
Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson




 ,
, 
Abstract
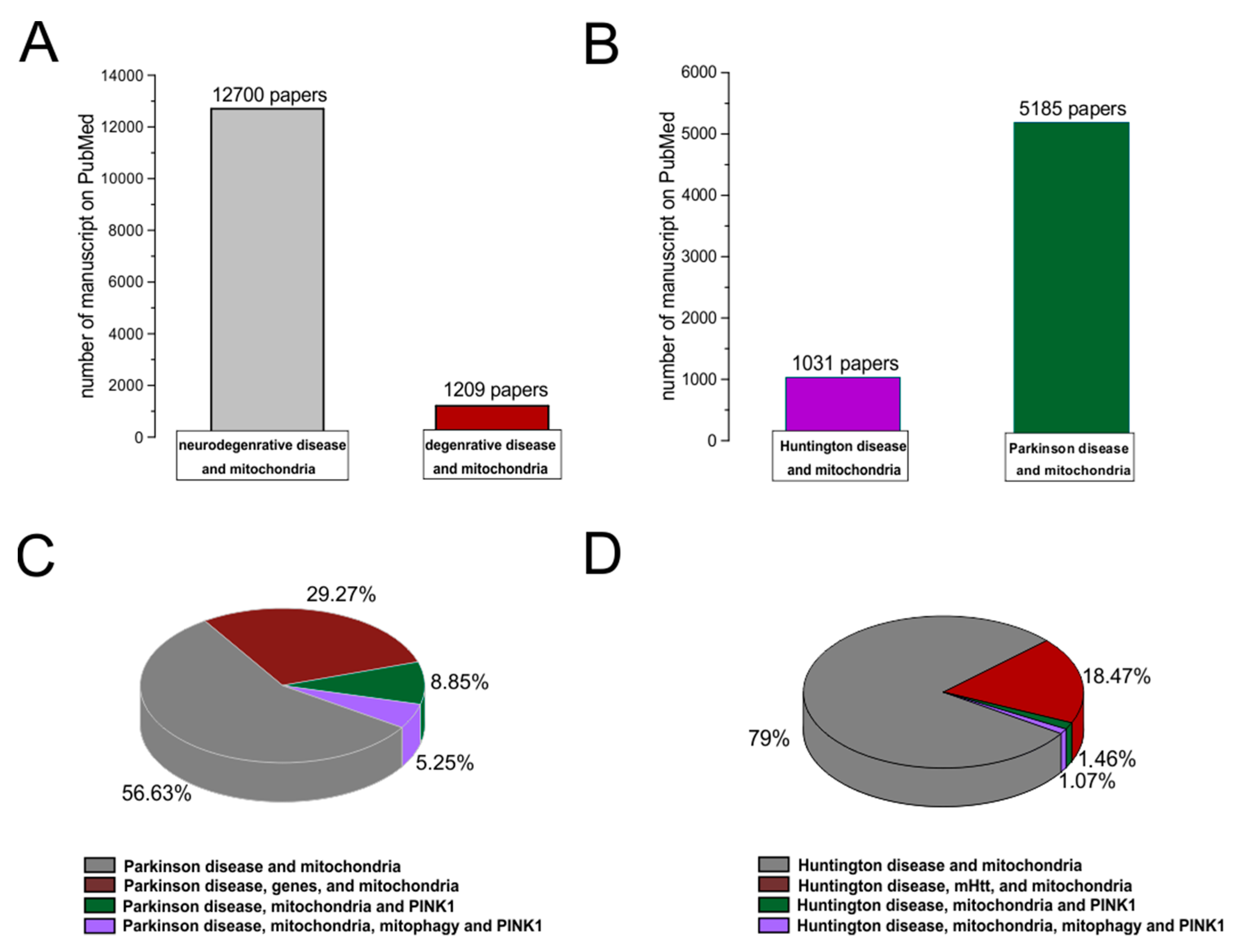
:1. Introduction
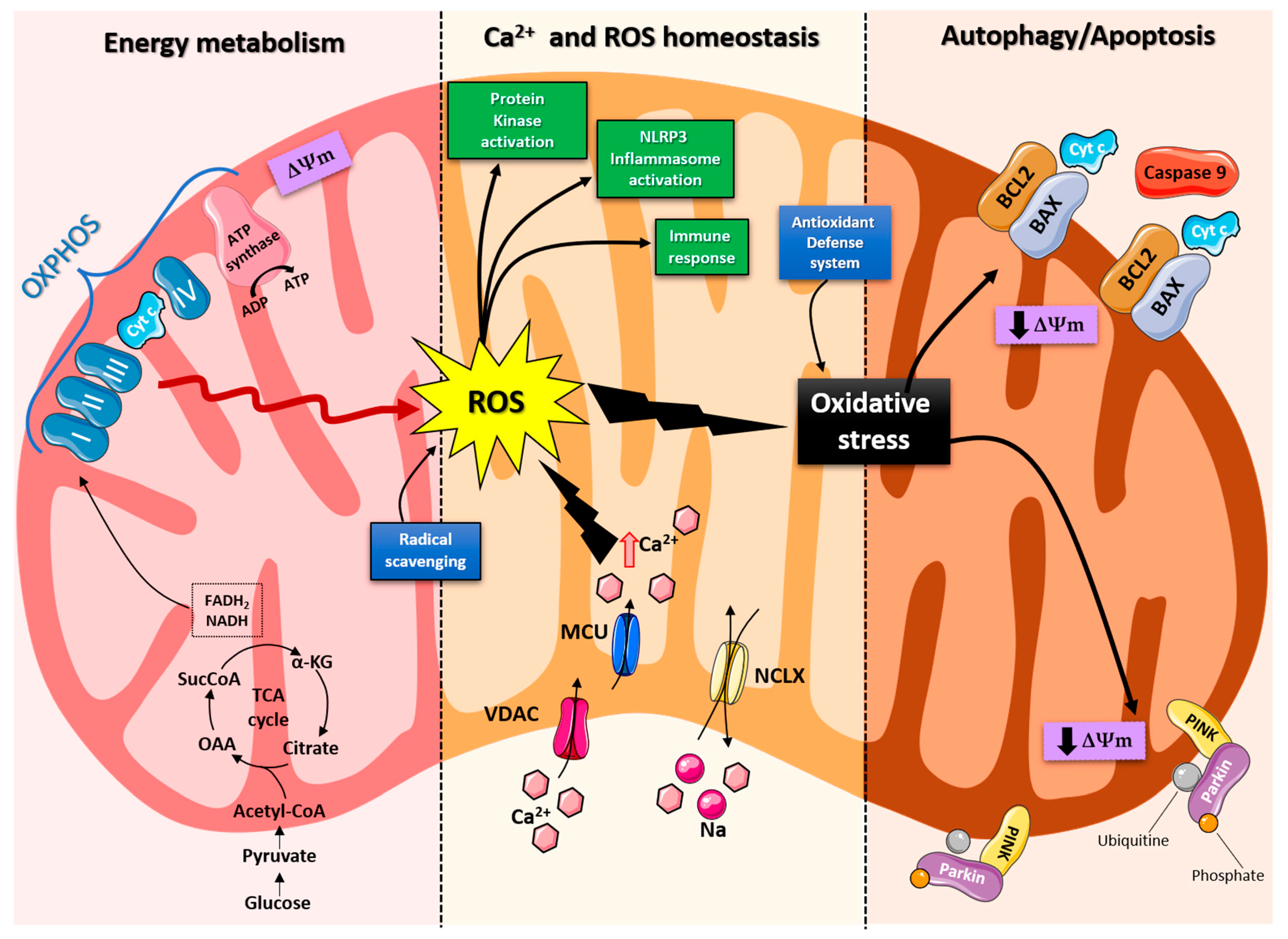
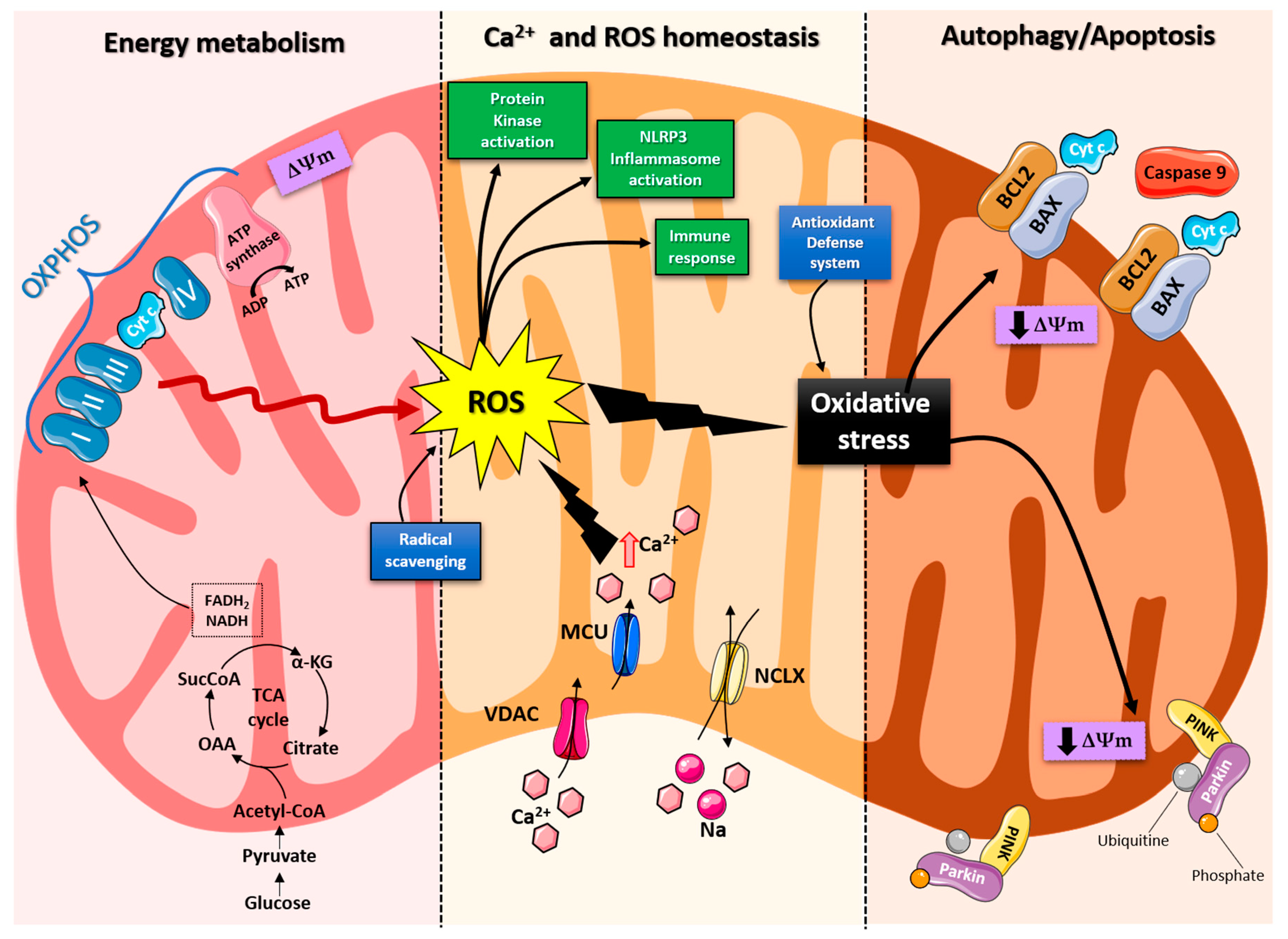
2. Role of Mitochondria in Brain Energy Metabolism, Calcium Homeostasis, and Signal Transduction
3. Role of Mitochondria in Neurodegenerative Disease
4. Mitochondria Bioenergy in Parkinson’s Disease and Huntington Disease in Rodents Animal Models
4.1. Parkinson Disease
4.1.1. Neurotoxin-Induced and Autosomal-Dominant PD Models
4.1.2. Autosomal-Recessive PD Models
4.2. Huntington Disease
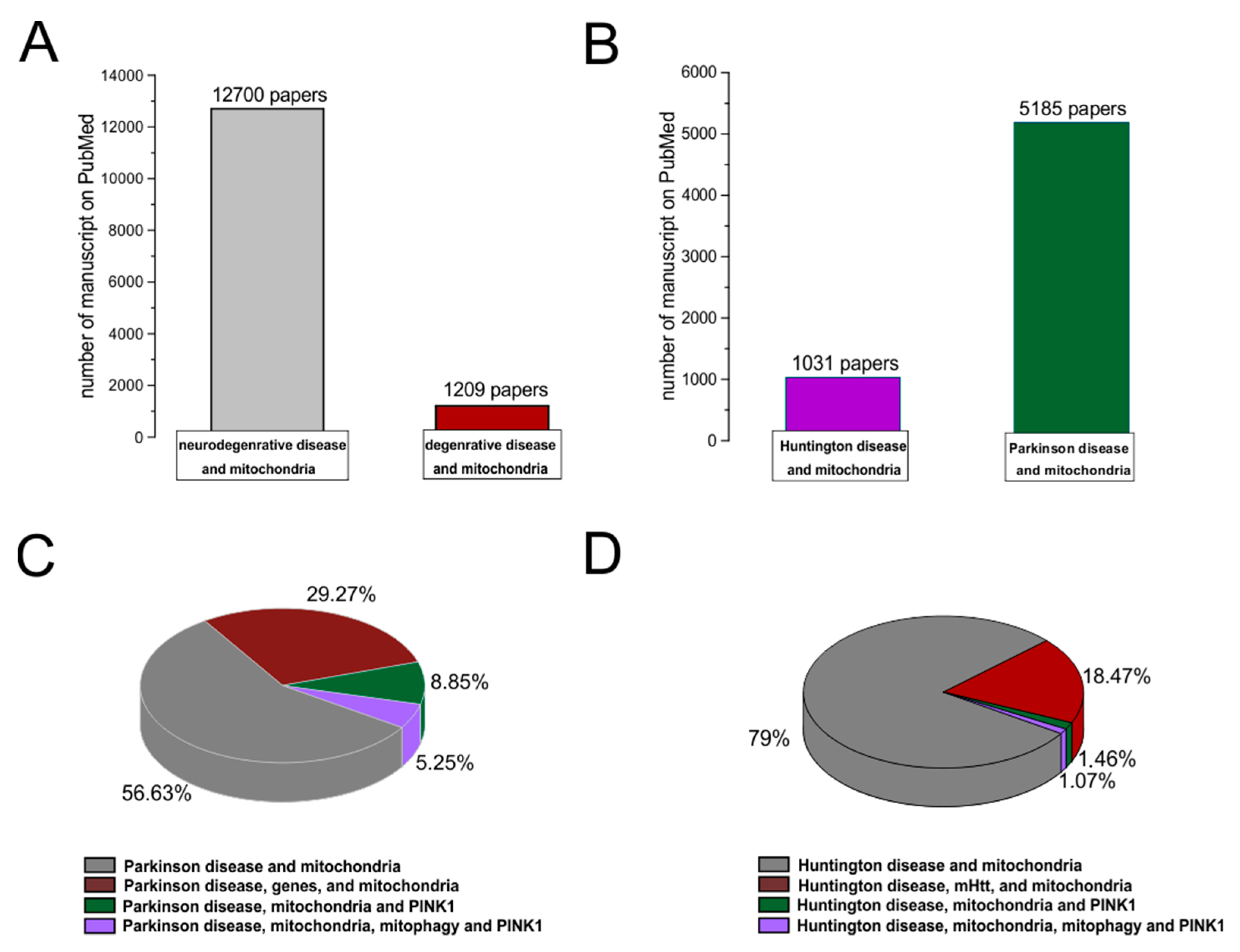
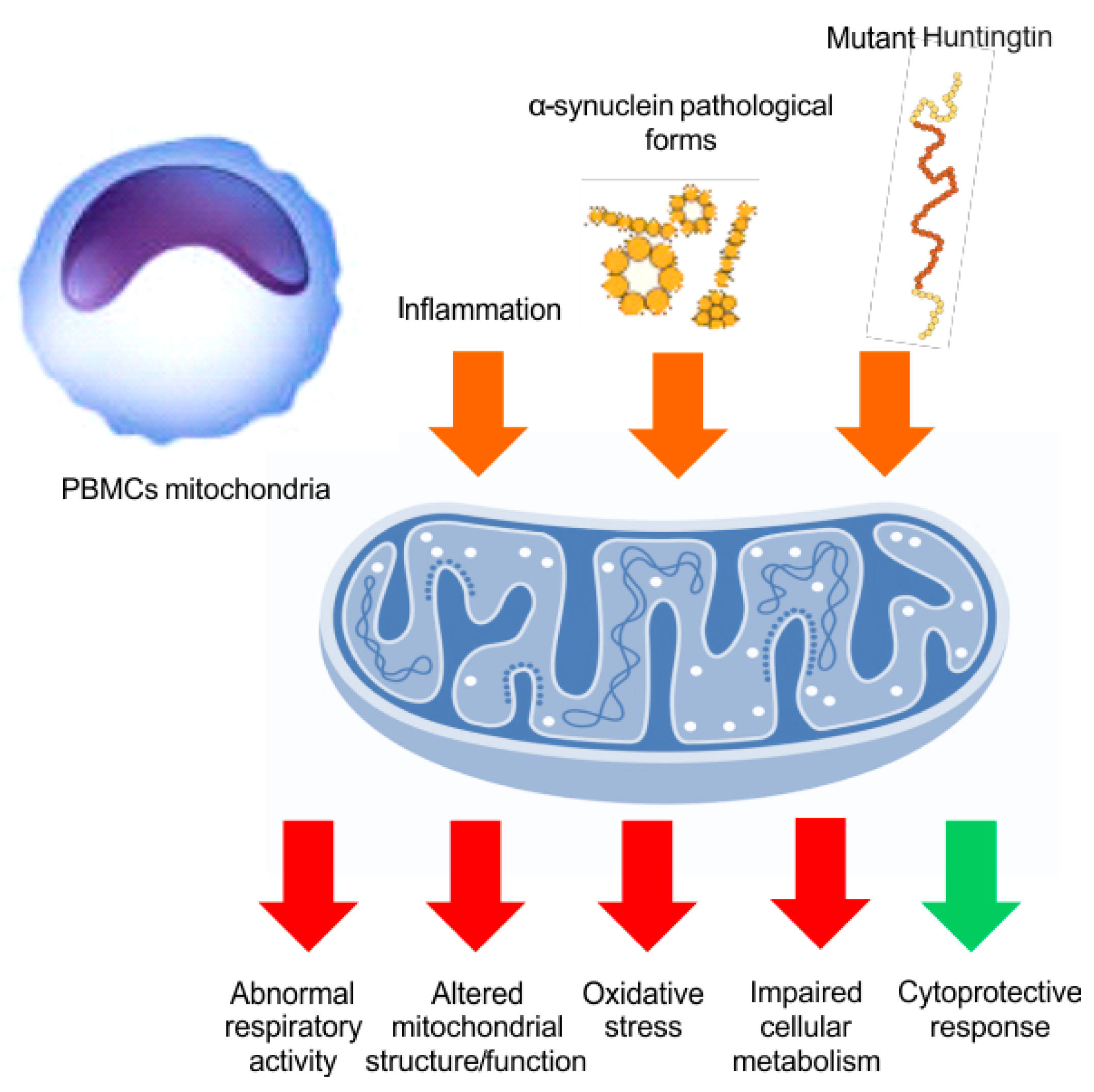
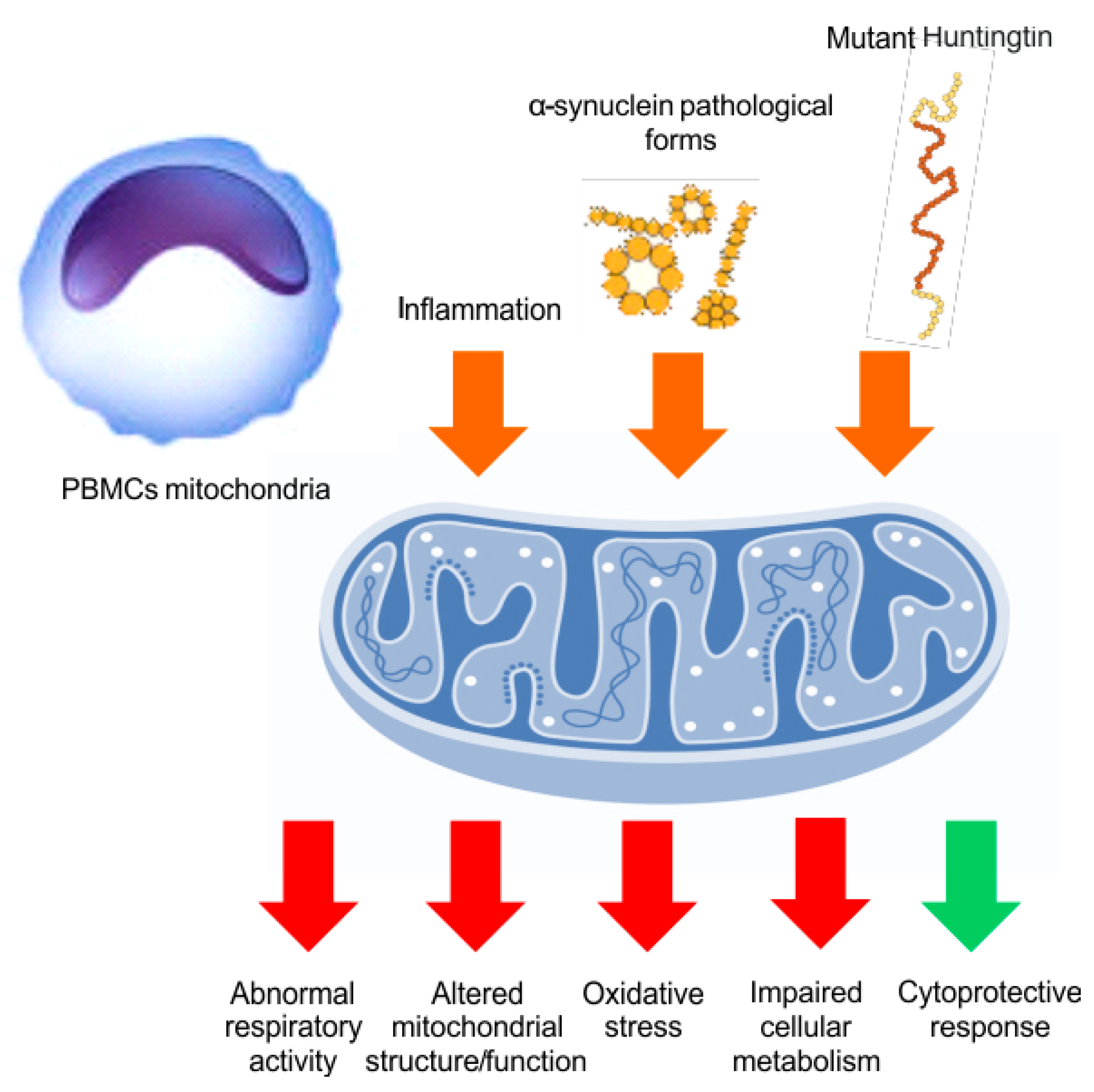
5. Mitochondrial Bioenergy in Parkinson’s Disease and Huntington Disease, Based on Human Evidences
6. Discussion
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A53T | α-synuclein with a PD-associated mutation |
| ADP | Adenosine diphosphate |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| ATP | Adenosine triphosphate |
| ATP5A | ATP synthase alpha-subunit gene |
| ATP5A1 | encodes for a subunit of ATP synthase (complex V) |
| BAC | bacterial artificial chromosome |
| BCL-2 | B-cell lymphoma 2 |
| Ca | calcium |
| COX4I1 | Cytochrome C Oxidase Subunit 4I1 |
| CSF | cerebrospinal fluid |
| DJ-1 | protein of the peptidase C56 family |
| DNA | deoxyribonucleic acid |
| Drp1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| ETC | electron transport chain |
| FADH2 | flavin adenine dinucleotides |
| Glu | glutamate |
| GR | glutathione reductase |
| GRX | glutaredoxin |
| GSH | glutathione reduced |
| GSSG | Glutathione oxidized |
| GTP | Guanosine-5′-triphosphate |
| HD | Huntington’s disease |
| Homer1 | Homer Scaffold Protein 1 |
| HTT | huntingtin |
| IL-18 | interleukin-18 |
| IL-1β | interleukin-1β |
| IMM | inner mitochondrial membrane |
| KO | knockout |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAO | Monoamine oxidases |
| MCU | mitochondrial calcium uniporter |
| Mfn1 | Mitofusin1 |
| Mfn2 | Mitofusin2 |
| MICU3 | mitochondrial calcium uptake family member3 |
| MPTP | 1-metil-4-fenil-1,2,3,6-tetraidropiridina |
| NADH | nicotinamide adenine dinucleotides |
| NCLX | mitochondrial Na+/Ca2+ exchanger |
| NDUFS3 | NADH: Ubiquinone Oxidoreductase Core Subunit S3 |
| NLR | Nod-like Receptor |
| NLRP3 | nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing 3 |
| NLRX1 | NLR Family Member X1 |
| NO | Nitric oxide |
| 3-NPA | 3-nitropropionic acid |
| Nrf2 | nuclear factor erythroid 2–related factor 2 |
| OMM | outer mitochondrial membrane |
| OXPHOS | mitochondrial oxidative phosphorylation |
| PBMCs | Peripheral blood mononucleate cells |
| PD | Parkinson’s disease |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced kinase 1 |
| PRKAG2 | protein kinase AMP-activated non-catalytic subunit gamma 2 |
| Rab5 | Ras-related protein5 |
| ROS | generation of free radical species |
| SDHB | Succinate Dehydrogenase Complex, Subunit B |
| SNCA | alpha-synuclein gene |
| SNpc | Substantia nigra pars compacta |
| SPN | medium spine neuron |
| TCA | tricarboxylic acid |
| Trx | Thioredoxin |
| TTR | Transthyretin |
| VDAC | Voltage-dependent anion channel |
| VDAC3 | Voltage-dependent anion-selective channel protein 3 |
| YAC | yeast artificial chromosome |
| ΔΨm | Mitochondrial membrane potential |
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and Molecular Mechanisms of Mitochondrial Function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzorno, J. Mitochondria-Fundamental to Life and Health. Integr. Med. 2014, 13, 8–15. [Google Scholar]
- Bruce, A. Molecular Biology of the Cell; W. W. Norton & Company: New York, NY, USA, 2022. [Google Scholar]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial Dysfunction in Neurodegenerative Diseases and the Potential Countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative Diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of Metabolic Dysfunction in Neurodegeneration: Focus on Mitochondrial Function and Calcium Signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Zhu, X.-H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative Imaging of Energy Expenditure in Human Brain. NeuroImage 2012, 60, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Vergara, R.C.; Jaramillo-Riveri, S.; Luarte, A.; Moënne-Loccoz, C.; Fuentes, R.; Couve, A.; Maldonado, P.E. The Energy Homeostasis Principle: Neuronal Energy Regulation Drives Local Network Dynamics Generating Behavior. Front. Comput. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.-J. Mitochondrial Dysfunction and Cell Death in Neurodegenerative Diseases through Nitroxidative Stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [Green Version]
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in Neuronal Health: From Energy Metabolism to Parkinson’s Disease. Adv. Biol. 2021, 5, 2100663. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; Cruz e Silva, O. Mitochondria, Energy, and Metabolism in Neuronal Health and Disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial Calcium Homeostasis as Potential Target for Mitochondrial Medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ Signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a Multi-Functional Mitochondrial Protein as a Pharmacological Target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M.; Hoogerheide, D.P. Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci. 2021, 22, 7358. [Google Scholar] [CrossRef]
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.-K. Dysfunction of Mitochondrial Ca2+ Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Gunter, K.K.; Gunter, T.E. A System for Producing and Monitoring in Vitro Calcium Pulses Similar to Those Observed in Vivo. Anal. Biochem. 1994, 219, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.-S.; Gunter, T.E. Mitochondrial Calcium Uptake from Physiological-Type Pulses of Calcium: A description of the rapid uptake mode. J. Biol. Chem. 1995, 270, 27510–27515. [Google Scholar] [CrossRef] [Green Version]
- Grienberger, C.; Konnerth, A. Imaging Calcium in Neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, O.H. Calcium Signal Compartmentalization. Biol. Res. 2002, 35, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Laude, A.J.; Simpson, A.W.M. Compartmentalized Signalling: Ca2+ Compartments, Microdomains and the Many Facets of Ca2+ Signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Panda, S.; Behera, S.; Alam, M.F.; Syed, G.H. Endoplasmic Reticulum & Mitochondrial Calcium Homeostasis: The Interplay with Viruses. Mitochondrion 2021, 58, 227–242. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-Mitochondria Tethering by PDZD8 Regulates Ca2+ Dynamics in Mammalian Neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R. Sources and Triggers of Oxidative Damage in Neurodegeneration. Free Radic. Biol. Med. 2021, 173, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Signalling. J. Cell Sci. 2012, 125 Pt 4, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordijk, P.L. Regulation of NADPH Oxidases: The Role of Rac Proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.-H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Breda, C.N.d.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as Central Hub of the Immune System. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Wu, S.; Raju, R. NLRX1 Regulation Following Acute Mitochondrial Injury. Front. Immunol. 2019, 10, 2431. [Google Scholar] [CrossRef] [Green Version]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; et al. NLRX1 Dampens Oxidative Stress and Apoptosis in Tissue Injury via Control of Mitochondrial Activity. J. Exp. Med. 2017, 214, 2405–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The Role of Mitochondria in NLRP3 Inflammasome Activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: An Immune Sensor of Cellular Stress and Infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Behrouzi, A.; Kelley, M.R.; Fehrenbacher, J.C. Oxidative DNA Damage: A Role in Altering Neuronal Function. J. Cell. Signal. 2022, 3, 160–166. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The Role of Nrf2 Signaling in Counteracting Neurodegenerative Diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial Autophagy—An Essential Quality Control Mechanism for Myocardial Homeostasis. Circ. J. Off. J. Jpn. Circ. Soc. 2013, 77, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial Dynamics and Apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.-H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and Roles of Mitochondrial Localisation and Dynamics in Neuronal Function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic Mitochondria in Synaptic Transmission and Organization of Vesicle Pools in Health and Disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef] [Green Version]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Jaiswal, M. Mitochondrial Calcium at the Synapse. Mitochondrion 2021, 59, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schapira, A.H. Mitochondria and Degenerative Disorders. Am. J. Med. Genet. 2001, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.N.; Zhou, M.Q.; Guo, J.; Zheng, H.J.; Tang, J.; Zhang, C.; Liu, Y.N.; Liu, W.J.; Wang, Y.X. Mitochondrial Dysfunction and Diabetic Nephropathy: Nontraditional Therapeutic Opportunities. J. Diabetes Res. 2021, 2021, 1010268. [Google Scholar] [CrossRef]
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects Beta-Cells from Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Lewin, R. Trail of Ironies to Parkinson’s Disease. Science 1984, 224, 1083–1085. [Google Scholar] [CrossRef]
- Hauser, D.N.; Hastings, T.G. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease and Monogenic Parkinsonism. Neurobiol. Dis. 2013, 51, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, J.D.; Cardoso, S.M. Mitochondrial Signaling on Innate Immunity Activation in Parkinson Disease. Curr. Opin. Neurobiol. 2023, 78, 102664. [Google Scholar] [CrossRef] [PubMed]
- Sagan, L. On the Origin of Mitosing Cells. J. Theor. Biol. 1967, 14, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P. Mitochondrial Bioenergetics and Neurodegeneration: A Paso Doble. Neural Regen. Res. 2021, 16, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, S.; Li, X.; Xu, Q.; Lin, Y.; Lin, F.; Yuan, M.; Zi, Y.; Cai, J. Progress in Parkinson’s Disease Animal Models of Genetic Defects: Characteristics and Application. Biomed. Pharmacother. 2022, 155, 113768. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.; Valente, E.M. PINK1 in the Limelight: Multiple Functions of an Eclectic Protein in Human Health and Disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial Import and Enzymatic Activity of PINK1 Mutants Associated to Recessive Parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef] [Green Version]
- Unoki, M.; Nakamura, Y. Growth-Suppressive Effects of BPOZ and EGR2, Two Genes Involved in the PTEN Signaling Pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. Parkinson’s Disease and Cancer: The Unexplored Connection. J. Natl. Cancer Inst. 2010, 102, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Masgras, I.; Laquatra, C.; Cannino, G.; Serapian, S.A.; Colombo, G.; Rasola, A. The Molecular Chaperone TRAP1 in Cancer: From the Basics of Biology to Pharmacological Targeting. Semin. Cancer Biol. 2021, 76, 45–53. [Google Scholar] [CrossRef]
- Exner, N.; Treske, B.; Paquet, D.; Holmström, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.-H.; Gasser, T.; et al. Loss-of-Function of Human PINK1 Results in Mitochondrial Pathology and Can Be Rescued by Parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired Dopamine Release and Synaptic Plasticity in the Striatum of PINK1-Deficient Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to Low-Dose Rotenone Precipitates Synaptic Plasticity Alterations in PINK1 Heterozygous Knockout Mice. Neurobiol. Dis. 2016, 91, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; D’Angelo, V.; Platania, P.; Di Lazzaro, G.; Scalise, S.; Salimei, C.; El Atiallah, I.; Colona, V.L.; Mercuri, N.B.; Bonsi, P.; et al. Ischemic Injury Precipitates Neuronal Vulnerability in Parkinson’s Disease: Insights from PINK1 Mouse Model Study and Clinical Retrospective Data. Park. Relat. Disord. 2020, 74, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; Tassone, A.; Meringolo, M.; Ponterio, G.; Madeo, G.; Pisani, A.; Bonsi, P.; Martella, G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 3407. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, F.; Valente, E.M.; Arena, G. Mechanisms of Neurodegeneration in Parkinson’s Disease: Keep Neurons in the PINK1. Mech. Ageing Dev. 2020, 189, 111277. [Google Scholar] [CrossRef]
- Zhi, L.; Qin, Q.; Muqeem, T.; Seifert, E.L.; Liu, W.; Zheng, S.; Li, C.; Zhang, H. Loss of PINK1 Causes Age-Dependent Decrease of Dopamine Release and Mitochondrial Dysfunction. Neurobiol. Aging 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Regulation of Neuronal Bioenergetics as a Therapeutic Strategy in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 1467–1482. [Google Scholar] [CrossRef]
- Xie, W.; Chung, K.K.K. Alpha-Synuclein Impairs Normal Dynamics of Mitochondria in Cell and Animal Models of Parkinson’s Disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef]
- Portz, P.; Lee, M.K. Changes in Drp1 Function and Mitochondrial Morphology Are Associated with the α-Synuclein Pathology in a Transgenic Mouse Model of Parkinson’s Disease. Cells 2021, 10, 885. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy in Vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. Available online: https://pubmed.ncbi.nlm.nih.gov/28341600/ (accessed on 6 March 2023). [CrossRef] [PubMed]
- Goldberg, J.A.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium Entry Induces Mitochondrial Oxidant Stress in Vagal Neurons at Risk in Parkinson’s Disease. Nat. Neurosci. 2012, 15, 1414–1421. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.-M.; Lo, S.-C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 Activation via Mitochondria Is Required for Long-Term Depression and AMPA Receptor Internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopert, P.; Patel, M. Brain Mitochondria from DJ-1 Knockout Mice Show Increased Respiration-Dependent Hydrogen Peroxide Consumption. Redox Biol. 2014, 2, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Liu, H.; Liu, S.; Kang, Y.; Nie, Z.; Lei, H. Altered Prefrontal Neurochemistry in the DJ-1 Knockout Mouse Model of Parkinson’s Disease: Complementary Semi-Quantitative Analyses with in Vivo Magnetic Resonance Spectroscopy and MALDI-MSI. Anal. Bioanal. Chem. 2022, 414, 7977–7987. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Moraes, C.T. Reactive Oxygen Species and Mitochondrial Diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione Metabolism in Brain Metabolic Interaction between Astrocytes and Neurons in the Defense against Reactive Oxygen Species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Darios, F. Parkin Prevents Mitochondrial Swelling and Cytochrome c Release in Mitochondria-Dependent Cell Death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal Dopaminergic Deficits and Hypokinesia Caused by Inactivation of the Familial Parkinsonism-Linked Gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Tkac, I.; Dubinsky, J.M.; Keene, C.D.; Gruetter, R.; Low, W.C. Neurochemical Changes in Huntington R6/2 Mouse Striatum Detected by in Vivo 1H NMR Spectroscopy. J. Neurochem. 2007, 100, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Ghiglieri, V.; Bagetta, V.; Calabresi, P.; Picconi, B. Functional Interactions within Striatal Microcircuit in Animal Models of Huntington’s Disease. Neuroscience 2012, 211, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.; Ferreira, I.L.; Maranga, C.; Beatriz, M.; Mota, S.I.; Sereno, J.; Castelhano, J.; Abrunhosa, A.; Oliveira, F.; De Rosa, M.; et al. Mitochondrial and Redox Modifications in Early Stages of Huntington’s Disease. Redox Biol. 2022, 56, 102424. [Google Scholar] [CrossRef] [PubMed]
- Herbst, E.a.F.; Holloway, G.P. Exercise Training Normalizes Mitochondrial Respiratory Capacity within the Striatum of the R6/1 Model of Huntington’s Disease. Neuroscience 2015, 303, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective Effects of Phenylbutyrate in the N171-82Q Transgenic Mouse Model of Huntington’s Disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Nithianantharajah, J.; Hannan, A.J. Dysregulation of Synaptic Proteins, Dendritic Spine Abnormalities and Pathological Plasticity of Synapses as Experience-Dependent Mediators of Cognitive and Psychiatric Symptoms in Huntington’s Disease. Neuroscience 2013, 251, 66–74. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Campanelli, F.; Marino, G.; Natale, G.; Picconi, B.; Calabresi, P. Corticostriatal Synaptic Plasticity Alterations in the R6/1 Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. Res. 2019, 97, 1655–1664. [Google Scholar] [CrossRef]
- Wellington, C.L.; Ellerby, L.M.; Hackam, A.S.; Margolis, R.L.; Trifiro, M.A.; Singaraja, R.; McCutcheon, K.; Salvesen, G.S.; Propp, S.S.; Bromm, M.; et al. Caspase Cleavage of Gene Products Associated with Triplet Expansion Disorders Generates Truncated Fragments Containing the Polyglutamine Tract. J. Biol. Chem. 1998, 273, 9158–9167. [Google Scholar] [CrossRef] [Green Version]
- Schirinzi, T.; Salvatori, I.; Zenuni, H.; Grillo, P.; Valle, C.; Martella, G.; Mercuri, N.B.; Ferri, A. Pattern of Mitochondrial Respiration in Peripheral Blood Cells of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10863. [Google Scholar] [CrossRef]
- Anderson, K.E.; Marshall, F.J. Behavioral Symptoms Associated with Huntington’s Disease. Adv. Neurol. 2005, 96, 197–208. [Google Scholar]
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s Disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, M.U.; Sehar, N.; Dar, N.J.; Khan, A.; Arafah, A.; Rashid, S.; Rashid, S.M.; Ganaie, M.A. Mitochondrial Dysfunctions, Oxidative Stress and Neuroinflammation as Therapeutic Targets for Neurodegenerative Diseases: An Update on Current Advances and Impediments. Neurosci. Biobehav. Rev. 2023, 144, 104961. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, C.M. Mitochondria in Huntington’s Disease: Implications in Pathogenesis and Mitochondrial-Targeted Therapeutic Strategies. Neural Regen. Res. 2023, 18, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Kawsar, M.; Taz, T.A.; Paul, B.K.; Ahmed, K.; Habib, M.A.; Bhuyian, T. Identification of Vital Regulatory Genes with Network Pathways among Huntington’s, Parkinson’s, and Alzheimer’s Diseases. Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 50. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and Age-Related Diseases: From Mechanisms to Therapeutic Strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-Induced Senescence Results in Marked Metabolic and Bioenergetic Alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-L.; Feng, S.-T.; Wang, Y.-T.; Yuan, Y.-H.; Li, Z.-P.; Chen, N.-H.; Wang, Z.-Z.; Zhang, Y. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson’s Disease. Cell. Mol. Neurobiol. 2022, 42, 1321–1339. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial Dysfunction and Mitophagy in Parkinson’s: From Familial to Sporadic Disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H. Impaired Mitochondrial Dynamics and Function in the Pathogenesis of Parkinson’s Disease. Exp. Neurol. 2009, 218, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease—Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, W.D.; Boyson, S.J.; Parks, J.K. Abnormalities of the Electron Transport Chain in Idiopathic Parkinson’s Disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Thirugnanam, T.; Santhakumar, K. Chemically Induced Models of Parkinson’s Disease. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2022, 252, 109213. [Google Scholar] [CrossRef]
- Bové, J.; Perier, C. Neurotoxin-Based Models of Parkinson’s Disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial Dysfunction in Parkinson’s Disease: Molecular Mechanisms and Pathophysiological Consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef] [Green Version]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef] [Green Version]
- Imbriani, P.; Schirinzi, T.; Meringolo, M.; Mercuri, N.B.; Pisani, A. Centrality of Early Synaptopathy in Parkinson’s Disease. Front. Neurol. 2018, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Imbriani, P.; Sciamanna, G.; Santoro, M.; Schirinzi, T.; Pisani, A. Promising Rodent Models in Parkinson’s Disease. Park. Relat. Disord. 2018, 46 (Suppl. S1), S10–S14. [Google Scholar] [CrossRef] [PubMed]
- Innos, J.; Hickey, M.A. Using Rotenone to Model Parkinson’s Disease in Mice: A Review of the Role of Pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; Martella, G.; Bonsi, P.; Pisani, A. Oxidative Stress and Synaptic Dysfunction in Rodent Models of Parkinson’s Disease. Neurobiol. Dis. 2022, 173, 105851. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Ribich, S.; Hyman, B.T. Subcellular Localization of Alpha-Synuclein in Primary Neuronal Cultures: Effect of Missense Mutations. J. Neural Transm. Suppl. 2000, 7, 53–63. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Uéda, K.; Yu, S.; Yang, H. Semi-Quantitative Analysis of Alpha-Synuclein in Subcellular Pools of Rat Brain Neurons: An Immunogold Electron Microscopic Study Using a C-Terminal Specific Monoclonal Antibody. Brain Res. 2008, 1244, 40–52. [Google Scholar] [CrossRef]
- Cole, N.B.; Dieuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial Translocation of Alpha-Synuclein Is Promoted by Intracellular Acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a Gene for Parkinson’s Disease to Chromosome 4q21-Q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal Models of α-Synucleinopathy for Parkinson Disease Drug Development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s Disease Alpha-Synuclein Transgenic Mice Develop Neuronal Mitochondrial Degeneration and Cell Death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Merino-Galán, L.; Jimenez-Urbieta, H.; Zamarbide, M.; Rodríguez-Chinchilla, T.; Belloso-Iguerategui, A.; Santamaria, E.; Fernández-Irigoyen, J.; Aiastui, A.; Doudnikoff, E.; Bézard, E.; et al. Striatal Synaptic Bioenergetic and Autophagic Decline in Premotor Experimental Parkinsonism. Brain J. Neurol. 2022, 145, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; García-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-Alpha-Synuclein Overexpression Impairs Dopamine Signaling and Striatal Synaptic Plasticity in Old Mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef] [PubMed]
- Durante, V.; de Iure, A.; Loffredo, V.; Vaikath, N.; De Risi, M.; Paciotti, S.; Quiroga-Varela, A.; Chiasserini, D.; Mellone, M.; Mazzocchetti, P.; et al. Alpha-Synuclein Targets GluN2A NMDA Receptor Subunit Causing Striatal Synaptic Dysfunction and Visuospatial Memory Alteration. Brain J. Neurol. 2019, 142, 1365–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozzi, A.; de Iure, A.; Bagetta, V.; Tantucci, M.; Durante, V.; Quiroga-Varela, A.; Costa, C.; Di Filippo, M.; Ghiglieri, V.; Latagliata, E.C.; et al. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting With GluN2D N-Methyl-D-Aspartate Receptor Subunit. Biol. Psychiatry 2016, 79, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, A.; Sciaccaluga, M.; Loffredo, V.; Megaro, A.; Ledonne, A.; Cardinale, A.; Federici, M.; Bellingacci, L.; Paciotti, S.; Ferrari, E.; et al. Dopamine-Dependent Early Synaptic and Motor Dysfunctions Induced by α-Synuclein in the Nigrostriatal Circuit. Brain J. Neurol. 2021, 144, 3477–3491. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [Green Version]
- Alves Da Costa, C.; Paitel, E.; Vincent, B.; Checler, F. Alpha-Synuclein Lowers P53-Dependent Apoptotic Response of Neuronal Cells: Abolishment by 6-Hydroxydopamine and Implication for Parkinson’s Disease. J. Biol. Chem. 2002, 277, 50980–50984. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Mattson, M.P. Caspase and Calpain Substrates: Roles in Synaptic Plasticity and Cell Death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal Caspase-3 Signaling: Not Only Cell Death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Snigdha, S.; Smith, E.D.; Prieto, G.A.; Cotman, C.W. Caspase-3 Activation as a Bifurcation Point between Plasticity and Cell Death. Neurosci. Bull. 2012, 28, 14–24. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 Acts in Parallel to the PINK1/Parkin Pathway to Control Mitochondrial Function and Autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trancikova, A.; Tsika, E.; Moore, D.J. Mitochondrial Dysfunction in Genetic Animal Models of Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 896–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.-F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 Signalling Rescues Amyloid Pathology and Mitochondrial Dysfunction in Alzheimer’s Disease. Brain J. Neurol. 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between Early-Onset Parkinson’s Disease and Mutations in the Parkin Gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Moore, D.J. Parkin: A Multifaceted Ubiquitin Ligase. Biochem. Soc. Trans. 2006, 34 Pt 5, 749–753. [Google Scholar] [CrossRef]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 Form a Ubiquitin E3 Ligase Complex Promoting Unfolded Protein Degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Periquet, M.; Corti, O.; Jacquier, S.; Brice, A. Proteomic Analysis of Parkin Knockout Mice: Alterations in Energy Metabolism, Protein Handling and Synaptic Function. J. Neurochem. 2005, 95, 1259–1276. [Google Scholar] [CrossRef]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Jo, A.; Lee, Y.; Kam, T.-I.; Kang, S.-U.; Neifert, S.; Karuppagounder, S.S.; Khang, R.; Kang, H.; Park, H.; Chou, S.-C.; et al. PARIS Farnesylation Prevents Neurodegeneration in Models of Parkinson’s Disease. Sci. Transl. Med. 2021, 13, eaax8891. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin-Induced Mitophagy in the Pathogenesis of Parkinson Disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in Parkin-Deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [Green Version]
- Itier, J.-M.; Ibanez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin Gene Inactivation Alters Behaviour and Dopamine Neurotransmission in the Mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Pisani, A.; Karouani, M.; Haburcak, M.; Martella, G.; Tscherter, A.; Platania, P.; Wu, B.; Pothos, E.N.; Shen, J. Impaired Dopamine Release and Synaptic Plasticity in the Striatum of Parkin −/− Mice. J. Neurochem. 2009, 110, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Cortese, G.P.; Zhu, M.; Williams, D.; Heath, S.; Waites, C.L. Parkin Deficiency Reduces Hippocampal Glutamatergic Neurotransmission by Impairing AMPA Receptor Endocytosis. J. Neurosci. 2016, 36, 12243–12258. [Google Scholar] [CrossRef] [Green Version]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced Parkin Levels Favor ER-Mitochondria Crosstalk and Guarantee Ca(2+) Transfer to Sustain Cell Bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef]
- Bianchi, K.; Rimessi, A.; Prandini, A.; Szabadkai, G.; Rizzuto, R. Calcium and Mitochondria: Mechanisms and Functions of a Troubled Relationship. Biochim. Biophys. Acta 2004, 1742, 119–131. [Google Scholar] [CrossRef]
- Jo, D.; Song, J. Irisin Acts via the PGC-1α and BDNF Pathway to Improve Depression-like Behavior. Clin. Nutr. Res. 2021, 10, 292–302. [Google Scholar] [CrossRef]
- Zhou, H.; Falkenburger, B.H.; Schulz, J.B.; Tieu, K.; Xu, Z.; Xia, X.G. Silencing of the Pink1 Gene Expression by Conditional RNAi Does Not Induce Dopaminergic Neuron Death in Mice. Int. J. Biol. Sci. 2007, 3, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Madeo, G.; Schirinzi, T.; Martella, G.; Latagliata, E.C.; Puglisi, F.; Shen, J.; Valente, E.M.; Federici, M.; Mercuri, N.B.; Puglisi-Allegra, S.; et al. PINK1 Heterozygous Mutations Induce Subtle Alterations in Dopamine-Dependent Synaptic Plasticity. Mov. Disord. 2014, 29, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, K.D.; De Silva, S.; Sheth, N.P.; Ramboz, S.; Beck, M.J.; Quang, C.; Switzer, R.C.; Ahmad, S.O.; Sunkin, S.M.; Walker, D.; et al. Phenotypic Characterization of Recessive Gene Knockout Rat Models of Parkinson’s Disease. Neurobiol. Dis. 2014, 70, 190–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Ottemann, B.M.; Emanuel, K.; Fox, H.S. Loss of Pink1 Modulates Synaptic Mitochondrial Bioenergetics in the Rat Striatum Prior to Motor Symptoms: Concomitant Complex I Respiratory Defects and Increased Complex II-Mediated Respiration. Proteom. Clin. Appl. 2016, 10, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, L.M.; Purnell, P.R.; Boska, M.D.; Fox, H.S. Early Expression of Parkinson’s Disease-Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol. Neurobiol. 2016, 53, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creed, R.B.; Goldberg, M.S. Analysis of α-Synuclein Pathology in PINK1 Knockout Rat Brains. Front. Neurosci. 2018, 12, 1034. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 Causes Mitochondrial Functional Defects and Increased Sensitivity to Oxidative Stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.-W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial Localization of the Parkinson’s Disease Related Protein DJ-1: Implications for Pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edzamko, N.; Zhou, J.; Huang, Y.; Halliday, G.M. Parkinson’s Disease-Implicated Kinases in the Brain; Insights into Disease Pathogenesis. Front. Mol. Neurosci. 2014, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J.I.; Park, S.K.; Lim, K.; et al. DJ-1 Null Dopaminergic Neuronal Cells Exhibit Defects in Mitochondrial Function and Structure: Involvement of Mitochondrial Complex I Assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 Gene Deletion Reveals That DJ-1 Is an Atypical Peroxiredoxin-like Peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Hirrlinger, J. Glutathione Pathways in the Brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial Localization of DJ-1 Leads to Enhanced Neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s Disease-Linked Gene DJ-1 Perturbs Mitochondrial Dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.-Y.; Lee, K.-W.; Woo, J.-M.; Junn, E.; Mouradian, M.M. DJ-1 Induces Thioredoxin 1 Expression through the Nrf2 Pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [Green Version]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the Mechanism of Pathogenesis of Parkinson’s Disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Tong, Y.; Gautier, C.A.; Shen, J. Absence of Nigral Degeneration in Aged Parkin/DJ-1/PINK1 Triple Knockout Mice. J. Neurochem. 2009, 111, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Farshim, P.P.; Bates, G.P. Mouse Models of Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 97–120. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an Animal Model for the Study of Huntington’s Disease. Nat. Rev. Neurosci. 2013, 14, 708–721. [Google Scholar] [CrossRef]
- Yu, Z.-X.; Li, S.-H.; Evans, J.; Pillarisetti, A.; Li, H.; Li, X.-J. Mutant Huntingtin Causes Context-Dependent Neurodegeneration in Mice with Huntington’s Disease. J. Neurosci. 2003, 23, 2193–2202. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant Huntingtin Directly Increases Susceptibility of Mitochondria to the Calcium-Induced Permeability Transition and Cytochrome c Release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.V.; Gutekunst, C.-A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early Mitochondrial Calcium Defects in Huntington’s Disease Are a Direct Effect of Polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suárez-Fariñas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic Behavioral Evaluation of Huntington’s Disease Transgenic and Knock-in Mouse Models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased Oxidative Damage to DNA in a Transgenic Mouse Model of Huntington’s Disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H. Mitochondrial Dysfunction and Free Radical Damage in the Huntington R6/2 Transgenic Mouse. Ann. Neurol. 2000, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.H.; Willert, C.W.; Andersen, J.V.; Madsen, M.; Waagepetersen, H.S.; Skotte, N.H.; Nørremølle, A. Progressive Mitochondrial Dysfunction of Striatal Synapses in R6/2 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2022, 11, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Milnerwood, A.J.; Raymond, L.A. Corticostriatal Synaptic Function in Mouse Models of Huntington’s Disease: Early Effects of Huntingtin Repeat Length and Protein Load. J. Physiol. 2007, 585 Pt 3, 817–831. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive Dysfunction in Huntington’s Disease: Humans, Mouse Models and Molecular Mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef] [Green Version]
- Rosenstock, T.R.; Bertoncini, C.R.A.; Teles, A.V.; Hirata, H.; Fernandes, M.J.S.; Smaili, S.S. Glutamate-Induced Alterations in Ca2+ Signaling Are Modulated by Mitochondrial Ca2+ Handling Capacity in Brain Slices of R6/1 Transgenic Mice. Eur. J. Neurosci. 2010, 32, 60–70. [Google Scholar] [CrossRef]
- Clark, J.B. N-Acetyl Aspartate: A Marker for Neuronal Loss or Mitochondrial Dysfunction. Dev. Neurosci. 1998, 20, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.G.; Klivenyi, P.; Kustermann, E.; Andreassen, O.A.; Ferrante, R.J.; Rosen, B.R.; Beal, M.F. Nonlinear Decrease over Time in N-Acetyl Aspartate Levels in the Absence of Neuronal Loss and Increases in Glutamine and Glucose in Transgenic Huntington’s Disease Mice. J. Neurochem. 2000, 74, 2108–2119. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Jenkins, B.G.; Ferrante, K.L.; Thomas, M.; Friedlich, A.; Browne, S.E.; Schilling, G.; Borchelt, D.R.; et al. Creatine Increase Survival and Delays Motor Symptoms in a Transgenic Animal Model of Huntington’s Disease. Neurobiol. Dis. 2001, 8, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, B.G.; Andreassen, O.A.; Dedeoglu, A.; Leavitt, B.; Hayden, M.; Borchelt, D.; Ross, C.A.; Ferrante, R.J.; Beal, M.F. Effects of CAG Repeat Length, HTT Protein Length and Protein Context on Cerebral Metabolism Measured Using Magnetic Resonance Spectroscopy in Transgenic Mouse Models of Huntington’s Disease. J. Neurochem. 2005, 95, 553–562. [Google Scholar] [CrossRef]
- Browne, S.E. Mitochondria and Huntington’s Disease Pathogenesis: Insight from Genetic and Chemical Models. Ann. N. Y. Acad. Sci. 2008, 1147, 358–382. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine Improves Mitochondrial Function and Ameliorates Behavioral Deficits in the R6/1 Mouse Model of Huntington’s Disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermel, E.; Gafni, J.; Propp, S.S.; Leavitt, B.R.; Wellington, C.L.; Young, J.E.; Hackam, A.S.; Logvinova, A.V.; Peel, A.L.; Chen, S.F.; et al. Specific Caspase Interactions and Amplification Are Involved in Selective Neuronal Vulnerability in Huntington’s Disease. Cell Death Differ. 2004, 11, 424–438. [Google Scholar] [CrossRef] [Green Version]
- Ona, V.O.; Li, M.; Vonsattel, J.P.; Andrews, L.J.; Khan, S.Q.; Chung, W.M.; Frey, A.S.; Menon, A.S.; Li, X.J.; Stieg, P.E.; et al. Inhibition of Caspase-1 Slows Disease Progression in a Mouse Model of Huntington’s Disease. Nature 1999, 399, 263–267. [Google Scholar] [CrossRef]
- Carroll, J.B.; Southwell, A.L.; Graham, R.K.; Lerch, J.P.; Ehrnhoefer, D.E.; Cao, L.-P.; Zhang, W.-N.; Deng, Y.; Bissada, N.; Henkelman, R.M.; et al. Mice Lacking Caspase-2 Are Protected from Behavioral Changes, but Not Pathology, in the YAC128 Model of Huntington Disease. Mol. Neurodegener. 2011, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Avenali, M.; Cerri, S.; Ongari, G.; Ghezzi, C.; Pacchetti, C.; Tassorelli, C.; Valente, E.M.; Blandini, F. Profiling the Biochemical Signature of GBA-Related Parkinson’s Disease in Peripheral Blood Mononuclear Cells. Mov. Disord. 2021, 36, 1267–1272. [Google Scholar] [CrossRef]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic Activation of Nrf2 Pathway in Parkinson’s Disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.-P.; Ngoei, K.R.W.; et al. Immortalized Parkinson’s Disease Lymphocytes Have Enhanced Mitochondrial Respiratory Activity. Dis. Models Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haylett, W.; Swart, C.; van der Westhuizen, F.; van Dyk, H.; van der Merwe, L.; van der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered Mitochondrial Respiration and Other Features of Mitochondrial Function in Parkin-Mutant Fibroblasts from Parkinson’s Disease Patients. Park. Dis. 2016, 2016, 1819209. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.M.A.; Kondratyeva, O.; Mommaerts, K.; Ostaszewski, M.; Sokolowska, K.; Baumuratov, A.S.; Longhino, L.; Poulain, J.F.; Grossmann, D.; Balling, R.; et al. Fibroblast Mitochondria in Idiopathic Parkinson’s Disease Display Morphological Changes and Enhanced Resistance to Depolarization. Sci. Rep. 2020, 10, 1569. [Google Scholar] [CrossRef] [Green Version]
- Fais, M.; Dore, A.; Galioto, M.; Galleri, G.; Crosio, C.; Iaccarino, C. Parkinson’s Disease-Related Genes and Lipid Alteration. Int. J. Mol. Sci. 2021, 22, 7630. [Google Scholar] [CrossRef]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial Dysfunction and Increased Glycolysis in Prodromal and Early Parkinson’s Blood Cells. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1580–1590. [Google Scholar] [CrossRef]
- Havelund, J.F.; Heegaard, N.H.H.; Færgeman, N.J.K.; Gramsbergen, J.B. Biomarker Research in Parkinson’s Disease Using Metabolite Profiling. Metabolites 2017, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Willkommen, D.; Lucio, M.; Moritz, F.; Forcisi, S.; Kanawati, B.; Smirnov, K.S.; Schroeter, M.; Sigaroudi, A.; Schmitt-Kopplin, P.; Michalke, B. Metabolomic Investigations in Cerebrospinal Fluid of Parkinson’s Disease. PLoS ONE 2018, 13, e0208752. [Google Scholar] [CrossRef] [Green Version]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.-I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased Long-Chain Acylcarnitines from Insufficient β-Oxidation as Potential Early Diagnostic Markers for Parkinson’s Disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Schirinzi, T.; Di Lazzaro, G.; Sancesario, G.M.; Summa, S.; Petrucci, S.; Colona, V.L.; Bernardini, S.; Pierantozzi, M.; Stefani, A.; Mercuri, N.B.; et al. Young-Onset and Late-Onset Parkinson’s Disease Exhibit a Different Profile of Fluid Biomarkers and Clinical Features. Neurobiol. Aging 2020, 90, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Vasco, G.; Zanni, G.; Petrillo, S.; Piemonte, F.; Castelli, E.; Bertini, E.S. Serum Uric Acid in Friedreich Ataxia. Clin. Biochem. 2018, 54, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Di Lazzaro, G.; Colona, V.L.; Imbriani, P.; Alwardat, M.; Sancesario, G.M.; Martorana, A.; Pisani, A. Assessment of Serum Uric Acid as Risk Factor for Tauopathies. J. Neural Transm. 2017, 124, 1105–1108. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Ueno, S.-I.; Hatano, T.; Okuzumi, A.; Saiki, S.; Oji, Y.; Mori, A.; Koinuma, T.; Fujimaki, M.; Takeshige-Amano, H.; Kondo, A.; et al. Nonmercaptalbumin as an Oxidative Stress Marker in Parkinson’s and PARK2 Disease. Ann. Clin. Transl. Neurol. 2020, 7, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, Y.; Saigoh, K.; Saito, Y.; Ogawa, I.; Mitsui, Y.; Hamada, Y.; Samukawa, M.; Suzuki, H.; Kuwahara, M.; Hirano, M.; et al. Diagnosis of Parkinson’s Disease and the Level of Oxidized DJ-1 Protein. Neurosci. Res. 2018, 128, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Sancesario, G.M.; Di Lazzaro, G.; Grillo, P.; Biticchi, B.; Giannella, E.; Alwardat, M.; Pieri, M.; Bernardini, S.; Mercuri, N.B.; Pisani, A.; et al. Biofluids Profile of α-Klotho in Patients with Parkinson’s Disease. Park. Relat. Disord. 2021, 90, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Espejo, E.; Rodriguez de Fonseca, F.; Suárez, J.; Martín de Pablos, Á. Cerebrospinal Fluid Lactoperoxidase Level Is Enhanced in Idiopathic Parkinson’s Disease, and Correlates with Levodopa Equivalent Daily Dose. Brain Res. 2021, 1761, 147411. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zheng, J.; Ma, J.; Wang, Z.; Shi, X.; Li, M.; Huang, S.; Hu, S.; Zhao, Z.; Li, D. Increased Plasma Heme Oxygenase-1 Levels in Patients With Early-Stage Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 621508. [Google Scholar] [CrossRef]
- Shamir, R.; Klein, C.; Amar, D.; Vollstedt, E.-J.; Bonin, M.; Usenovic, M.; Wong, Y.C.; Maver, A.; Poths, S.; Safer, H.; et al. Analysis of Blood-Based Gene Expression in Idiopathic Parkinson Disease. Neurology 2017, 89, 1676–1683. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jędrak, P.; Mozolewski, P.; Węgrzyn, G.; Więckowski, M.R. Mitochondrial Alterations Accompanied by Oxidative Stress Conditions in Skin Fibroblasts of Huntington’s Disease Patients. Metab. Brain Dis. 2018, 33, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanisova, M.; Stufkova, H.; Kohoutova, M.; Rakosnikova, T.; Krizova, J.; Klempir, J.; Rysankova, I.; Roth, J.; Zeman, J.; Hansikova, H. Mitochondrial Organization and Structure Are Compromised in Fibroblasts from Patients with Huntington’s Disease. Ultrastruct. Pathol. 2022, 46, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A.; Orth, M. Mitochondrial Biology and the Identification of Biomarkers of Huntington’s Disease. Neurodegener. Dis. Manag. 2020, 10, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.; Wu, Y.-R.; Cheng, M.-L.; Liu, J.-L.; Lee, Y.-M.; Lee, P.-W.; Soong, B.-W.; Chiu, D.T.-Y. Increased Oxidative Damage and Mitochondrial Abnormalities in the Peripheral Blood of Huntington’s Disease Patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Zaganjor, E. Mitochondrial Efficiency Directs Cell Fate. Nat. Cell Biol. 2022, 24, 125–126. [Google Scholar] [CrossRef]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- McAdam, E.; Brem, R.; Karran, P. Oxidative Stress-Induced Protein Damage Inhibits DNA Repair and Determines Mutation Risk and Therapeutic Efficacy. Mol. Cancer Res. MCR 2016, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, Oxidative Stress and Aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the Human Mitochondrial Transcription Factor A Gene by Nuclear Respiratory Factors: A Potential Regulatory Link between Nuclear and Mitochondrial Gene Expression in Organelle Biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, W.M.; Hollenbeck, P.J. The Axonal Transport of Mitochondria. J. Cell Sci. 2012, 125 Pt 9, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, T.L. Mitochondrial Trafficking in Neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [Green Version]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Keating, D.J. Mitochondrial Dysfunction, Oxidative Stress, Regulation of Exocytosis and Their Relevance to Neurodegenerative Diseases. J. Neurochem. 2008, 104, 298–305. [Google Scholar] [CrossRef]
- Schirinzi, T.; Madeo, G.; Martella, G.; Maltese, M.; Picconi, B.; Calabresi, P.; Pisani, A. Early Synaptic Dysfunction in Parkinson’s Disease: Insights from Animal Models: Early Synaptic Dysfunction in PD. Mov. Disord. 2016, 31, 802–813. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Doczi, J.; Töröcsik, B.; Bossy-Wetzel, E.; Adam-Vizi, V. Mitochondrial Swelling Measurement in Situ by Optimized Spatial Filtering: Astrocyte-Neuron Differences. Biophys. J. 2008, 95, 2583–2598. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, J.D.B.; Bullen, A.; Mann, Z.F. Mitochondrial Form and Function in Hair Cells. Heart Res. 2023, 428, 108660. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, A.A.; McMurray, C.T. The Chicken or the Egg: Mitochondrial Dysfunction as a Cause or Consequence of Toxicity in Huntington’s Disease. Mech. Ageing Dev. 2017, 161 Pt A, 181–197. [Google Scholar] [CrossRef]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.-H.N.; Huang, C.-S.; Chuang, H.-H.; Lai, H.-J.; Yang, C.-K.; Yang, Y.-C.; Kuo, C.-C. An Electrophysiological Perspective on Parkinson’s Disease: Symptomatic Pathogenesis and Therapeutic Approaches. J. Biomed. Sci. 2021, 28, 85. [Google Scholar] [CrossRef]
- Jamwal, S.; Kumar, P. Insight Into the Emerging Role of Striatal Neurotransmitters in the Pathophysiology of Parkinson’s Disease and Huntington’s Disease: A Review. Curr. Neuropharmacol. 2019, 17, 165–175. [Google Scholar] [CrossRef]
- Terreros-Roncal, J.; Moreno-Jiménez, E.P.; Flor-García, M.; Rodríguez-Moreno, C.B.; Trinchero, M.F.; Cafini, F.; Rábano, A.; Llorens-Martín, M. Impact of Neurodegenerative Diseases on Human Adult Hippocampal Neurogenesis. Science 2021, 374, 1106–1113. [Google Scholar] [CrossRef]
- Levine, M.S.; Cepeda, C.; Hickey, M.A.; Fleming, S.M.; Chesselet, M.-F. Genetic Mouse Models of Huntington’s and Parkinson’s Diseases: Illuminating but Imperfect. Trends Neurosci. 2004, 27, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Mitochondria Alteration | Molecules | References | |
|---|---|---|---|---|
| PD | α-synuclein A53T Mouse (Tg) | EN-mt | ↓ Drp1, ↓ Mfn1 | [90] |
| mtDNA damage | c-caspase-3 and p53 | [91] | ||
| ↑ Mfn1, ↓ Mfn2 | [92] | |||
| A53T-hα-syn | mtUA | ↑ PRKAG2, ↑ TTR | [93] | |
| DE-autophagic/endocytic DA fibres | [93] | |||
| altered TCA cycle at striatal synapses | [93] | |||
| PINK1 KO mouse | ↑ number of larger mt | [94] | ||
| ↓ respiratory complex I, II, III activity, age dependent; ↓ CAA and TCA cycle activity; ↑ protein oxidation | _ | [94] | ||
| PINK1 KO rat | ↓ ATP production | ↑DRP1 | [95] | |
| defects complex I | ↑ O2 consumption | [95] | ||
| increased complex II | [95] | |||
| bioinformatic analysis, PGC1A, PG1B, TFAM, GF1R, INSR, pathways were deactivated | [95] | |||
| DJ-1 mouse KO | ↓ aconitase | [96,97,98,99] | ||
| activity; ↑ROS | ||||
| production | ||||
| ↑ Ca | [96,97,98,99] | |||
| ↑ GSH level and ↑ GSH/glutamate ↑ Glu | [96,97,98,99] | |||
| ↑ TCA cycle, H2O2 consumption ↑ mitochondrial Trx activity, ↑ GSH and ↑ GSSG, ↑ GRX ↓ GR | [96,97,98,99] | |||
| Parkin mouse KO | DP, Cell Stress Chaperones and UPP components | [100] | ||
| ↓ subunits of complexes I ↓subunits IV | ↓ peroxide reductases | [101] | ||
| ↓ antioxidant capacity ↓ protein of lipid peroxidation | [101] | |||
| HD | R6/1 mouse | ↑ (ΔΨm) | ↑ Ca2+, ↑ NAD(P)H | [102] |
| R6/2 mouse | ↑ OH(8)dG | [103] | ||
| ↓ in NAA | [104] | |||
| ↑ glutamine ↑ glucose | [105] | |||
| ↑ creatine | [106] | |||
| ↑ GPC, ↑ glutamine and ↑ glutathione ↓ AA decreased at 8 weeks | [106] | |||
| reduction in mt complex IV activities (12 weeks) | ↑ iNOS and ↑nitrotyrosine | [107] | ||
| ↓ aconitase cerebral cortex | ||||
| ↓ decrease in mitochondrial mass | synaptosomal ↑ ROS production and ↑ antioxidant in striatum | [108] | ||
| YAC128 mouse | ↑ basal and maximal mitochondrial respiration | ↑ [64Cu]-ATSM | [109] | |
| ↑ ATP production, and ↑ complex II and III | [109] | |||
| ↑ oxygen consumption rate | [109] | |||
| ↓ Ca handling | [109] | |||
| YAC72 mouse | ↑ caspase-2 | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. Int. J. Mol. Sci. 2023, 24, 7221. https://doi.org/10.3390/ijms24087221
Tassone A, Meringolo M, Ponterio G, Bonsi P, Schirinzi T, Martella G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. International Journal of Molecular Sciences. 2023; 24(8):7221. https://doi.org/10.3390/ijms24087221
Chicago/Turabian StyleTassone, Annalisa, Maria Meringolo, Giulia Ponterio, Paola Bonsi, Tommaso Schirinzi, and Giuseppina Martella. 2023. "Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson" International Journal of Molecular Sciences 24, no. 8: 7221. https://doi.org/10.3390/ijms24087221
APA StyleTassone, A., Meringolo, M., Ponterio, G., Bonsi, P., Schirinzi, T., & Martella, G. (2023). Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. International Journal of Molecular Sciences, 24(8), 7221. https://doi.org/10.3390/ijms24087221