
Defective Mitochondrial Dynamics and Protein Degradation Pathways Underlie Cadmium-Induced Neurotoxicity and Cell Death in Huntington’s Disease Striatal Cells
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
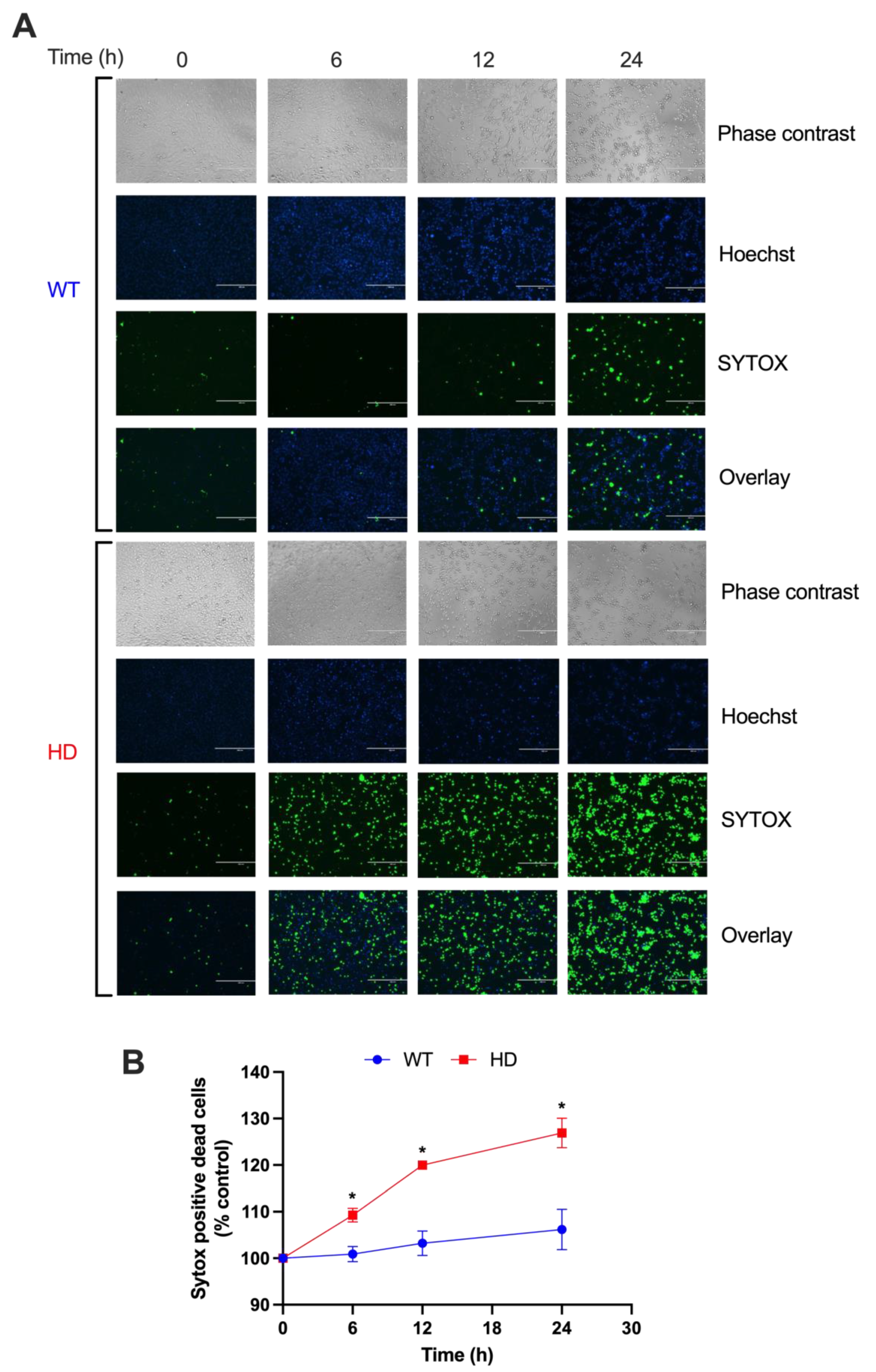
2.1. HD Cells Are More Sensitive to Early Cd-Induced Neurotoxicity and Exhibit Greater Cell Death
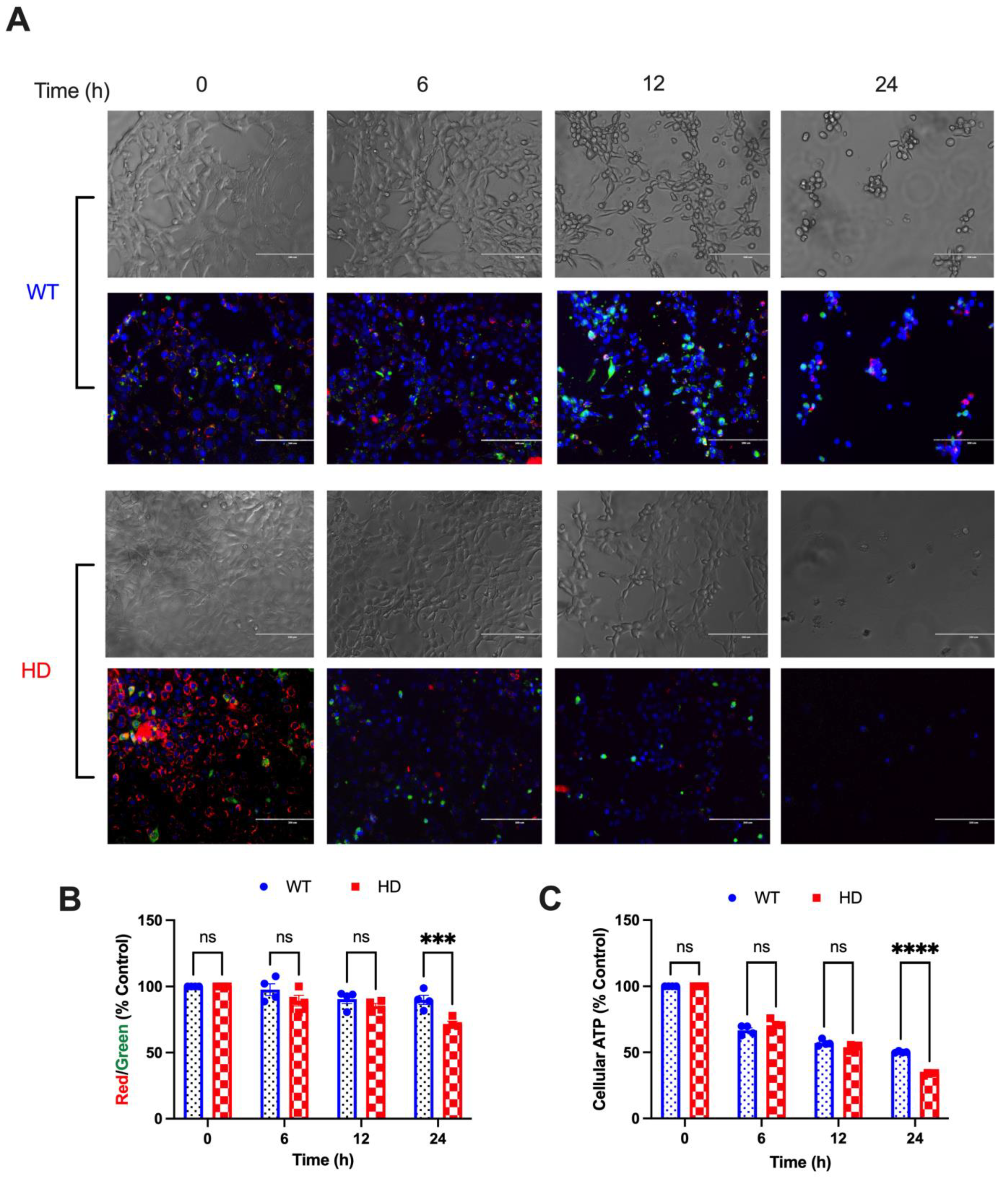
2.2. HD Cells Are More Susceptible to Cd-Induced Mitochondrial Dysfunction Compared with WT
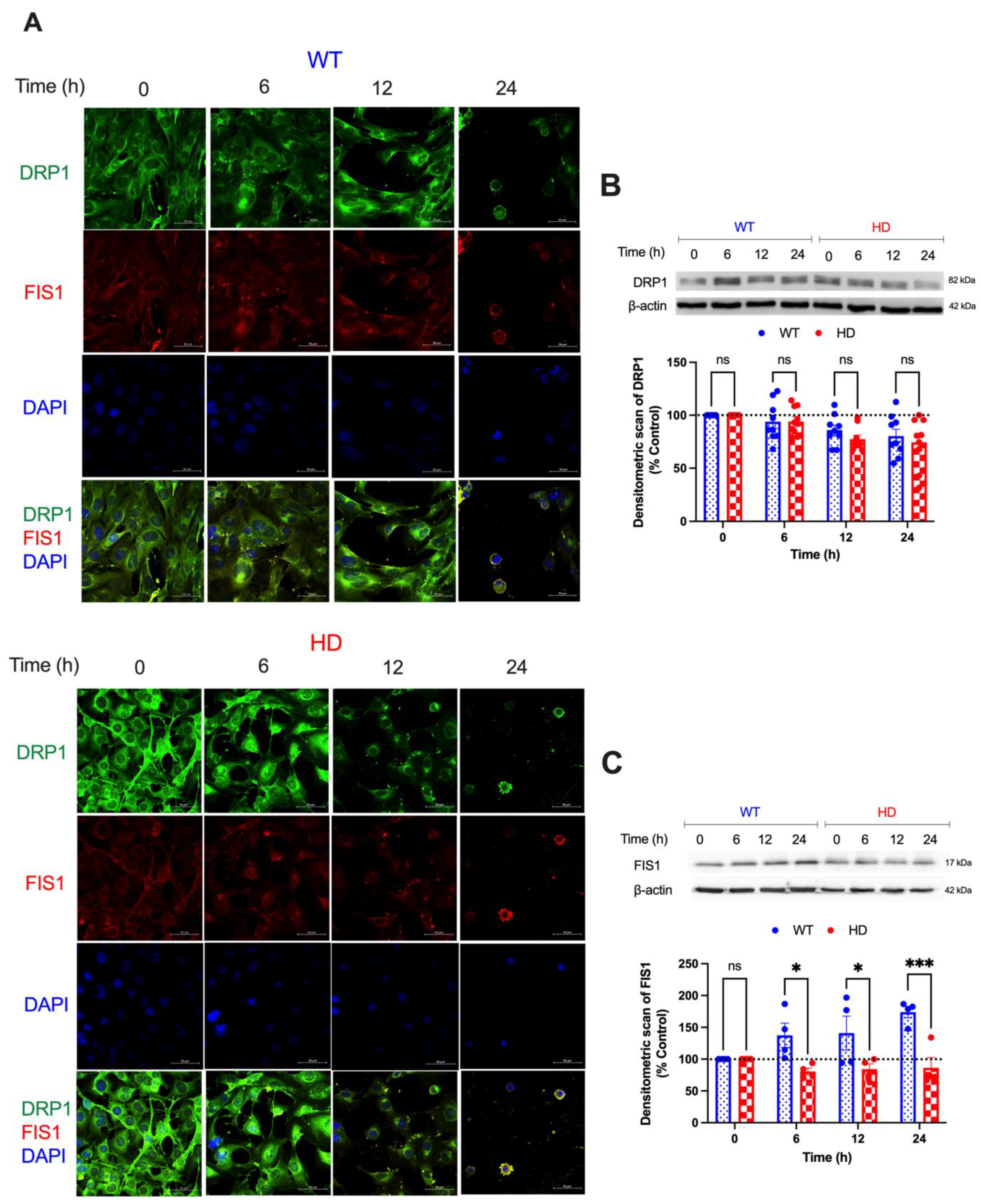
2.3. HD Promotes Cd-Induced Alterations in Mitochondrial Fission Proteins
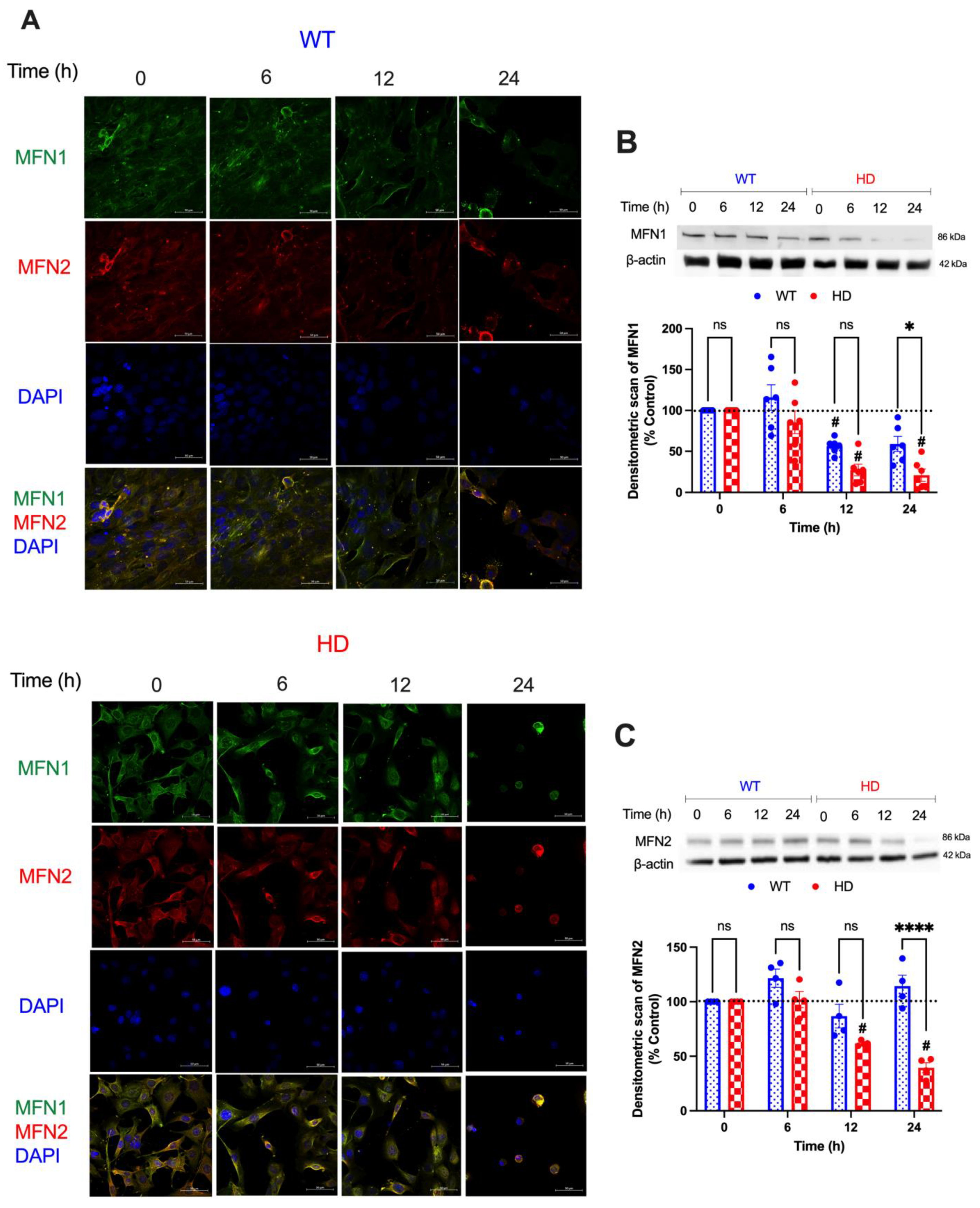
2.4. Mitochondrial Fusion Proteins Are Modulated by Acute Cd Exposure
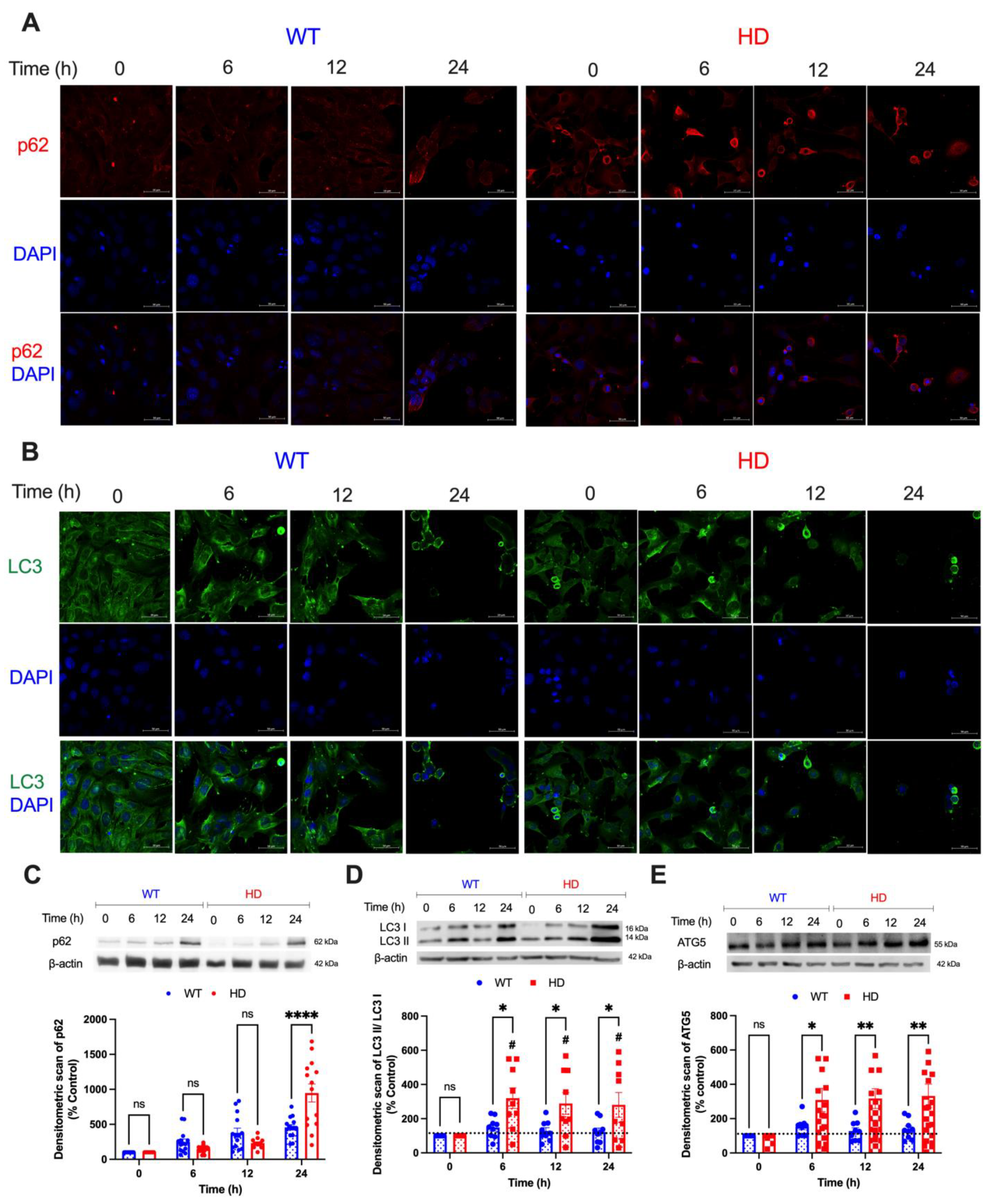
2.5. Key Autophagic Markers Are Upregulated in HD Cells upon Cd Exposure
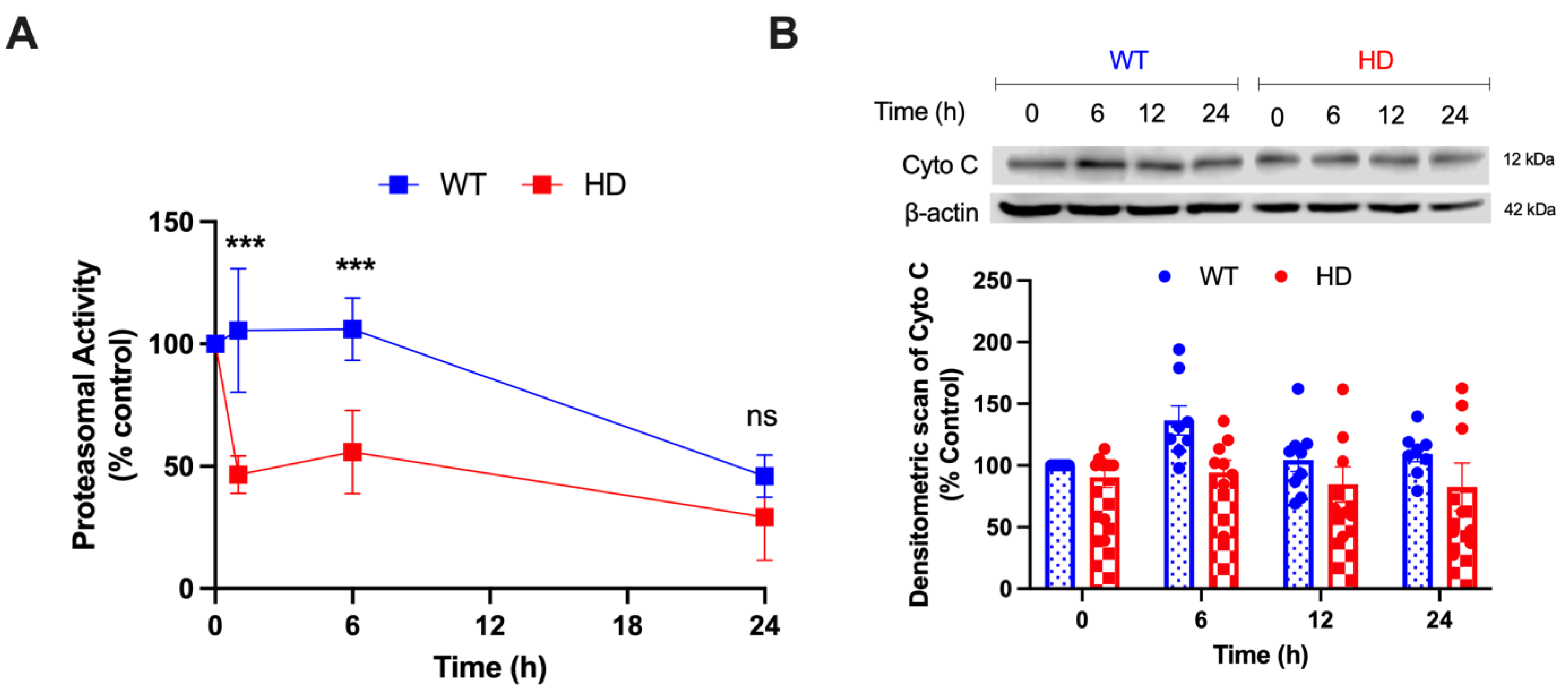
2.6. HD and Cd Cooperatively Reduce Proteasomal Activity and Augments Mitochondrial Mediated Cell Death
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Death Assay
4.4. Mitochondrial Membrane Potential
4.5. ATP Assay
4.6. Immunocytochemistry (ICC)
4.7. Immunoblotting
4.8. Proteasomal Activity Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghosh, R.; Tabrizi, S.J. Clinical features of huntington’s disease. Adv. Exp. Med. Biol. 2018, 1049, 1–28. [Google Scholar]
- Gusella, J.F.; MacDonald, M.E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000, 1, 109–115. [Google Scholar] [CrossRef]
- Wanker, E.E. Protein aggregation and pathogenesis of Huntington’s disease: Mechanisms and correlations. Biol. Chem. 2000, 381, 937–942. [Google Scholar] [CrossRef]
- Henshall, T.L.; Tucker, B.; Lumsden, A.L.; Nornes, S.; Lardelli, M.T.; Richards, R.I. Selective Neuronal Requirement for Huntingtin in the Developing Zebrafish. Hum. Mol. Genet. 2009, 18, 4830–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.H.J. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant Huntingtin Impairs Axonal Trafficking in Mammalian Neurons In Vivo and In Vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernández, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011, 25, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the synapse. Front. Cell. Neurosci. 2021, 15, 689332. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Möncke-Buchner, E.; Reich, S.; Mücke, M.; Reuter, M.; Messer, W.; Wanker, E.E.; Krüger, D.H. Counting CAG repeats in the Huntington’s disease gene by restriction endonuclease EcoP15I cleavage. Nucleic Acids Res. 2002, 30, e83. [Google Scholar] [CrossRef] [Green Version]
- Zala, D.; Benchoua, A.; Brouillet, E.; Perrin, V.; Gaillard, M.C.; Zurn, A.D.; Aebischer, P.; Déglon, N. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol. Dis. 2005, 20, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Wegrzynowicz, M.; Holt, H.K.; Friedman, D.B.; Bowman, A.B. Changes in the striatal proteome of YAC128Q mice exhibit gene-environment interactions between mutant huntingtin and manganese. J. Proteome Res. 2012, 11, 1118–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwakye, G.F.; Jiménez, J.A.; Thomas, M.G.; Kingsley, B.A.; McIIvin, M.; Saito, M.A.; Korley, E.M. Heterozygous huntingtin promotes cadmium neurotoxicity and neurodegeneration in striatal cells via altered metal transport and protein kinase C delta dependent oxidative stress and apoptosis signaling mechanisms. Neurotoxicology 2019, 70, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of Copper Ion Mediated Huntington’s Disease Progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.B.; Li, D.; Wegrzynowicz, M.; Vadodaria, B.K.; Anderson, J.G.; Kwakye, G.F.; Aschner, M.; Erikson, K.M.; Bowman, A.B. Disease-toxicant screen reveals a neuroprotective interaction between Huntington’s disease and manganese exposure. J. Neurochem. 2010, 112, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.; Hornburg, D.; Antonin, M.; Krahmer, N.; Collado, J.; Schaffer, M.; Zubaite, G.; Lüchtenborg, C.; Sachsenheimer, T.; Brügger, B.; et al. Molecular and structural architecture of polyQ aggregates in yeast. Proc. Natl. Acad. Sci. USA 2018, 115, E3446–E3453. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C.; Martin, S.J. Mitochondrial Fission/Fusion Dynamics and Apoptosis. Mitochondrion 2010, 10, 640–648. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M.A. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The functional impact of mitochondrial structure across subcellular scales. Front. Physiol. 2020, 11, 541040. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Ohsumi, Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007, 581, 2156–2161. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Poliakoff, E.; Sollom, A.C.; Howard, E.; Craufurd, D.; Snowden, J.S. Automaticity and attention in Huntington’s disease: When two hands are not better than one. Neuropsychologia 2010, 48, 171–178. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic Proteasomes Cannot Digest Polyglutamine Sequences and Release Them during Degradation of Polyglutamine-Containing Proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Van Dellen, A.; Grote, H.E.; Hannan, A.J. Gene-Environment Interactions, Neuronal Dysfunction and Pathological Plasticity in Huntington’s Disease. Clin. Exp. Pharmacol. Physiol. 2005, 32, 1007–1019. [Google Scholar] [CrossRef]
- Mo, C.; Hannan, A.J.; Renoir, T. Environmental factors as modulators of neurodegeneration: Insights from gene-environment interactions in Huntington’s disease. Neurosci. Biobehav. Rev. 2015, 52, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Van Dellen, A.; Hannan, A.J. Genetic and Environmental Factors in the Pathogenesis of Huntington’s Disease. Neurogenetics 2004, 5, 9–17. [Google Scholar] [CrossRef]
- Dominah, G.A.; McMinimy, R.A.; Kallon, S.; Kwakye, G.F. Acute exposure to chlorpyrifos caused NADPH oxidase mediated oxidative stress and neurotoxicity in a striatal cell model of Huntington’s disease. Neurotoxicology 2017, 60, 54–69. [Google Scholar] [CrossRef]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayán, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar] [PubMed] [Green Version]
- Sudarsky, L.; Myers, R.H.; Walshe, T.M. Huntington’s disease in monozygotic twins reared apart. J. Med. Genet. 1983, 20, 408–411. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.D.; Omenn, G.S. Monozygotic twins with Huntington’s disease in a family expressing the rigid variant. Neurology 1975, 25, 1126–1129. [Google Scholar] [CrossRef]
- Georgiou, N.; Bradshaw, J.L.; Chiu, E.; Tudor, A.; O’Gorman, L.; Phillips, J.G. Differential clinical and motor control function in a pair of monozygotic twins with Huntington’s disease. Mov. Disord. 1999, 14, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Henshall, T.L.; Dayan, S.; Lardelli, M.T.; Richards, R.I. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum. Mol. Genet. 2007, 16, 1905–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfalzer, A.C.; Wilcox, J.M.; Codreanu, S.G.; Totten, M.; Bichell, T.J.; Halbesma, T.; Umashanker, P.; Yang, K.L.; Parmalee, N.L.; Sherrod, S.D.; et al. Huntington’s disease genotype suppresses global manganese-responsive processes in pre-manifest and manifest YAC128 mice. Met. Integr. Biometal Sci. 2020, 12, 1118–1130. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain A J. Neurol. 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Res 2016, 5, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Kwakye, G.F.; Paoliello, M.M.B.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Manganese-Induced Parkinsonism and Parkinson’s Disease: Shared and Distinguishable Features. Int. J. Environ. Res. Public Health 2015, 2, 7519–7540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Leavitt, B.R. Iron dysregulation in Huntington’s disease. J. Neurochem. 2014, 130, 328–350. [Google Scholar] [CrossRef]
- Patel, R.; Aschner, M. Commonalities between Copper Neurotoxicity and Alzheimer’s Disease. Toxics 2021, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Kubier, A.; Wilkin, R.T.; Pichler, T. Cadmium in soils and groundwater: A review. Appl. Geochem. 2019, 108, 104388. [Google Scholar] [CrossRef]
- Tucker, P. Cadmium toxicity. In Case Studies in Environmental Medicine (CSEM); Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2008. [Google Scholar]
- Proshad, R.; Zhang, D.; Uddin, M.; Wu, Y. Presence of cadmium and lead in tobacco and soil with ecological and human health risks in Sichuan province, China. Environ. Sci. Pollut. Res. Int. 2020, 27, 18355–18370. [Google Scholar] [CrossRef]
- Henson, M.C.; Chedrese, P.J. Endocrine disruption by cadmium, a common environmental toxicant with paradoxical effects on reproduction. Exp. Biol. Med. 2004, 229, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Du, Y. Cadmium and its neurotoxic effects. Oxidative Med. Cell. Longev. 2013, 2013, 898034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzocco, J.C.; Jagadapillai, R.; Gozal, E.; Kong, M.; Xu, Q.; Barnes, G.N.; Freedman, J.H. Disruption of essential metal homeostasis in the brain by cadmium and high-fat diet. Toxicol. Rep. 2020, 7, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.-O.; Wang, X.; Hitron, J.A.; Zhang, Z.; Cheng, S.; Budhraja, A.; Ding, S.; Lee, J.-C.; Shi, X. Cadmium Induces Autophagy through ROS-Dependent Activation of the LKB1-AMPK Signaling in Skin Epidermal Cells. Toxicol. Appl. Pharmacol. 2011, 255, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Satarug, S.; Moore, M. Adverse Health Effects of Chronic Exposure to Low-Level Cadmium in Foodstuffs and Cigarette Smoke. Environ. Health Perspect. 2004, 112, 1099–1103. [Google Scholar] [CrossRef] [Green Version]
- Ehret, J.C.; Day, P.S.; Wiegand, R.; Wojcieszek, J.; Chambers, R.A. Huntington disease as a dual diagnosis disorder: Data from the National Research Roster for Huntington disease patients and families. Drug Alcohol Depend. 2007, 86, 283–286. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.N.; Jhnson GV, W. The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J. Bioenerg. Biomembr. 2010, 42, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Mendezarmenta, M. Brain Regional Lipid Peroxidation and Metallothionein Levels of Developing Rats Exposed to Cadmium and Dexamethasone. Toxicol. Lett. 2003, 144, 151–157. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Ortega, Z.; Lucas, J.J. Ubiquitin-proteasome system involvement in Huntington’s disease. Front. Mol. Neurosci. 2014, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [Green Version]
- DelRaso, N.J.; Foy, B.D.; Gearhart, J.M.; Frazier, J.M. Cadmium uptake kinetics in rat hepatocytes: Correction for albumin binding. Toxicol. Sci. 2003, 72, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Shukla, G.S. Ontogenic profile of brain lipids following perinatal exposure to cadmium. J. Appl. Toxicol. 1996, 16, 227–233. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The effects of cadmium toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Kaul, S.; Kanthasamy, A.; Kitazawa, M.; Anantharam, V.; Kanthasamy, A.G. Caspase-3 dependent proteolytic activation of protein kinase Cδ mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: Relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 2003, 18, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Increased mitochondrial fission and neuronal dysfunction in Huntingtons disease: Implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov. Today 2014, 19, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant Huntingtin Binds the Mitochondrial Fission GTPase Dynamin-Related Protein-1 and Increases Its Enzymatic Activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, E.A.; Glazunov, V.V.; Korotkov, S.M. Cd2+-promoted mitochondrial permeability transition: A comparison with other heavy metals. Acta Biochim. Pol. 2004, 51, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.-G.; Hirst, J. The enigma of the respiratory chain supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Zhang, C.; Sun, Y.C.; Zhang, Q.; Lv, M.W.; Guo, K.; Li, J.L. Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci. Total Environ. 2019, 689, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.E.; Sarkar, S.; Rubinsztein, D.C. The ubiquitin proteasome system in Huntington’s disease and the spinocerebellar ataxias. BMC Biochem. 2007, 8 (Suppl. 1), S2. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H.; Moraes, C.T. Extended polyglutamine repeats trigger a feedback loop involving the mitochondrial complex III, the proteasome and huntingtin aggregates. Hum. Mol. Genet. 2007, 16, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Zainelli, G.M.; Ross, C.A.; Troncoso, J.C.; Muma, N.A. Transglutaminase cross-links in intranuclear inclusions in Huntington disease. J. Neuropathol. Exp. Neurol. 2003, 62, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Hong, S.; Faustman, E.M. Cadmium-induced activation of stress signaling pathways, disruption of ubiquitin-dependent protein degradation and apoptosis in primary rat Sertoli cell-gonocyte cocultures. Toxicol. Sci. 2008, 104, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Heng, M.Y.; Detloff, P.J.; Paulson, H.L.; Albin, R.L. Early alterations of autophagy in Huntington disease-like mice. Autophagy 2010, 6, 1206–1208. [Google Scholar] [CrossRef] [Green Version]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef]
- Trettel, F.; Rigamonti, D.; Hilditch-Maguire, P.; Wheeler, V.C.; Sharp, A.H.; Persichetti, F.; Cattaneo, E.; MacDonald, M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000, 9, 2799–2809. [Google Scholar] [CrossRef] [Green Version]
- Kwakye, G.F.; Li, D.; Bowman, A.B. Novel high-throughput assay to assess cellular manganese levels in a striatal cell line model of Huntington’s disease confirms a deficit in manganese accumulation. Neurotoxicology 2011, 32, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Bryan, M.R.; Uhouse, M.A.; Nordham, K.D.; Joshi, P.; Rose, D.I.; O’Brien, M.T.; Aschner, M.; Bowman, A.B. Phosphatidylinositol 3 kinase (PI3K) modulates manganese homeostasis and manganese-induced cell signaling in a murine striatal cell line. Neurotoxicology 2018, 64, 185–194. [Google Scholar] [CrossRef]
- Bryan, M.R.; O’Brien, M.T.; Nordham, K.D.; Rose, D.I.; Foshage, A.M.; Joshi, P.; Nitin, R.; Uhouse, M.A.; Di Pardo, A.; Zhang, Z.; et al. Acute manganese treatment restores defective autophagic cargo loading in Huntington’s disease cell lines. Hum. Mol. Genet. 2019, 28, 3825–3841. [Google Scholar] [CrossRef]
- Bryan, M.R.; Nordham, K.D.; Rose, D.I.; O’Brien, M.T.; Joshi, P.; Foshage, A.M.; Gonçalves, F.M.; Nitin, R.; Uhouse, M.A.; Aschner, M.; et al. Manganese Acts upon Insulin/IGF Receptors to Phosphorylate AKT and Increase Glucose Uptake in Huntington’s Disease Cells. Mol. Neurobiol. 2020, 57, 1570–1593. [Google Scholar] [CrossRef] [PubMed]
- Rigg, N.; Abu-Hijleh, F.A.; Patel, V.; Mishra, R.K. Ketamine-induced neurotoxicity is mediated through endoplasmic reticulum stress in vitro in STHdhQ7/Q7 cells. Neurotoxicology 2022, 91, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.J.; Murray, M.R.; Gosink, E.C.; Russell, H.R.; Srinivasan, A.; Kapsetaki, M.; Korsmeyer, S.J.; McKinnon, P.J. Atm and Bax cooperate in ionizing radiation-induced apoptosis in the central nervous system. Proc. Natl. Acad. Sci. USA 2000, 97, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamitsuka, P.J.; Ghanem, M.M.; Ziar, R.; McDonald, S.E.; Thomas, M.G.; Kwakye, G.F. Defective Mitochondrial Dynamics and Protein Degradation Pathways Underlie Cadmium-Induced Neurotoxicity and Cell Death in Huntington’s Disease Striatal Cells. Int. J. Mol. Sci. 2023, 24, 7178. https://doi.org/10.3390/ijms24087178
Kamitsuka PJ, Ghanem MM, Ziar R, McDonald SE, Thomas MG, Kwakye GF. Defective Mitochondrial Dynamics and Protein Degradation Pathways Underlie Cadmium-Induced Neurotoxicity and Cell Death in Huntington’s Disease Striatal Cells. International Journal of Molecular Sciences. 2023; 24(8):7178. https://doi.org/10.3390/ijms24087178
Chicago/Turabian StyleKamitsuka, Paul J., Marwan M. Ghanem, Rania Ziar, Sarah E. McDonald, Morgan G. Thomas, and Gunnar F. Kwakye. 2023. "Defective Mitochondrial Dynamics and Protein Degradation Pathways Underlie Cadmium-Induced Neurotoxicity and Cell Death in Huntington’s Disease Striatal Cells" International Journal of Molecular Sciences 24, no. 8: 7178. https://doi.org/10.3390/ijms24087178