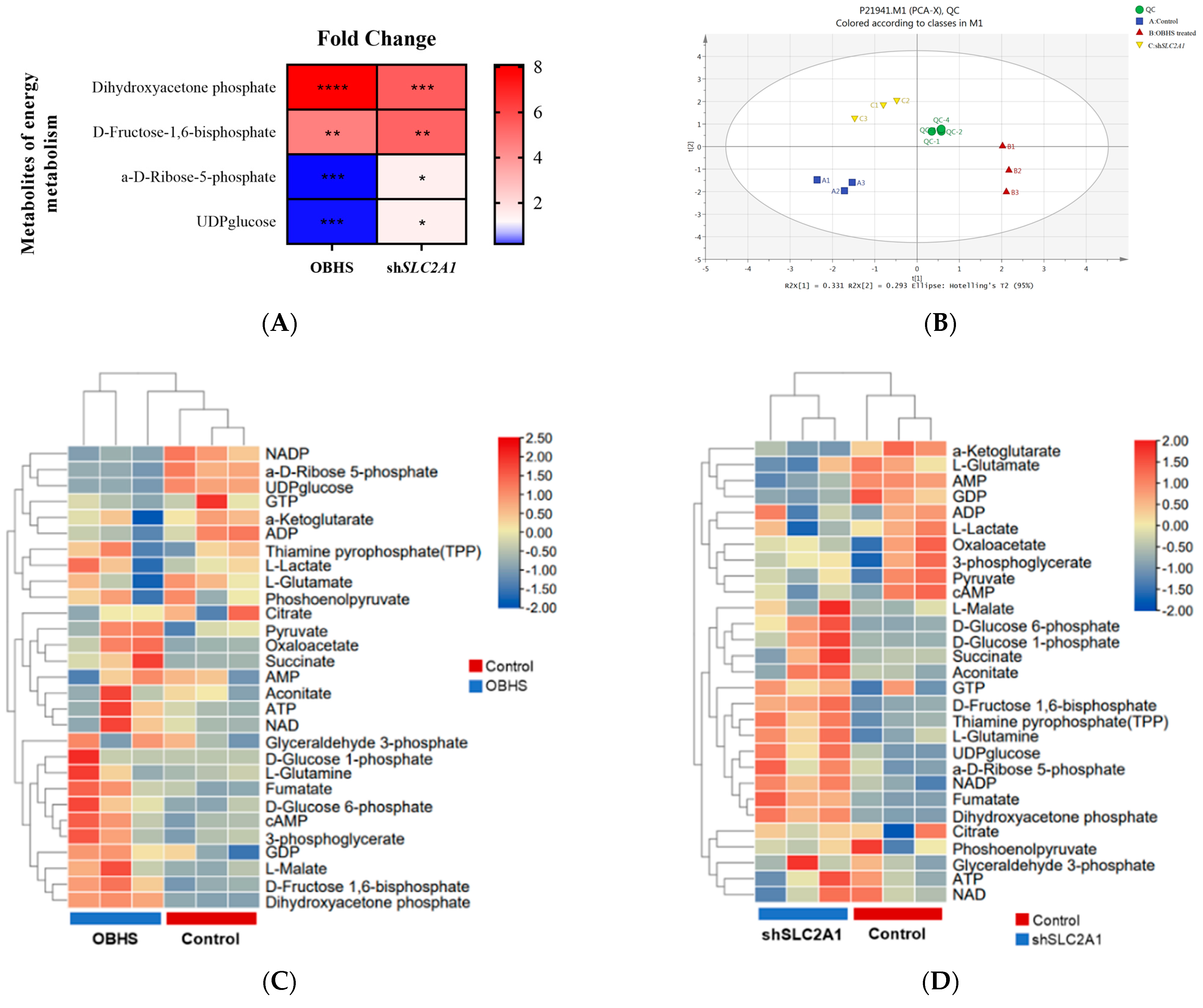
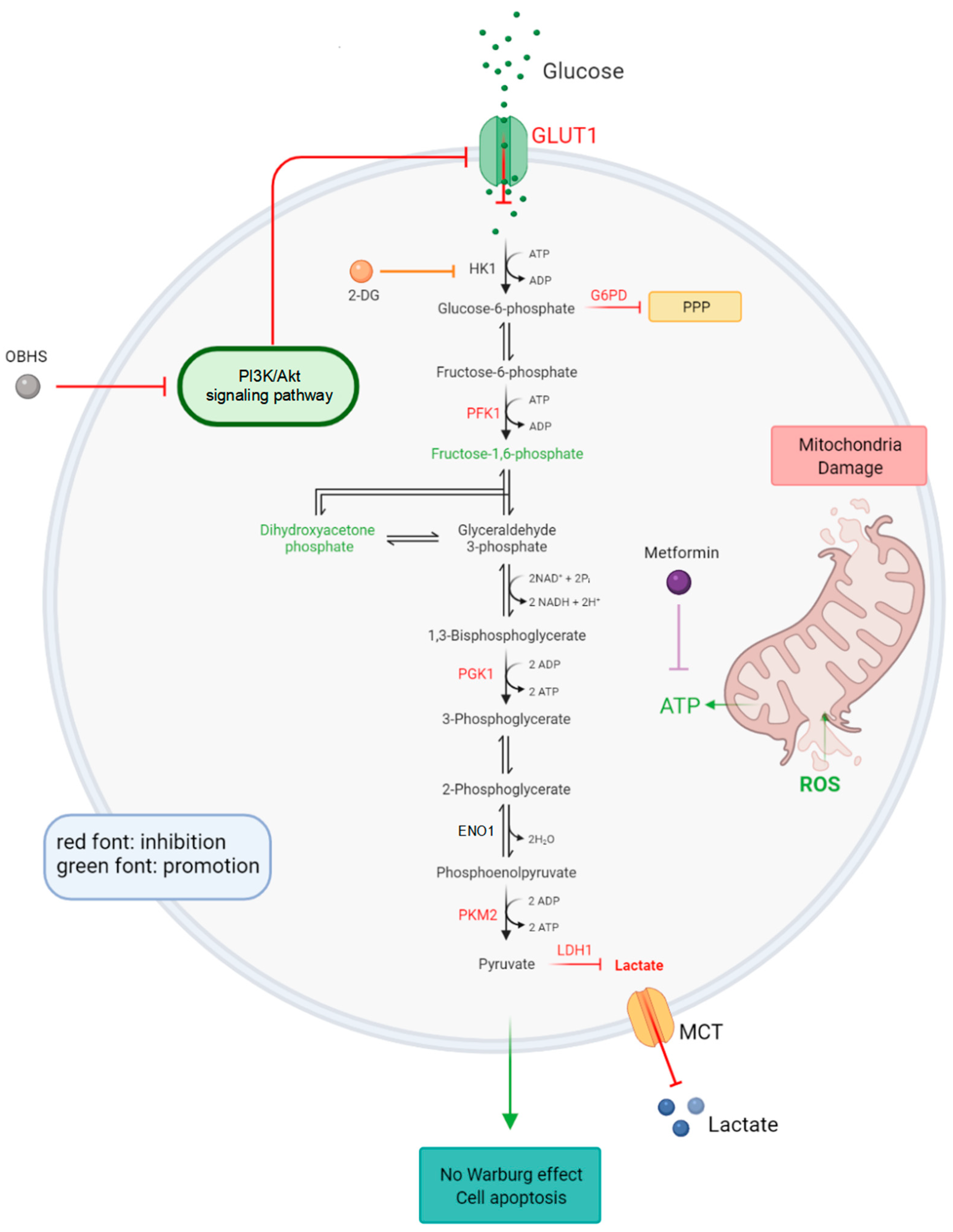
2.3. OBHS Suppressed Energy Metabolism by Altering the Expression Levels of Associated Metabolic Intermediates
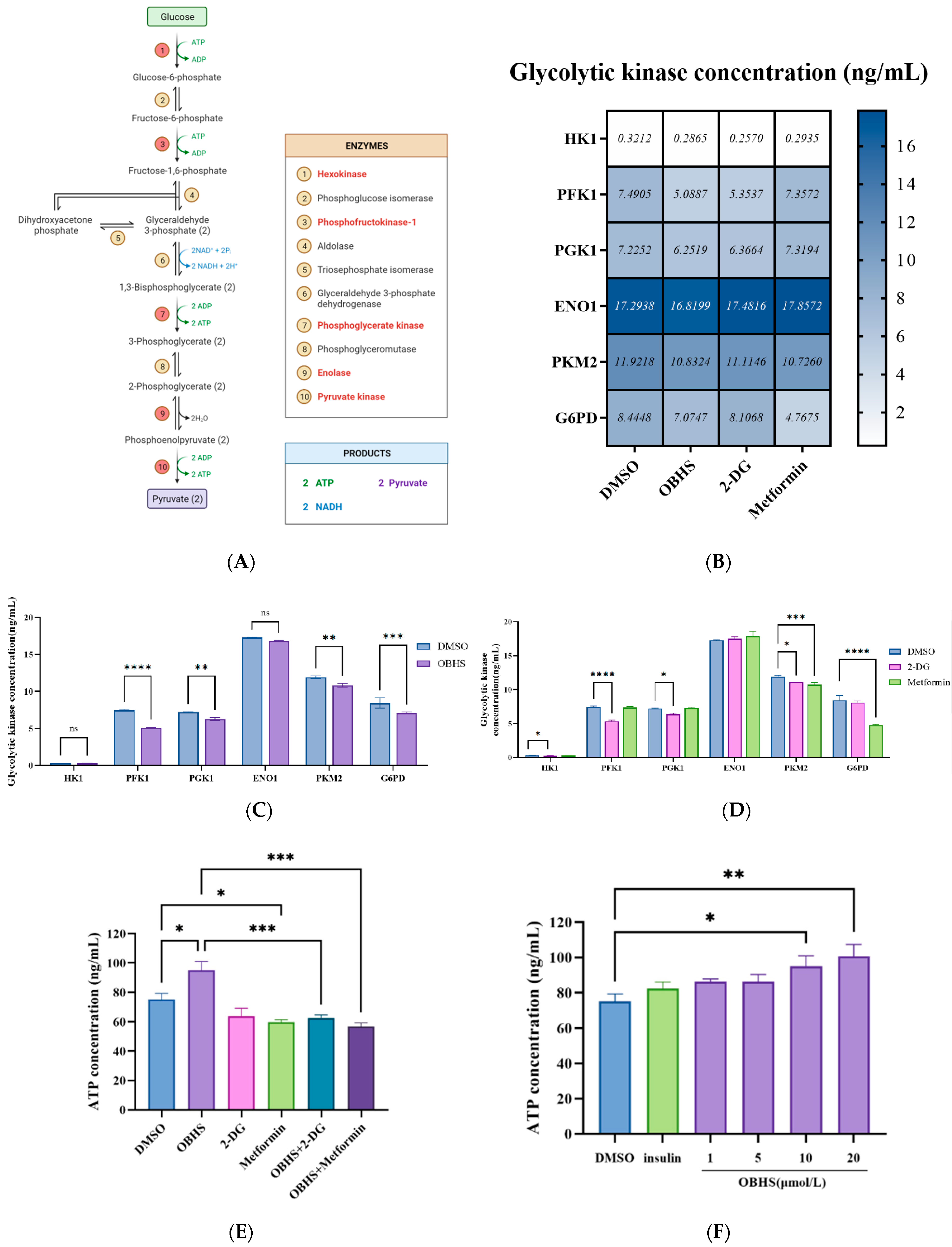
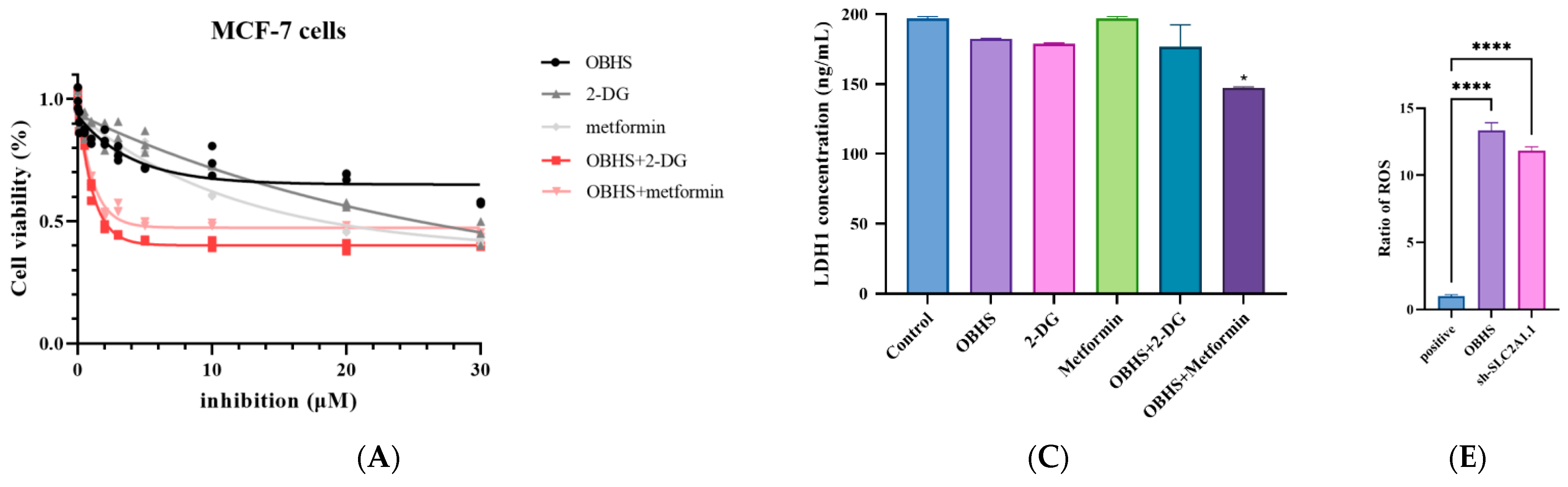
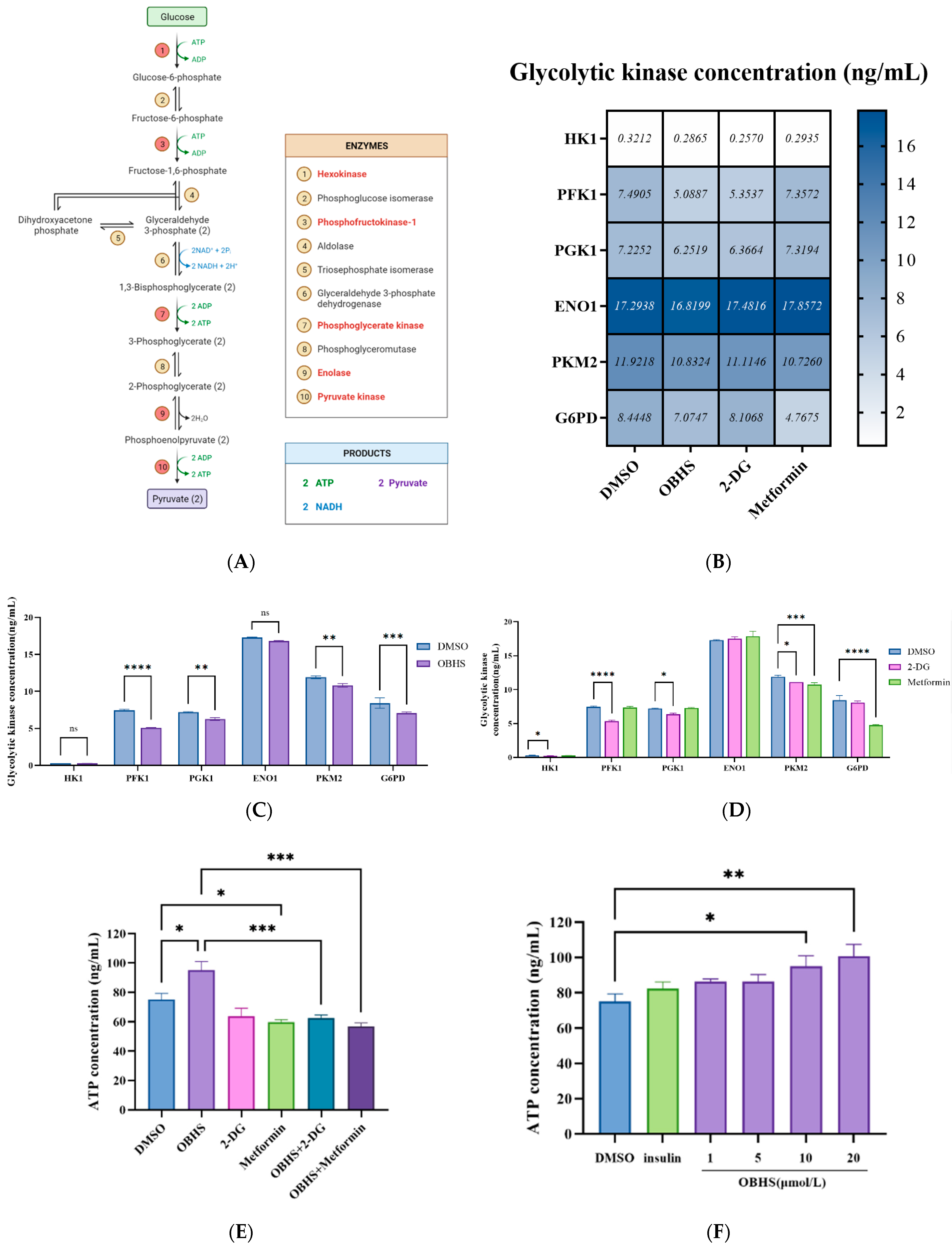
2.4. Anti-Glucometabolic Effect of OBHS in MCF-7 Cells Occurred via the Reprogramming of the Glycolytic Metabolism Pathway in Breast Cancer Cells
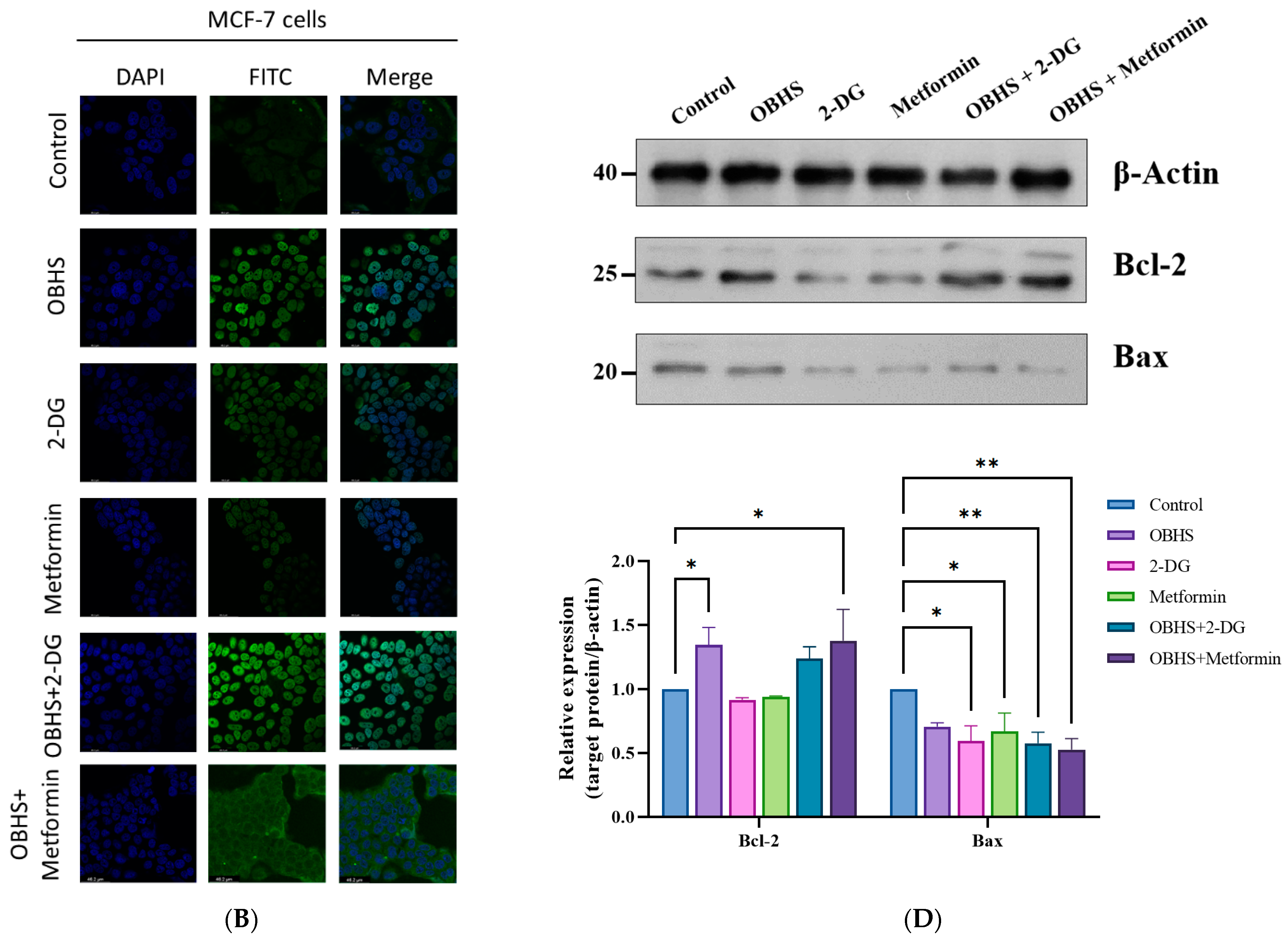
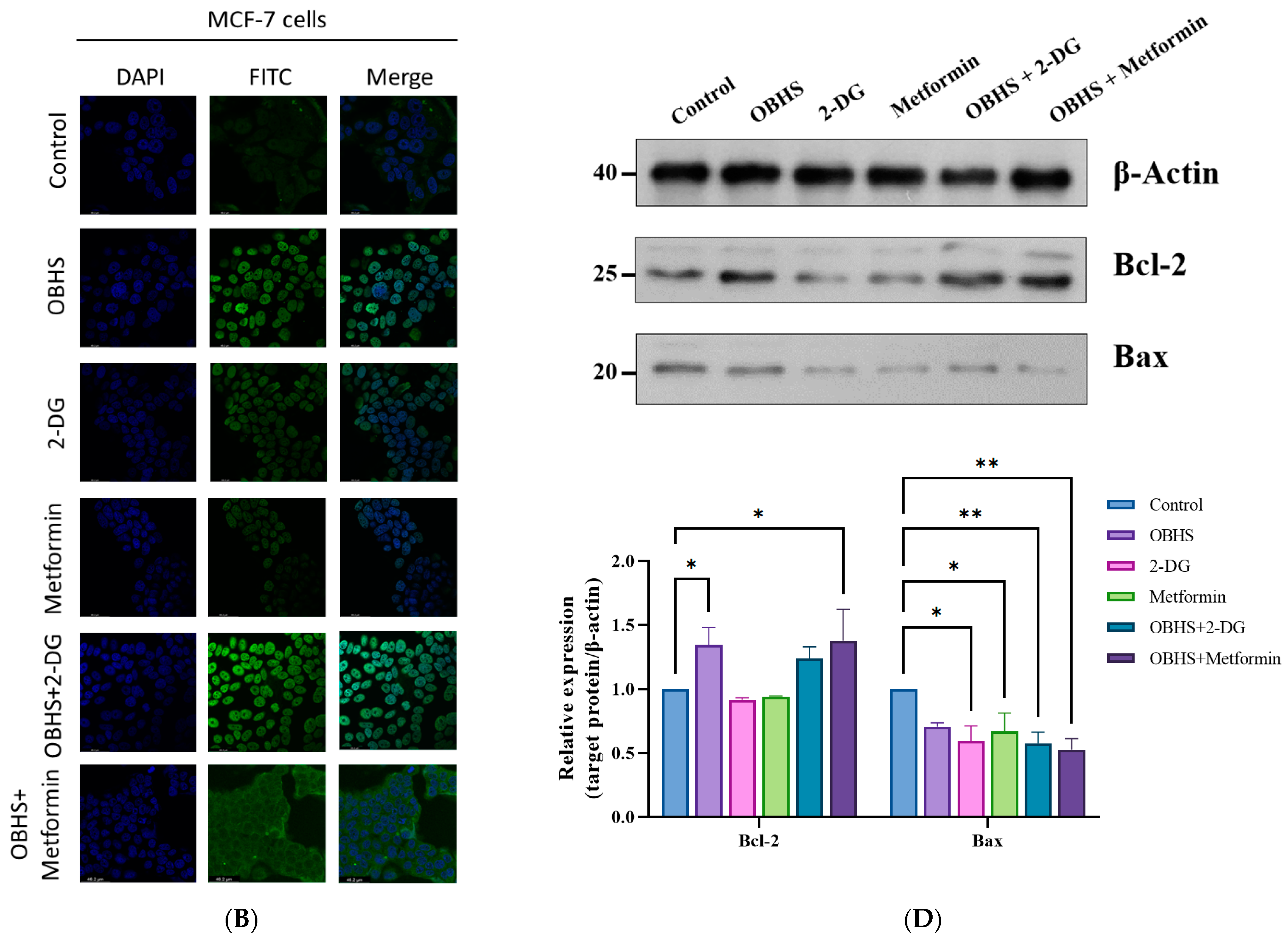
2.5. Enhanced ROS Fluxes Imply Mitochondria Toxicity by OBHS and Lactate-Inducted Cell Apoptosis
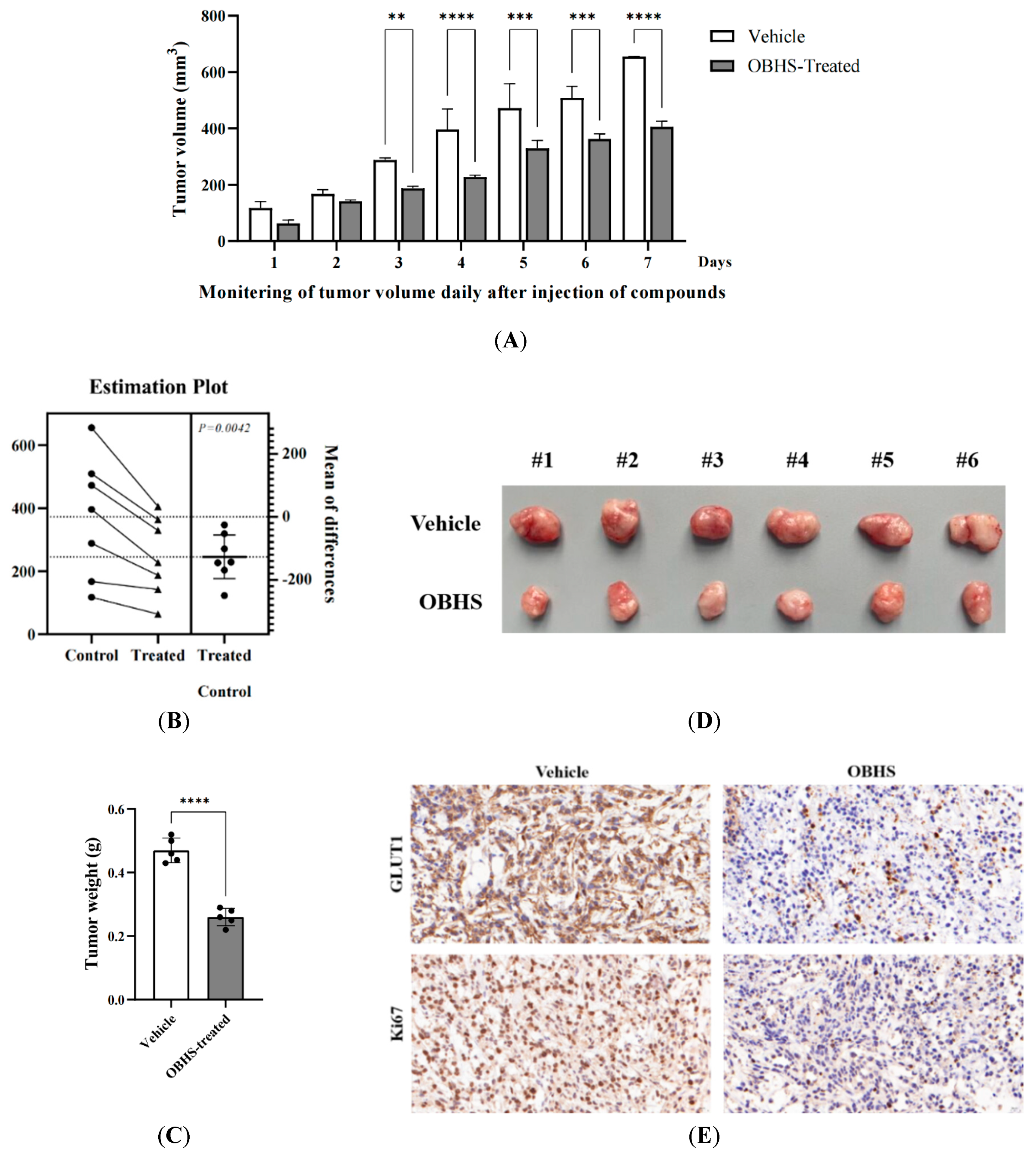
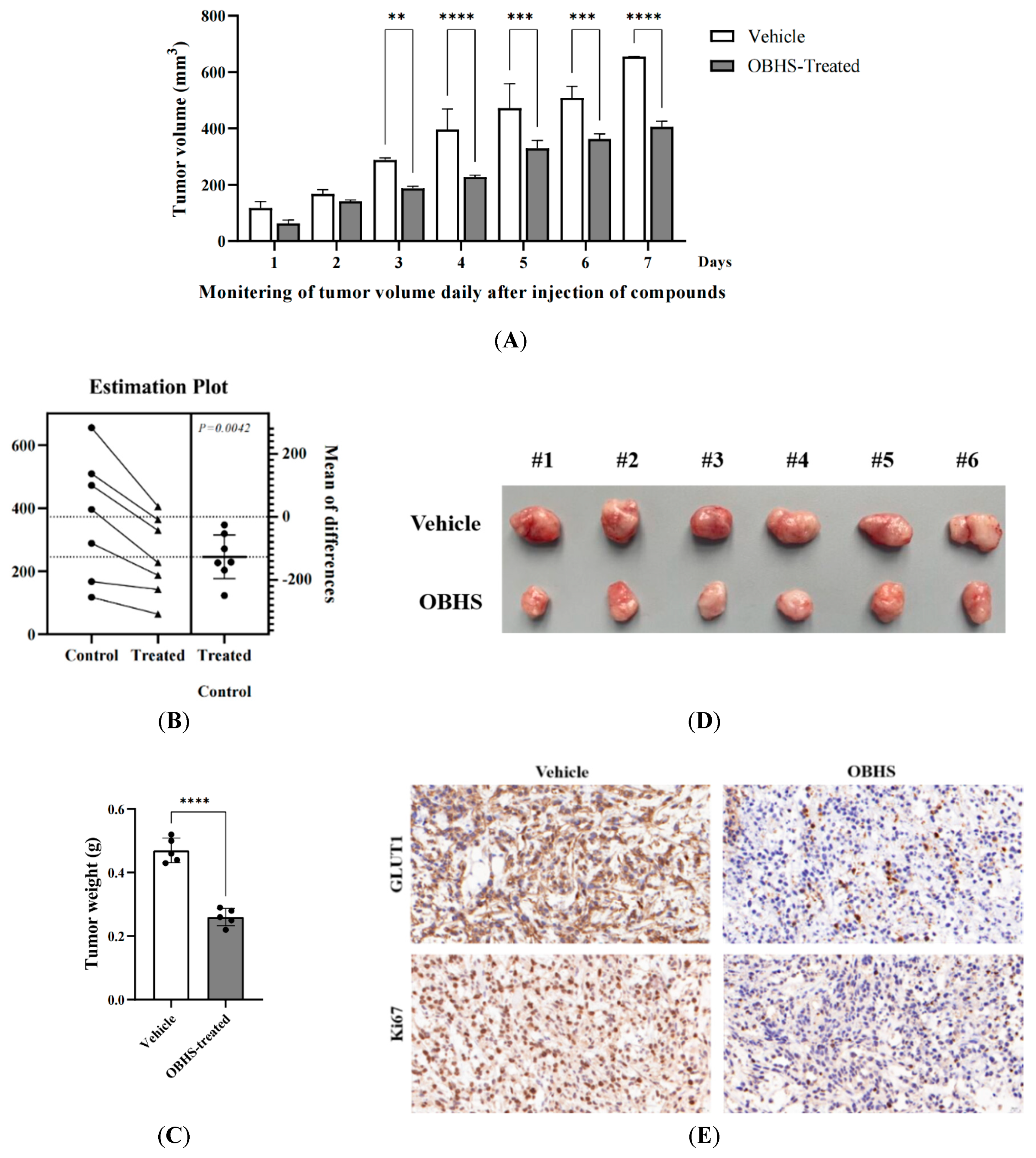
2.6. OBHS Had Suppression Effect on Breast Cancer Progression and Proliferation, and It Reprogrammed the Metabolic Pathways In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Viability Studies
4.4. Survival Analysis
4.5. Western Blot Analysis
4.6. Real-time Quantitative PCR (RT-qPCR)
4.7. Glucose Consumption and Uptake Assay
4.8. Quantification of Enzymatic Production
4.9. Quantification of Intracellular and Extracellular Lactate and ATP Production
4.10. Reactive Oxygen Species (ROS) Determination
4.11. TdT-Mediated dUTP Nick-End Labeling (TUNEL) Staining
4.12. Transfection
4.13. MCF-7 Xenograft Mouse Model
4.14. Immunohistochemistry
4.15. Bioinformatic Studies
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Zhao, B.; Kang, J.; Li, S.; Wu, H. Trend of disease burden and risk factors of breast cancer in developing countries and territories, from 1990 to 2019: Results from the Global Burden of Disease Study 2019. Front. Public Health 2022, 10, 1078191. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.M.; Webb-Vargas, Y.; Wheeler, W.; Gail, M.H. Proportion of U.S. trends in breast cancer incidence attributable to long-term changes in risk factor distributions. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1214–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Riondino, S.; Buonomo, O.; Palmirotta, R.; Guadagni, F.; Roselli, M. Type 2 diabetes and breast cancer: The interplay between impaired glucose metabolism and oxidant stress. Oxid. Med. Cell. Longev. 2015, 2015, 183928. [Google Scholar] [CrossRef] [Green Version]
- Lambe, M.; Wigertz, A.; Garmo, H.; Walldius, G.; Jungner, I.; Hammar, N. Impaired glucose metabolism and diabetes and the risk of breast, endometrial, and ovarian cancer. Cancer Causes Control. 2011, 22, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. CA Cancer J. Clin. 2010, 60, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.X.; Zhu, H.H.; Zhu, Y.M. Diabetes and cancer: Associations, mechanisms, and implications for medical practice. World J. Diabetes 2014, 5, 372–380. [Google Scholar] [CrossRef]
- Miao, Z.F.; Xu, H.; Xu, Y.Y.; Wang, Z.N.; Zhao, T.T.; Song, Y.X.; Xu, H.M. Diabetes mellitus and the risk of gastric cancer: A meta-analysis of cohort studies. Oncotarget 2017, 8, 44881–44892. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Arizmendi-Izazaga, A.; Navarro-Tito, N.; Jiménez-Wences, H.; Mendoza-Catalán, M.A.; Martínez-Carrillo, D.N.; Zacapala-Gómez, A.E.; Olea-Flores, M.; Dircio-Maldonado, R.; Torres-Rojas, F.I.; Soto-Flores, D.G.; et al. Metabolic Reprogramming in Cancer: Role of HPV 16 Variants. Pathogens 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Unterlass, J.E.; Curtin, N.J. Warburg and Krebs and related effects in cancer. Expert Rev. Mol. Med. 2019, 21, e4. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.B.; Comninos, J.S.; Stossi, F.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Synthesis and evaluation of estrogen receptor ligands with bridged oxabicyclic cores containing a diarylethylene motif: Estrogen antagonists of unusual structure. J. Med. Chem. 2005, 48, 7261–7274. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, C.; Li, C.; Qiao, J.; Zhang, F.; Huang, M.; Ren, W.; Dong, C.; Huang, J.; Zhou, H.B. Discovery of novel SERMs with a ferrocenyl entity based on the oxabicyclo[2.2.1]heptene scaffold and evaluation of their antiproliferative effects in breast cancer cells. Org. Biomol. Chem. 2012, 10, 9689–9699. [Google Scholar] [CrossRef]
- Tang, C.; Li, C.; Zhang, S.; Hu, Z.; Wu, J.; Dong, C.; Huang, J.; Zhou, H.B. Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. J. Med. Chem. 2015, 58, 4550–4572. [Google Scholar] [CrossRef]
- Wu, J.; Yan, J.; Fang, P.; Zhou, H.B.; Liang, K.; Huang, J. Three-dimensional oxabicycloheptene sulfonate targets the homologous recombination and repair programmes through estrogen receptor α antagonism. Cancer Lett. 2020, 469, 78–88. [Google Scholar] [CrossRef]
- Giammarioli, A.M.; Gambardella, L.; Barbati, C.; Pietraforte, D.; Tinari, A.; Alberton, M.; Gnessi, L.; Griffin, R.J.; Minetti, M.; Malorni, W. Differential effects of the glycolysis inhibitor 2-deoxy-D-glucose on the activity of pro-apoptotic agents in metastatic melanoma cells, and induction of a cytoprotective autophagic response. Int. J. Cancer 2012, 131, E337–E347. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Hosio, M.; Urpilainen, E.; Hautakoski, A.; Marttila, M.; Arffman, M.; Sund, R.; Ahtikoski, A.; Puistola, U.; Läärä, E.; Karihtala, P.; et al. Association of antidiabetic medication and statins with survival from ductal and lobular breast carcinoma in women with type 2 diabetes. Sci. Rep. 2021, 11, 10445. [Google Scholar] [CrossRef] [PubMed]
- Hosio, M.; Urpilainen, E.; Hautakoski, A.; Marttila, M.; Arffman, M.; Sund, R.; Ahtikoski, A.; Puistola, U.; Karihtala, P.; Jukkola, A.; et al. Survival after breast cancer in women with type 2 diabetes using antidiabetic medication and statins: A retrospective cohort study. Acta Oncol. 2020, 59, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.C.; Krishan, A.; Lampidis, T.J. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother. Pharm. 2004, 53, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Mehrens, H.; Wang, P.; Kim, S.G. Glucose metabolism-weighted imaging with chemical exchange-sensitive MRI of 2-deoxyglucose (2DG) in brain: Sensitivity and biological sources. Neuroimage 2016, 143, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.Z.; Sutula, T.P.; Rutecki, P.A. 2-Deoxy-d-glucose reduces epileptiform activity by presynaptic mechanisms. J. Neurophysiol. 2019, 121, 1092–1101. [Google Scholar] [CrossRef]
- Zhu, G.; Guo, N.; Yong, Y.; Xiong, Y.; Tong, Q. Effect of 2-deoxy-D-glucose on gellan gum biosynthesis by Sphingomonas paucimobilis. Bioprocess Biosyst. Eng. 2019, 42, 897–900. [Google Scholar] [CrossRef]
- O’Neill, S.; Porter, R.K.; McNamee, N.; Martinez, V.G.; O’Driscoll, L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci. Rep. 2019, 9, 3788. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef]
- Spencer, N.Y.; Stanton, R.C. The Warburg Effect, Lactate, and Nearly a Century of Trying to Cure Cancer. Semin. Nephrol. 2019, 39, 380–393. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Robertson-Tessi, M.; Gillies, R.J.; Gatenby, R.A.; Anderson, A.R. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 2015, 75, 1567–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Alò, P.L.; Visca, P.; Botti, C.; Galati, G.M.; Sebastiani, V.; Andreano, T.; Di Tondo, U.; Pizer, E.S. Immunohistochemical expression of human erythrocyte glucose transporter and fatty acid synthase in infiltrating breast carcinomas and adjacent typical/atypical hyperplastic or normal breast tissue. Am. J. Clin Pathol. 2001, 116, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Rivenzon-Segal, D.; Rushkin, E.; Polak-Charcon, S.; Degani, H. Glucose transporters and transport kinetics in retinoic acid-differentiated T47D human breast cancer cells. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E508–E519. [Google Scholar] [CrossRef]
- Avril, N.; Menzel, M.; Dose, J.; Schelling, M.; Weber, W.; Jänicke, F.; Nathrath, W.; Schwaiger, M. Glucose metabolism of breast cancer assessed by 18F-FDG PET: Histologic and immunohistochemical tissue analysis. J. Nucl. Med. 2001, 42, 9–16. [Google Scholar]
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002, 524, 199–203. [Google Scholar] [CrossRef]
- Godoy, A.; Ulloa, V.; Rodríguez, F.; Reinicke, K.; Yañez, A.J.; García Mde, L.; Medina, R.A.; Carrasco, M.; Barberis, S.; Castro, T.; et al. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell. Physiol. 2006, 207, 614–627. [Google Scholar] [CrossRef]
- Doege, H.; Bocianski, A.; Joost, H.G.; Schürmann, A. Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem. J. 2000, 350 Pt 3, 771–776. [Google Scholar] [CrossRef]
- Dawson, P.A.; Mychaleckyj, J.C.; Fossey, S.C.; Mihic, S.J.; Craddock, A.L.; Bowden, D.W. Sequence and functional analysis of GLUT10: A glucose transporter in the Type 2 diabetes-linked region of chromosome 20q12-13.1. Mol. Genet. Metab. 2001, 74, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Park, C.K.; Park, M.; Kim, W.K.; Cho, A.; Kim, H. Clinicopathologic Features and Molecular Characteristics of Glucose Metabolism Contributing to ¹⁸F-fluorodeoxyglucose Uptake in Gastrointestinal Stromal Tumors. PLoS ONE 2015, 10, e0141413. [Google Scholar] [CrossRef]
- Matsui, C.; Takatani-Nakase, T.; Maeda, S.; Nakase, I.; Takahashi, K. Potential Roles of GLUT12 for Glucose Sensing and Cellular Migration in MCF-7 Human Breast Cancer Cells Under High Glucose Conditions. Anticancer. Res. 2017, 37, 6715–6722. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, Z.; Chen, J.; Yi, J.; Liu, Z.; Zhao, H.; Wei, H. Glycometabolic adaptation mediates the insensitivity of drug-resistant K562/ADM leukaemia cells to adriamycin via the AKT-mTOR/c-Myc signalling pathway. Mol. Med. Rep. 2017, 15, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.L.; Chen, W.K.; Chen, X.Y.; Lu, H.; Sun, Z.R.; Yu, Q.; Sun, P.F.; Xu, Y.J.; Zhu, M.M.; Jiang, N.; et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1α pathway suppression. Nat. Commun. 2020, 11, 1869. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, H.; Che, N.; Zheng, Y.; Fan, W.; Li, M.; Li, X.; Xuan, Y. HBXIP accelerates glycolysis and promotes cancer angiogenesis via AKT/mTOR pathway in bladder cancer. Exp. Mol. Pathol. 2021, 121, 104665. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feng, R.; Wang, D.; Huo, T.; Jiang, H. Triclosan-induced glycolysis drives inflammatory activation in microglia via the Akt/mTOR/HIF 1α signaling pathway. Ecotoxicol. Environ. Saf. 2021, 224, 112664. [Google Scholar] [CrossRef] [PubMed]
- Holloway, R.W.; Marignani, P.A. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers 2021, 13, 2922. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Voelker, H.U.; Kapp, M.; Krockenberger, M.; Dietl, J.; Kammerer, U. Glycolytic phenotype in breast cancer: Activation of Akt, up-regulation of GLUT1, TKTL1 and down-regulation of M2PK. J. Cancer Res. Clin. Oncol. 2010, 136, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Kim, H.; Nam, K.; Shin, I. Silencing of Glut1 induces chemoresistance via modulation of Akt/GSK-3β/β-catenin/survivin signaling pathway in breast cancer cells. Arch. Biochem. Biophys. 2017, 636, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 2011, 16, 12–19. [Google Scholar] [CrossRef]
- Dockx, Y.; Vangestel, C.; De Bruycker, S.; Van den Wyngaert, T.; Huizing, M.; Staelens, S.; Stroobants, S. 18F-FDG and 18F-FLT Uptake Profiles for Breast Cancer Cell Lines Treated with Targeted PI3K/Akt/mTOR Therapies. Cancer Biother. Radiopharm. 2023, 38, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Hosios, A.M.; Manning, B.D. Cancer signaling drives cancer metabolism: AKT and the warburg effect. Cancer Res. 2021, 81, 4896–4898. [Google Scholar] [CrossRef]
- Yecies, J.L.; Manning, B.D. mTOR links oncogenic signaling to tumor cell metabolism. J. Mol. Med. 2011, 89, 221–228. [Google Scholar] [CrossRef]
- Chen, L.; Bai, Y.; Everaert, N.; Li, X.; Tian, G.; Hou, C.; Zhang, D. Effects of protein phosphorylation on glycolysis through the regulation of enzyme activity in ovine muscle. Food Chem. 2019, 293, 537–544. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annibaldi, A.; Widmann, C. Glucose metabolism in cancer cells. Curr. Opin. Clin. Nutr. Metab Care 2010, 13, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [CrossRef]
- You, M.; Jin, J.; Liu, Q.; Xu, Q.; Shi, J.; Hou, Y. PPARα Promotes Cancer Cell Glut1 Transcription Repression. J. Cell Biochem. 2017, 118, 1556–1562. [Google Scholar] [CrossRef]
- Melvin, J.C.; Garmo, H.; Holmberg, L.; Hammar, N.; Walldius, G.; Jungner, I.; Lambe, M.; Van Hemelrijck, M. Glucose and lipoprotein biomarkers and breast cancer severity using data from the Swedish AMORIS cohort. BMC Cancer 2017, 17, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, T.; Kang, X.; Liu, Z.; Zhao, S.; Ma, W.; Xuan, Q.; Liu, H.; Wang, Z.; Zhang, Q. Altered glycometabolism affects both clinical features and prognosis of triple-negative and neoadjuvant chemotherapy-treated breast cancer. Tumour Biol. 2016, 37, 8159–8168. [Google Scholar] [CrossRef]
- Sakashita, M.; Aoyama, N.; Minami, R.; Maekawa, S.; Kuroda, K.; Shirasaka, D.; Ichihara, T.; Kuroda, Y.; Maeda, S.; Kasuga, M. Glut1 expression in T1 and T2 stage colorectal carcinomas: Its relationship to clinicopathological features. Eur. J. Cancer 2001, 37, 204–209. [Google Scholar] [CrossRef]
- Hong, S.Y.; Yu, F.X.; Luo, Y.; Hagen, T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell. Signal. 2016, 28, 377–383. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [Green Version]
- Avanzato, D.; Pupo, E.; Ducano, N.; Isella, C.; Bertalot, G.; Luise, C.; Pece, S.; Bruna, A.; Rueda, O.M.; Caldas, C.; et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res. 2018, 78, 3432–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, M.; Abdullah, N.; Thowfeik, F.S.; Altorki, N.K.; McGraw, T.E. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. eLife 2017, 6, e26896. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, H.; Pal, S.; Sabnam, S.; Pal, A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. 2020, 241, 117148. [Google Scholar] [CrossRef]
- Rashmi, K.C.; Harsha Raj, M.; Paul, M.; Girish, K.S.; Salimath, B.P.; Aparna, H.S. A new pyrrole based small molecule from Tinospora cordifolia induces apoptosis in MDA-MB-231 breast cancer cells via ROS mediated mitochondrial damage and restoration of p53 activity. Chem. Biol. Interact. 2019, 299, 120–130. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, T.; Zhu, X.; Yang, C.; Wang, Y.; Zhou, N.; Ju, B.; Zhou, T.; Deng, G.; Qiu, C. Hyperoside Induces Breast Cancer Cells Apoptosis via ROS-Mediated NF-κB Signaling Pathway. Int. J. Mol. Sci. 2019, 21, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashourpour, M.; Mostafavi Hosseini, F.; Amini, M.; Saeedian Moghadam, E.; Kazerouni, F.; Arman, S.Y.; Shahsavari, Z. Pyrazole Derivatives Induce Apoptosis via ROS Generation in the Triple Negative Breast Cancer Cells, MDA-MB-468. Asian Pac. J. Cancer Prev. 2021, 22, 2079–2087. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Li, Q.; Fan, Y.; Fang, P.; Zhou, H.; Huang, J. OBHS Drives Abnormal Glycometabolis Reprogramming via GLUT1 in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 7136. https://doi.org/10.3390/ijms24087136
Wang K, Li Q, Fan Y, Fang P, Zhou H, Huang J. OBHS Drives Abnormal Glycometabolis Reprogramming via GLUT1 in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(8):7136. https://doi.org/10.3390/ijms24087136
Chicago/Turabian StyleWang, Kexin, Qiuzi Li, Yufeng Fan, Pingping Fang, Haibing Zhou, and Jian Huang. 2023. "OBHS Drives Abnormal Glycometabolis Reprogramming via GLUT1 in Breast Cancer" International Journal of Molecular Sciences 24, no. 8: 7136. https://doi.org/10.3390/ijms24087136
APA StyleWang, K., Li, Q., Fan, Y., Fang, P., Zhou, H., & Huang, J. (2023). OBHS Drives Abnormal Glycometabolis Reprogramming via GLUT1 in Breast Cancer. International Journal of Molecular Sciences, 24(8), 7136. https://doi.org/10.3390/ijms24087136