
PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway
Abstract
:1. Introduction
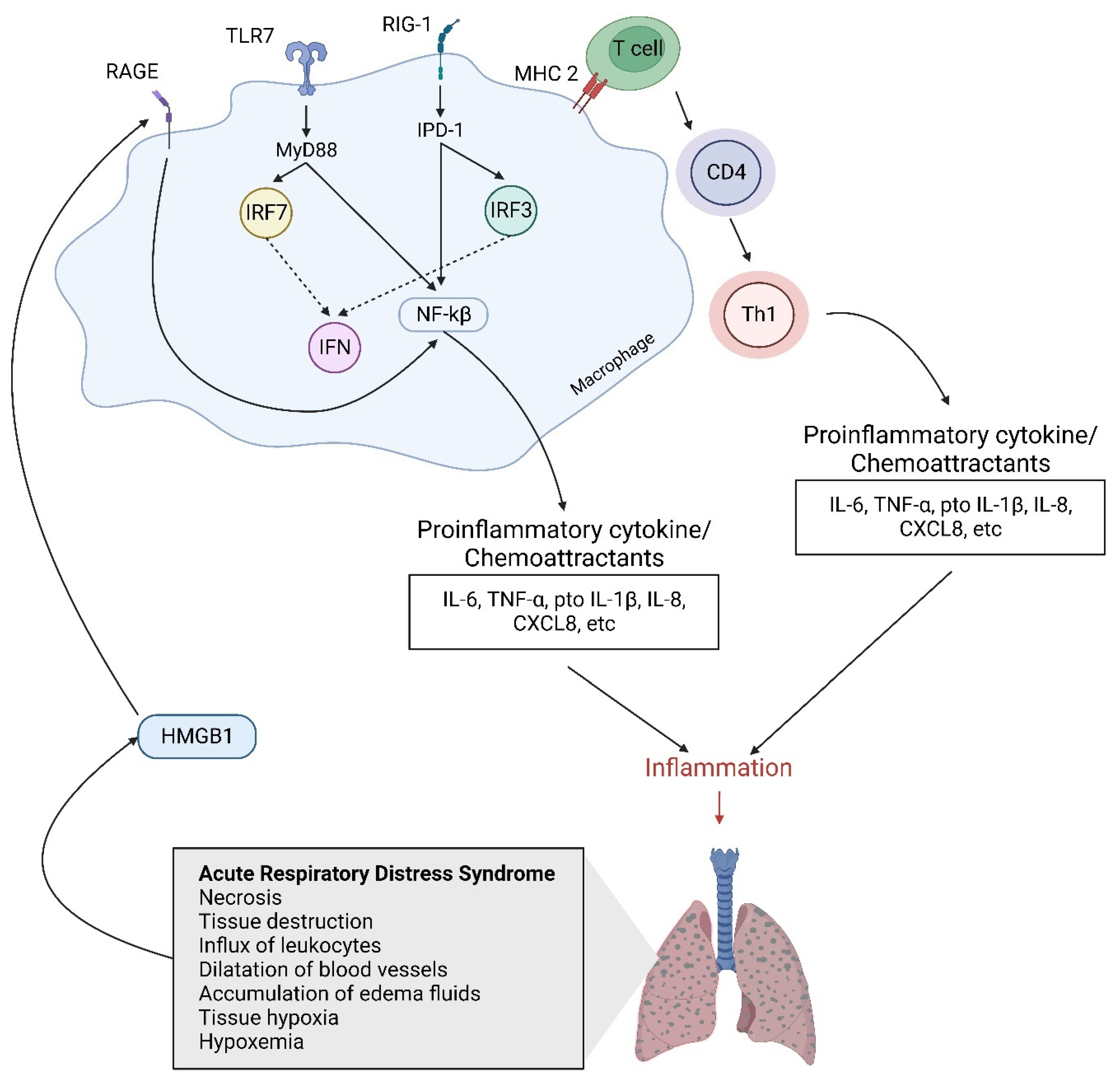
2. Host Immune Response and Cellular Signaling Pathway Activation Induced by Influenza Virus
3. Cytokine Storm and ALI/ARDS Induced by Influenza Virus Infection
4. Mesenchymal Stem Cells
4.1. Allogeneic MSCs
4.2. MSCs Mode and Delivery
4.3. The Mechanisms of Paracrine, Endocrine, and Cell Engraftment of MSCs
4.4. MSCs and Niche Inflammation
4.5. The Immunobiology and Communication between MSCs and Damaged Tissue
4.6. The Immunosuppression Property of MSCs
4.7. The Immunomodulation Roles of Exogenous MSCs in Lung Injury
4.8. The Effect of Exogenous Mesenchymal Stem Cells on Influenza-Virus-Induced Lung Injury
5. Prostaglandin E2 Acts as an Immunosuppressive Properties of Mesenchymal Stromal Cells
6. PGE2 Promotes Immunoregulation through Activation of Wnt-β-Catenin Signaling
6.1. β-Catenin Protein Activates Wnt-β-Catenin Signaling
6.1.1. Wnt Signaling and Regulation
6.1.2. Canonical Signaling Pathway
6.2. PGE2 Elevates the Accumulation of β-Catenin in Lung Injury
6.3. Cross-Regulation between β-Catenin and NF-κB Signaling Pathways
7. The Mechanisms of Immunoregulation after Exogenous MSCs Administration on Virus-Induced Acute Lung Injury
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALI | Acute lung injury |
| Allo-MSCs | Allogeneic MSCs |
| Ang-1 | Angiopoietin-1 |
| AP1 | Activator protein 1 |
| APC | Adenomatous polyposis coli |
| Aqp5 | Aquaporin 5 |
| ARDS | Acute respiratory distress syndrome |
| Auto-MSCs | Autologous MSCs |
| BASCs | Bronchioalveolar stem cells |
| BM-MSCs | Bone-marrow-derived mesenchymal stem cells |
| CK1α | Casein kinase 1α |
| COX-2 | Cyclooxygenase-2 |
| CXCR4+ | Chemokine receptor 4+ |
| DAMPs | Danger-associated molecular patterns |
| DCs | Dendritic cells |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| FARDS | Fulminant ARDS |
| FGF | Fibroblast growth factor |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| GSK-3β | Glycogen synthase kinase-3β |
| HA | Hemagglutinin |
| HGF | Hepatocyte growth factor |
| HMCs | Human mesangial cells |
| HSCs | Hematopoietic stem cells |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IGF-1 | Insulin growth factor-1 |
| Iκβ | Inhibitory kappa beta |
| iNOS | Inducible nitric oxide synthase |
| IRF | Interferon regulatory factor |
| JNK | Jun N kinase |
| KGF | Keratinocyte growth factor |
| LEF | Lymphoid enhancer factors |
| LPS | Lipopolysaccharide |
| MDA 5 | Melanoma differentiation-associated gene 5 |
| MSCs | Mesenchymal stem cells |
| NA | Neuraminidase |
| NLR | Nucleotide-binding domain-leucine-rich repeat-containing |
| PAMPs | Pathogen-associated molecular patterns |
| PDGF | Platelet-derived growth factor |
| PGE2 | Prostaglandin E2 |
| PRRs | Pattern-recognition receptors |
| PI3K | Phosophoinositide 3-kinase |
| RAGE | Receptor for advanced glycation endproducts |
| RIG-1 | Retinol acid inducible gene 1 |
| RNA | Ribonucleic acid |
| RNP | Ribonucleate protein |
| SARS | Severe acute respiratory syndrome |
| SCA 1 | Stem cell antigen 1 |
| SCF | Stem cell factor |
| SDF-1 | Stromal-derived factor-1 |
| SSEA-1 | Stage-specific embryonic antigen 1 |
| ssRNA | Single-stranded RNA |
| TBK | TANK binding kinase |
| TCF | T-cell factor |
| TGF-β | Transforming growth factor-β |
| TLR | Toll-like receptor |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
References
- Dushianthan, A.; Grocott, M.P.; Postle, A.D.; Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad. Med. J. 2011, 87, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, L.B.; Matthay, M.A. The Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, V.; Vlachou, A.; Ghannadian, S.; Simonetti, U.; Slutsky, A.S.; Zhang, H. Acute respiratory distress syndrome: New definition, current and future therapeutic options. J. Thorac. Dis. 2013, 5, 326–334. [Google Scholar] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.T.; Ewig, S.; Rodloff, A.C.; Müller, E.E. Acute respiratory distress syndrome and pneumonia: A comprehensive review of clinical data. Clin. Infect. Dis. 2006, 43, 748–756. [Google Scholar] [CrossRef]
- Sugamata, R.; Dobashi, H.; Nagao, T.; Yamamoto, K.-I.; Nakajima, N.; Sato, Y.; Aratani, Y.; Oshima, M.; Sata, T.; Kobayashi, K.; et al. Contribution of neutrophil-derived myeloperoxidase in the early phase of fulminant acute respiratory distress syndrome induced by influenza virus infection. Microbiol. Immunol. 2012, 56, 171–182. [Google Scholar] [CrossRef]
- World Health Organization. Disease Outbreak News; Avian Influenza A (H5N1)—Cambodia. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON445 (accessed on 20 March 2023).
- Bi, Z.; Hong, W.; Que, H.; He, C.; Ren, W.; Yang, J.; Lu, T.; Chen, L.; Lu, S.; Peng, X.; et al. Inactivated SARS-CoV-2 induces acute respiratory distress syndrome in human ACE2-transgenic mice. Signal Transduct. Target. Ther. 2021, 6, 349–360. [Google Scholar] [CrossRef]
- Barbeta, E.; Motos, A.; Torres, A.; Ceccato, A.; Ferrer, M.; Cilloniz, C.; Bueno, L.; Badia, J.R.; Castro, P.; Ferrando, C.; et al. SARS-CoV-2-Induced Acute Respiratory Distress Syndrome: Pulmonary Mechanics and Gas-Exchange Abnormalities. Ann. Am. Thorac. Soc. 2020, 17, 1164–1168. [Google Scholar] [CrossRef]
- Pfortmueller, C.A.; Spinetti, T.; Urman, R.D.; Luedi, M.M.; Schefold, J.C. COVID-19-associated acute respiratory distress syndrome (CARDS): Current knowledge on pathophysiology and ICU treatment—A narrative review. Best Pract. Res. Clin. Anaesthesiol. 2021, 35, 351–368. [Google Scholar] [CrossRef]
- Esteban, A.; Frutos-Vivar, F.; Muriel, A.; Ferguson, N.D.; Peñuelas, O.; Abraira, V.; Raymondos, K.; Rios, F.; Nin, N.; Apezteguía, C.; et al. Evolution of Mortality over Time in Patients Receiving Mechanical Ventilation. Am. J. Respir. Crit. Care Med. 2013, 188, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Santesso, N.; Mustafa, R.; Brozek, J.; Chen, Y.L.; Hopkins, J.P.; Cheung, A.; Hovhannisyan, G.; Ivanova, L.; Flottorp, S.A.; et al. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann. Intern. Med. 2012, 156, 512–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanunchai, M.; Hongeng, S.; Thitithanyanont, A. Mesenchymal Stromal Cells and Viral Infection. Stem Cells Int. 2015, 2015, 860950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Zhang, L.; Liu, F.; Shen, J. Therapeutic Benefits of Mesenchymal Stem Cells in Acute Respiratory Distress Syndrome: Potential Mechanisms and Challenges. J. Inflamm. Res. 2022, 15, 5235–5246. [Google Scholar] [CrossRef] [PubMed]
- Lohan, P.; Coleman, C.M.; Murphy, J.M.; Griffin, M.D.; Ritter, T.; E Ryan, A. Changes in immunological profile of allogeneic mesenchymal stem cells after differentiation: Should we be concerned? Stem Cell Res. Ther. 2014, 5, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Thavapalachandran, S.; Le, T.Y.L.; Romanazzo, S.; Rashid, F.N.; Ogawa, M.; Kilian, K.A.; Brown, P.; Pouliopoulos, J.; Barry, A.M.; Fahmy, P.; et al. Pluripotent stem cell-derived mesenchymal stromal cells improve cardiac function and vascularity after myocardial infarction. Cytotherapy 2021, 23, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- El-Sawah, S.G.; Rashwan, H.M.; Althobaiti, F.; Aldhahrani, A.; Fayad, E.; Shabana, E.S.; El-Hallous, E.I.; Amen, R.M. AD-MSCs and BM-MSCs Ameliorationg Effects on The Metabolic and Hepato-Renal Abnormalities in Type 1 Diabetic Rats. Saudi J. Biol. Sci. 2022, 29, 1053–1060. [Google Scholar] [CrossRef]
- Luo, F.; Jiang, W.; Xu, Y.; Liu, X.-M.; Wang, W.; Zhang, W.; Luo, C. The Mechanisms Involved in Mesenchymal Stem Cell Alleviation of Sepsis-Induced Acute Lung Injury in Mice: A Pilot Study. Curr. Ther. Res. 2020, 93, 100593. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.-L.; Ma, H.-C.; Tang, Z.-T.; Ding, H.-R.; Shi, X.-L. Mesenchymal stem cells increase heme oxygenase 1-activated autophagy in treatment of acute liver failure. Biochem. Biophys. Res. Commun. 2019, 508, 682–689. [Google Scholar] [CrossRef]
- Zhou, T.; Liao, C.; Lin, S.; Lin, W.; Zhong, H.; Huang, S. The Efficacy of Mesenchymal Stem Cells in Therapy of Acute Kidney Injury Induced by Ischemia-Reperfusion in Animal Models. Stem Cells Int. 2020, 2020, 1873921. [Google Scholar] [CrossRef]
- Gad, E.S.; Salama, A.A.A.; El-Shafie, M.F.; Arafa, H.M.M.; Abdelsalam, R.M.; Khattab, M. The Anti-Fibrotic and Anti-Inflammatory Potential of Bone Marrow-Derived Mesenchymal Stem Cells and Nindetanib in Bleomycin-Induced Lung Fibrosis in Rats. Inflammation 2020, 43, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Dilogo, I.H.; Aditianingsih, D.; Sugiarto, A.; Burhan, E.; Damayanti, T.; Sitompul, P.A.; Mariana, N.; Antarianto, R.D.; Liem, I.K.; Kispa, T.; et al. Umbilical cord mesenchymal stromal cells as critical COVID-19 adjuvant therapy: A randomized controlled trial. Stem Cells Transl. Med. 2021, 10, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Zhu, Y.G.; Jia, X.M.; Liu, D.; Qu, J.M. Adipose-Derived Mesenchymal Stem Cells Ameliorating Pseudomonas Aeruginosa-Induced Acute Lung Infection via Inhibition of NLRC4 Inflammasome. Front. Cell. Infect. Microbiol 2021, 10, 581535. [Google Scholar] [CrossRef]
- Jankauskaite, L.; Schmoldt, C.; Lohmeyer, J.; Herold, S. Therapeutic potential of murine bone marrow derived mesenchymal stem cells in influenza virus-induced pneumonia. Pneumologie 2014, 68, A25. [Google Scholar] [CrossRef]
- Gotts, J.E.; Abbott, J.; Matthay, M.A. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L395–L406. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.W.; Kuok, D.I.T.; Leung, C.Y.H.; Hui, K.P.Y.; Valkenburg, S.A.; Lau, E.H.Y.; Nicholls, J.M.; Fang, X.; Guan, Y.; Lee, J.W.; et al. Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 3621–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, D.; Liu, X.; Tang, S.; Wei, F. Human umbilical cord mesenchymal stem cells reduce systemic inflammation and attenuate LPS-induced acute lung injury in rats. J. Inflamm. 2012, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Yu, J.; Chen, Q.; Yan, H.; Du, H.; Luo, W. Regulation of pathophysiological and tissue regenerative functions of mediated via the WNT signaling pathway (Review). Mol. Med. Rep. 2021, 24, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Lin, Q.; Wu, L.; Chatla, S.; Chowdhury, F.A.; Atale, N.; Joseph, J.; Du, W. Hematopoietic stem cell regeneration through paracrine regulation of the Wnt5a/Prox1 signaling axis. J. Clin. Investig. 2022, 132, e155914. [Google Scholar] [CrossRef]
- Gürtler, L. Virolog of Human Influenza. In Influenza Report 2006; Kamps, B.S., Hoffmann, C., Presier, W., Eds.; Flying Publisher: Paris, France, 2006; pp. 87–91. Available online: https://www.paho.org/hq/dmdocuments/2010/Influenza%20Report%202006_Sebastian_2006.pdf (accessed on 9 December 2022).
- Owen, D.M.; Gale, M. Fighting the flu with inflammasome signaling. Immunity 2009, 30, 476–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of Single-Stranded RNA Viruses by Toll-Like Receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, I.K.; Iwasaki, A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. 2011, 32, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Gaur, P.; Munjhal, A.; Lal, S.K. Influenza virus and cell signaling pathways. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2011, 17, RA148–RA154. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhang, L.; Hong, T. Host cellular signaling induced by influenza virus. Sci. China Life Sci. 2011, 54, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Osterholm, M.T. Preparing for the next pandemic. N. Engl. J. Med. 2005, 352, 1839–1842. [Google Scholar] [CrossRef] [Green Version]
- Pizzolla, A.; Smith, J.M.; Brooks, A.G.; Reading, P.C. Pattern recognition receptor immunomodulation of innate immunity as a strategy to limit the impact of influenza virus. J. Leukoc. Biol. 2017, 101, 851–861. [Google Scholar] [CrossRef]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines induced during influenza virus infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef]
- Freeman, C.M.; Martinez, F.J.; Han, M.K.; Ames, T.M.; Chensue, S.W.; Todt, J.C.; Arenberg, D.A.; Meldrum, C.A.; Getty, C.; McCloskey, L. Lung dendritic cell expression of maturation molecules increases with worsening chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Hsu, A.C.-Y.; Pang, Z.; Pan, H.; Zuo, X.; Wang, G.; Zheng, J.; Wang, F. Role of the Innate Cytokine Storm Induced by the Influenza A Virus. Viral Immunol. 2019, 32, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Maines, T.R.; Szretter, K.J.; Perrone, L.; Belser, J.A.; Bright, R.A.; Zeng, H.; Tumpey, T.M.; Katz, J.M. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol. Rev. 2008, 225, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolle, L.B.; Standiford, T.J. Danger-associated molecular patterns (DAMPs) in acute lung injury. J. Pathol. 2013, 229, 145–146. [Google Scholar] [CrossRef] [Green Version]
- van Zoelen, M.A.D.; van der Sluijs, K.F.; Achouiti, A.; Florquin, S.; Braun-Pater, J.M.; Yang, H.; Nawroth, P.P.; Tracey, K.J.; Bierhaus, A.; van der Poll, T. Receptor for Advanced Glycation end Products Is Detrimental during Influenza A Virus Pneumonia. Virology 2009, 391, 265–273. [Google Scholar] [CrossRef] [Green Version]
- van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced gkycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int. 2018, 2018, 3057624. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.I.; Frolova, G.P. Heterotopic of Bone Marrow. Analysis of Precursor Cells for Osteogenic and Hematopoietic Tissues. Transplantation 1968, 6, 230–247. [Google Scholar] [CrossRef]
- Haynesworth, S.E.; Goshima, J.; Goldberg, V.M.; Caplan, A.I. Characterization of cells with osteogenic potential from human marrow. Bone 1992, 13, 81–88. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells: The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, D.J.; Kolls, J.K.; Ortiz, L.; Panoskaltsis-Mortari, A.; Prockop, D.J. Stem Cells and Cell Therapies in Lung Biology and Lung Diseases. Proc. Am. Thorac. Soc. 2008, 5, 637–667. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.E.; Yoo, D.; Leroux, M.A.; Danilkovitch-Miagkova, A. Treatment of inflammatory diseases with mesenchymal stem cells. Inflamm. Allergy Drug Targets 2009, 8, 110–123. [Google Scholar] [CrossRef]
- Jung, H.G.; Ahn, E.-K.; Lee, J.-H.; Kim, Y.-H.; Leem, S.-H.; Heo, J.; Kim, H. Effects of harvesting sites and ages on adipose tissue-derived stem cells in rat. Tissue Eng. Regen. Med. 2014, 11, 137–142. [Google Scholar] [CrossRef]
- Cianfarani, F.; Toietta, G.; Di Rocco, G.; Cesareo, E.; Zambruno, G.; Odorisio, T. Diabetes impairs adipose tissue—Derived stem cell function and efficiency in promoting wound healing. Wound Repair Regen. 2013, 21, 545–553. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, X.; Wang, H.; Liu, X.; Zhang, T.; Wang, Y.; Hu, D. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res. Ther. 2015, 6, 234. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.D.; Ryan, A.E.; Alagesan, S.; Lohan, P.; Treacy, O.; Ritter, T. Anti-donor immune responses elicited by allogeneic mesenchymal stem cells: What have we learned so far? Immunol. Cell Biol. 2013, 91, 40–51. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Zhang, Y.; Li, X.; Deng, M.-X.; Gao, W.-X.; Yao, Y.; Chiu, S.-M.; Liang, X.; Gao, F.; Chan, C.W.; et al. Insensitivity of Human iPS Cells-Derived Mesenchymal Stem Cells to Interferon-γ-Induced HLA Expression Potentiates Repair Efficiency of Hind Limb Ischemia in Immune Humanized NOD Scid Gamma Mice. Stem Cells 2015, 33, 3452–3467. [Google Scholar] [CrossRef] [Green Version]
- Klyushnenkova, E.; Mosca, J.D.; Zernetkina, V.; Majumdar, M.K.; Beggs, K.J.; Simonetti, D.W.; Deans, R.J.; McIntosh, K.R. T cell responses to allogeneic human mesenchymal stem cells: Immunogenicity, tolerance, and supression. J. Biomed. Sci. 2005, 12, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Saldanha-Araujo, F. Mechanisms of T-Cell Immunosuppression by Mesenchymal Stromal Cells: What Do We Know So Far? Biomed Res. Int. 2014, 2014, 216806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtzberg, J.; Prockop, S.; Teira, P.; Bittencourt, H.; Lewis, V.; Chan, K.W.; Horn, B.; Yu, L.; Talano, J.A.; Nemecek, E.; et al. Allogeneic Human Mesenchymal Stem Cell Therapy (Remestemcel-L, Prochymal) as a Rescue Agent for Severe Refractory Acute Graft-Versus-Host Disease in Pediatric Patients. Biol. Blood Marrow Transplant. 2014, 20, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbano, P.; Cuccia, A.; Mazzanti, B.; Urbani, S.; Giusti, B.; Lapini, I.; Rossi, L.; Abbate, R.; Marseglia, G.; Nannetti, G.; et al. Use of donor bone marrow mesenchymal stem cells for treatment of skin allograft rejection in a preclinical rat model. Arch. Dermatol. Res. 2008, 300, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Urban, V.S.; Kiss, J.; Kovács, J.; Gócza, E.; Vas, V.; Monostori, E.; Uher, F. Mesenchymal stem cells cooperate with bone marrow cells in therapy of diabetes. Stem Cells 2008, 26, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.T.; Lin, P.; Caplan, A.I.; Dennis, J.E. MSCs: Delivery Routes and Engraftment, Cell-Targeting Strategies, and Immune Modulation. Stem Cells Int. 2013, 2013, 732742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, H.; Olson, S.D.; Kumar, A.; George, M.; Prabhakara, K.S.; Wenzel, P.; Bedi, S.; Toledano-Furman, N.E.; Triolo, F.; Kamhieh-Milz, J.; et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenge to Clinical Application. Front. Immunol. 2019, 10, 1645. [Google Scholar] [CrossRef]
- Moll, G.; Ankrum, J.A.; Kamhieh-Milz, J.; Bieback, K.; Ringdén, O.; Volk, H.D.; Geissler, S.; Reinke, P. Intravascular mesenchymal stromal/stemcell therapy product diversification: Time dor new clinical guidelines. Trends Mol. Med. 2019, 25, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–691. [Google Scholar] [CrossRef]
- Phinney, D.G.; Prockop, D.J. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair current views. Stem Cells 2007, 225, 2896–2902. [Google Scholar] [CrossRef]
- Kotton, D.N.; Fabian, A.J.; Mulligan, R.C. Failure of bone marrow to reconstitute lung epithelium. Am. J. Respir. Cell Mol. Biol. 2005, 33, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, M.; Xu, J.; Woods, C.R.; Mora, A.L.; Spears, W.; Roman, J.; Brigham, K.L. Bone marrow–derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 2005, 33, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowski, M.; Lyczek, A.; Engels, C.; Xu, J.; Lukomska, B.; Bulte, J.W.; Walczak, P. Cell size and velocity of injection are major determinants of the safety of intracarotid stem cell transplantation. J. Cereb. Blood Flow Metab. 2013, 33, 921–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Fang, X.; Krasnodembskaya, A.; Howard, J.P.; Matthay, M.A. Concise review: Mesenchymal stem cells for acute lung injury: Role of paracrine soluble factors. Stem Cells 2011, 29, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, L.A.; DuTreil, M.; Fattman, C.; Pandey, A.C.; Torres, G.; Go, K.; Phinney, D.G. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc. Natl. Acad. Sci. USA 2007, 104, 11002–11007. [Google Scholar] [CrossRef] [Green Version]
- Németh, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2—Dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Su, X.; Popov, B.; Lee, J.W.; Serikov, V.; Matthay, M.A. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J. Immunol. 2007, 179, 1855–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnodembskaya, A.; Song, Y.; Fang, X.; Gupta, N.; Serikov, V.; Lee, J.-W.; Matthay, M.A. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells 2010, 28, 2229–2238. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Fang, X.; Gupta, N.; Serikov, V.; Matthay, M.A. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc. Natl. Acad. Sci. USA 2009, 106, 16357–16362. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.H.J.; McCarter, S.D.; Deng, Y.; Parker, C.H.; Liles, W.C.; Stewart, D.J. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007, 4, e269. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Gambelli, F.; McBride, C.; Gaupp, D.; Baddoo, M.; Kaminski, N.; Phinney, D.G. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc. Natl. Acad. Sci. USA 2003, 100, 8407–8411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prockop, D.J.; Kota, D.J.; Bazhanov, N.; Reger, R.L. Evolving paradigms for repair of tissues by adult stem/progenitor cells (MSCs). J. Cell. Mol. Med. 2010, 14, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Theise, N.D.; Collector, M.I.; Henegariu, O.; Hwang, S.; Gardner, R.; Neutzel, S.; Sharkis, S.J. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 2001, 105, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Glennie, S.; Soeiro, I.; Dyson, P.J.; Lam, E.; Dazzi, F.; Lutsiak, M.E.C.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.S.; Schlom, J.; et al. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, I.; Ringdén, O.; Sundberg, B.; Le Blanc, K. Mesenchymal stem cells inhibit the formation of cytotoxic T lymphocytes, but not activated cytotoxic T lymphocytes or natural killer cells. Transplantation 2003, 76, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.X.; Zhang, Y.; Liu, B.; Zhang, S.-X.; Wu, Y.; Yu, X.-D.; Mao, N. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 2005, 105, 4120–4126. [Google Scholar] [CrossRef] [Green Version]
- Madrigal, M.; Rao, K.S.; Riordan, N.H. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J. Transl. Med. 2014, 12, 260. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef]
- Shi, Y.; Su, J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksh, D.; Song, L.; Tuan, R.S. Adult mesenchymal stem cells: Characterization, differentiation, and application in cell and gene therapy. J. Cell. Mol. Med. 2004, 8, 301–316. [Google Scholar] [CrossRef]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4 þ CD25(High) forkhead box P3 þ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef]
- Ylöstalo, J.H.; Bartosh, T.J.; Coble, K.; Prockop, D.J. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells 2012, 30, 2283–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaggiari, G.M.; Abdelrazik, H.; Becchetti, F.; Moretta, L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: Central role of MSC-derived prostaglandin E2. Blood 2009, 113, 6576–6583. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Han, Z.-B.; Liao, W.; Yang, S.G.; Yang, Z.; Yu, J.; Meng, L.; Wu, R.; Han, Z.C. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4 + CD25+ Forkhead Boxp3 (FOXP3) + regulatory T cells and balancing anti- and pro-inflammatory factors. Cell. Physiol. Biochem. 2011, 27, 587–596. [Google Scholar] [CrossRef]
- Mei, S.H.; Haitsma, J.J.; Dos Santos, C.C.; Deng, Y.; Lai, P.F.; Slutsky, A.S.; Liles, W.C.; Stewart, D.J. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am. J. Respir. Crit. Care Med. 2010, 182, 1047–1057. [Google Scholar] [CrossRef]
- Maron-Gutierrez, T.; Silva, J.D.; Asensi, K.D.; Bakker-Abreu, I.; Shan, Y.; Diaz, B.L.; Goldenberg, R.C.S.; Mei, S.H.J.; Stewart, D.J.; Morales, M.M.M.; et al. Effects of mesenchymal stem & cell therapy on the time course of pulmonary remodeling depend on the etiology of lung injury in mice. Crit. Care Med. 2013, 41, e319–e333. [Google Scholar]
- Shin, S.; Kim, Y.; Jeong, S.; Hong, S.; Kim, I.; Lee, W.; Choi, S. The therapeutic effect of human adult stem cells derived from adipose tissue in endotoxemic rat model. Int. J. Med. Sci. 2013, 10, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Danchuk, S.D.; Imhof, K.M.; A Semon, J.; A Scruggs, B.; Bonvillain, R.W.; Strong, A.L.; Gimble, J.M.; Betancourt, A.M.; E Sullivan, D.; et al. Comparison of the therapeutic effects of human and mouse adipose-derived stem cells in a murine model of lipopolysaccharide-induced acute lung injury. Stem Cell Res. Ther. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Choi, S.J.; Ahn, S.Y.; Sung, D.K.; Sung, S.I.; Yoo, H.S.; Oh, W.I.; Park, W.S. Timing of umbilical cord blood derived mesenchymal stem cells transplantation determines therapeutic efficacy in the neonatal hyperoxic lung injury. PLoS ONE 2013, 8, e52419. [Google Scholar] [CrossRef] [Green Version]
- Ornellas, D.S.; Maron-Gutierrez, T.; Ornellas, F.M.; Cruz, F.F.; Oliveira, G.P.; Lucas, I.H.; Fujisaki, L.; Oliveira, M.G.; Teodoro, W.R.; Capelozzi, V.L.; et al. Early and late effects of bone marrow-derived mononuclear cell therapy on lung and distal organs in experimental sepsis. Respir. Physiol. Neurobiol. 2011, 178, 304–314. [Google Scholar] [CrossRef]
- Nandra, K.K.; Takahashi, K.; Collino, M.; Benetti, E.; Wong, W.S.F.; Goh, F.Y.; Suzuki, K.; Patel, N.S.A.; Thiemermann, C. Acute treatment with bone marrow-derived mononuclear cells attenuates the organ injury/dysfunction induced by hemorrhagic shock in the rat. Shock 2012, 37, 592–598. [Google Scholar] [CrossRef]
- Gorman, E.; Shankar-Hari, M.; Hopkins, P.; Tunnicliffe, W.S.; Perkins, G.D.; Silversides, J.; McGuigan, P.; Krasnodembskaya, A.; Jackson, C.; Boyle, R.; et al. Repair of acute respiratory distress syndrome by stromal cell administration (REALIST) trial: A phase 1 trial. EClinicalMedicine 2021, 41, 101167. [Google Scholar] [CrossRef] [PubMed]
- Adas, G.; Cukurova, Z.; Yasar, K.K.; Yilmaz, R.; Isiksacan, N.; Kasapoglu, P.; Yesilbag, Z.; Koyuncu, I.; Karaoz, E. The Systematic Effect of Mesenchymal Stem Cell Therapy in Critical COVID-19 Patients: A Prospective Double Controlled Trial. Cell Transplant. 2021, 30, 9636897211024942. [Google Scholar] [CrossRef]
- Chen, J.; Hu, C.; Chen, L.; Tang, L.; Zhu, Y.; Xu, X.; Chen, L.; Gao, H.; Lu, X.; Yu, L.; et al. Clinical Study of Mesenchymal Stem Cell Treatment for Acute Respiratory Distress Syndrome Induced by Epidemic Influenza A (H7N9) Infection: A Hint for COVID-19 Treatment. Engineering 2020, 6, 1153–1161. [Google Scholar] [CrossRef]
- Yudhawati, R.; Amin, M.; Rantam, F.A.; Prasetya, R.R.; Dewantari, J.R.; Nastri, A.M.; Poetranto, E.D.; Wulandari, L.; Lusida, M.I.; Koesnowidagdo, S.; et al. Bone marrow-derived mesenchymal stem cells attenuate plmonary inflammation and lung damage caused by highly pathofenic avian influenza A/H5N1 virus in BALB/c mice. BMC Infect. Dis. 2020, 20, 823–838. [Google Scholar] [CrossRef]
- Bogatcheva, N.C.; Coleman, M.E. Concentrated Scretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice. Cells 2021, 10, 720. [Google Scholar] [CrossRef]
- Khatri, M.; Richardson, L.A.; Meulia, T. Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res. Ther. 2018, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, J.; Shi, W.; Chen, C.; Shao, Y.; Zhu, L.; Lu, W.; Han, X. Mesenchymal stromal cell treatment prevents H9N2 avian influenza virus-induced acute lung injury in mice. Stem Cell Res. Ther. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loy, H.; Kuok, D.I.; Hui, K.P.; Choi, M.H.; Yuen, W.; Nicholls, J.M.; Peiris, J.M.; Chan, M.C. Therapeutic Implications of Human Umbilical Cord Mesenchymal Stromal Cells in Attenuating Influenza A(H5N1) Virus–Associated Acute Lung Injury. J. Infect. Dis. 2019, 219, 189–196. [Google Scholar] [CrossRef]
- Mallis, P.; Michalopoulos, E.; Chatzistamatiou, T.; Giokas, C.S. Interplay between mesenchymal stromal cells and immune system: Clinical applications in immune-related diseases. Explor. Immunol. 2021, 1, 112–139. [Google Scholar] [CrossRef]
- Kota, D.J.; Prabhakara, K.S.; Toledano-Furman, N.; Bhattarai, D.; Chen, Q.; DiCarlo, B.; Smith, P.; Triolo, F.; Wenzel, P.L.; Cox, C.S.; et al. Prostaglandin E2 Indicates Therapeutic Efficacy of Mesenchymal Stem Cells in Experimental Traumatic Brain Injury. Stem Cells 2017, 35, 1416–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Han, Y.; Zhuang, Y.; Fu, J.; Liu, H.; Shi, Q.; Ju, X. Overexpression of COX-2 but not indoleamine 2,3-dioxygenase-1 enhances the immunosuppressive ability of human umbilical cord-derived mesenchymal stem cells. Int. J. Mol. Med. 2015, 35, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yañez, R.; Oviedo, A.; Aldea, M.; Bueren, J.A.; Lamana, M.L. Prostaglandin E2 plays a key role in the immunosuppressive properties of adipose and bone marrow tissue-derived mesenchymal stromal cells. Exp. Cell Res. 2010, 316, 3109–3123. [Google Scholar] [CrossRef]
- Legler, D.F.; Bruckne, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Han, J.; Xu, X.; Xu, J.; Liu, L.; Huang, Y.; Yang, Y.; Qiu, H. PGE2 Promotes the migration of mesenchymal stem cells through the activation of FAK and ERK1/2 pathway. Stem Cells Int. 2017, 2017, 8178643. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Dingledine, R. Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J. Pharmacol. Exp. Ther. 2013, 344, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.C.; Kim, H.S.; Shin, T.H.; Kang, I.; Lee, J.Y.; Kim, J.-J.; Kang, H.K.; Seo, Y.; Lee, S.; Yu, K.-R.; et al. PGE2 maintains self-renewal of human adult stem cells via EP2-mediated autocrine signaling and its production is regulated by cell-to-cell contact. Sci. Rep. 2016, 6, 26298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vancheri, C.; Mastruzzo, C.; Sortino, M.A.; Crimi, N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004, 25, 40–46. [Google Scholar] [CrossRef]
- Konya, V.; Maric, J.; Jandl, K.; Luschnig, P.; Aringer, I.; Lanz, I.; Platzer, W.; Theiler, A.; Bärnthaler, T.; Frei, R.; et al. Activation of EP4 receptors prevents endotoxin-induced neutrophil infiltration into the airways and enhances microvascular barrier function. Br. J. Pharmacol. 2015, 172, 4454–4468. [Google Scholar] [CrossRef] [Green Version]
- Fujino, H.; West, K.A.; Regan, J.W. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J. Biol. Chem. 2002, 277, 2614–2619. [Google Scholar] [CrossRef] [Green Version]
- Castellone, M.D.; De Falco, V.; Rao, D.M.; Bellelli, R.; Muthu, M.; Basolo, F.; Fusco, A.; Gutkind, J.S.; Santoro, M. The β catenin axis integrates multiple signals downstrem foe RET/Papillary thyroid carcinoma leading to cell proliferation. Cancer Res. 2009, 69, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, S.P.; Haq, S.; Force, T. Activation of β-catenin Signaling Pathways by Classical G-Protein-Coupled Receptors: Mechanisms and Consequences in Cycling and Non-cycling Cells. Cell Cycle 2006, 5, 2295–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Qu, Y.X.; Wang, B.; Shen, P.F.; Xu, J.D.; Chen, Y.X. COX-2/PGE2 facilitates fracture healing by activating the Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharmalogical Sci. 2019, 23, 9721–9728. [Google Scholar]
- Du, Q.; Geller, D.A. Cross-Regulation between Wnt and NF-β B Signaling Pathways. Immunopathol. Dis. Ther. 2010, 1, 151–181. [Google Scholar]
- Deng, J.; Miller, S.A.; Wang, H.Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.Y.; Hung, M.C. Beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer cell 2002, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Robert, M.E.; Duan, Y.; Rao, A.S.; He, T.C.; Chang, E.B.; Madara, J.L. Crosstalk between NFkB and β catenin pathways in bacterial colonized intestinal epithelial cell. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G129–G137. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Lu, R.; Wu, S.; Zhang, Y.-G.; Xia, Y.; Sartor, B.R.; Sun, J. Wnt2 inhibits enteric bacterial induce inflammation in intestinal epithelial cell. Inflamm. Bowel Dis. 2012, 18, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Silva-García, O.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M. The Wnt/β-Catenin Signaling Pathway Controls the Inflammatory Response in Infections Caused by Pathogenic Bacteria. Mediat. Inflamm. 2014, 2014, 310183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschman, H.R. Prostaglandin synthase 2. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1996, 1299, 125–140. [Google Scholar] [CrossRef]
- Hinz, B.; Brune, K. Cyclooxygenase-2–10 years later. J. Pharmacol. Exp. Ther. 2002, 300, 367–375. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryawanshi, A.; Tadagavadi, R.K.; Swafford, D.; Manicassamy, S. Modulation of Inflammatory Responses by Wnt/β-Catenin Signaling in Dendritic Cells: A Novel Immunotherapy Target for Autoimmunity and Cancer. Front. Immunol. 2016, 7, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Winter, T.J.J.; Nusse, R. Running Against the Wnt: How Wnt/β-Catenin Suppresses Adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales fromthe crypt: New insights into intestinal stem cells. Nat. Rev. 2019, 16, 19–34. [Google Scholar] [CrossRef]
- Silva-Garcia, O.; Valdez-Alarcόn, J.J.; Baizabal-Aguirre, V.M. Wnt/β-Catenin Signaling as a Molecular Target by Pathogenic Bacteria. Front. Immunol. 2019, 10, 2135. [Google Scholar] [CrossRef] [Green Version]
- Pokutta, S.; Weis, W.I. Structure of thr dimerization and β—Catenin binding region of α catenin. Mol. Cell 2000, 5, 533–543. [Google Scholar] [CrossRef]
- Fiedler, M.; Mendoza-Topaz, C.; Rutherford, T.J.; Mieszczanek, J.; Bienz, M. Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down-regulating β-catenin. Proc. Natl. Acad. Sci. USA 2011, 108, 1937–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt signalling—Two contrasting models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etheridge, S.L.; Spencer, G.J.; Heath, D.J.; Genever, P.G. Expression Profiling and Functional Analysis of Wnt Signaling Mechanisms in Mesenchymal Stem Cells. Stem Cells 2014, 22, 849–860. [Google Scholar] [CrossRef]
- Bärnthaler, T.; Maric, J.; Platzer, W.; Konya, V.; Theiler, A.; Hasenöhrl, C.; Gottschalk, B.; Trautmann, S.; Schreiber, Y.; Graier, W.F.; et al. The Role of PGE2 in Alveolar Epithelial and Lung Microvascular Endothelial Crosstalk. Sci. Rep. 2017, 7, 7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Jung, C.; Liu, C.; Sheng, H. Prostaglandin E2 Stimulates the B-Catenin/T Cell Factor-Dependent Transcription in Colon Cancer. J. Biol. Chem. 2005, 280, 26565–26572. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Chang, H.-M.; Cheng, J.-C.; Leung, P.C.K.; Sun, Y.-P. TGF-β1 induces COX-2 expression and pge2 production in human granulosa cells through smad signaling pathways. J. Clin. Endocrinol. Metab. 2014, 99, E1217–E1226. [Google Scholar] [CrossRef] [Green Version]
- Flozak, A.S.; Lam, A.P.; Russell, S.; Jain, M.; Peled, O.N.; Sheppard, K.A.; Beri, R.; Mutlu, G.M.; Budinger, G.S.; Gottardi, C.J. Beta-catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J. Biol. Chem. 2010, 285, 3157–3167. [Google Scholar] [CrossRef] [Green Version]
- Mutze, K.; Vierkotten, S.; Milosevic, J.; Eickelberg, O.; Königshoff, M. Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/β-catenin-driven transdifferentiation of murine alveolar epithelial cells. Dis. Model. Mech. 2015, 8, 877–890. [Google Scholar]
- Abraham, E.; Arcaroli, J.; Carmody, A.; Wang, H.; Tracey, K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000, 165, 2950–2954. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Hernandez, M.M.A. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Hoidal, J.R.; Tapan, K. Role of TNFα in pulmonary pathophysiology. Respir. Res. 2006, 7, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; A Amoscato, A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Author | Virus | MSCS Sources | Outcome of Infection | Study Design | References |
|---|---|---|---|---|---|---|
| 1 | Chen, J., et al. (2020) | Influenza A (H7N9) | Allogeneic, menstrual-blood-derived MSCs | Lower levels of PCT, serum creatinin (sCr), creatine kinase (CK), prothrombin time (PT), and D-dimer; hemoglobin (Hb) up-regulated; improvement on CCT | Human | [110] |
| 2 | Yudhawati, R., et al. (2020) | Avian influenza A/H5N1 of A/turkey/East Java/Av154/2013 | Bone-marrow-derived mesenchymal stem cells (BM-MSCs) | Lower levels of lung alveolar protein, PaO2/FiO2 ratio, and histopathological score; suppressed expressions of NF-κB, RAGE, TNFα, and IL-1β; Sftpc and Aqp5+ enhanced | In vivo (mice) | [111] |
| 3 | Bogatcheva and Coleman (2021) | Influenza A Virus H1N1 | Adipose stromal cells | Limits pulmonary histopathological changes, infiltration of inflammatory cells, protein leak, water accumulation, and arterial oxygen saturation (SpO2) reduction; significant suppression of IL-6 and MCP-1; viral antigen, BAL TNF-α, PDL1, and Angpt2 levels lowered | In vivo (mice) | [112] |
| 4 | Chan et al. (2016) | H5N1 influenza A viruses A/Hong Kong/483/97 (483/97), A/Hong Kong/486/97 (486/97), and A/Vietnam/3046/04 (3046/04) | Bone-marrow-derived human MSCs | Transporter proteins CFTR and α1Na,K-ATPase enhanced; Ang1 in MSCS supernatants, KGF secretion, CFTR protein expression increased; lower CD4+ T cells, NK cells, IP10, MCP-1, MCP-3, M1P-1α, RANTES, IL-4, IL-17, and TNF-α; more macrophages/monocytes | In vitro and in vivo | [27] |
| 5 | Khatri et al. (2018) | Influenza virus H1N1, H3N2, H9N5 | Isolated extracellular vesicles EVs from swine bone-marrow-derived MSCs | Reduced influenza virus replication; lower levels of TNF-α and CXCL10; cytokine IL-10 were higher | In vivo (pigs) | [113] |
| 6 | Li, Y., et al. (2016) | A/Hong Kong/2108/2003 [H9N2 (HK)] H9N2 virus | Adipocyte, chondrocytes, and osteocytes murine MSCs | Higher PaO2, SaO2 and pH values; PaCO2, GM-CSF, MCP-1, KC, MIP-1α, MIG, IL-1α, IL-6, IL-10, TNF-α, IFN-γ, ERK, JNK, CD14, and TLR4 expression were lowered | In vivo (mice) | [114] |
| 7 | Loy, H., et al. (2019) | Influenza A/Hong Kong/483/97(H5N1) | Human umbilical cord MSCs | In vitro: reduced IL-β, IFN-λ1, IL-6, IL-1β, IFN-γ-induced protein 10 (IP-10), MCP-1, and RANTES; enhances responses of IL-4, IL-10, IL-11, IL-13, IL-RA, IL-10, IL-11, and IL-RA. In vivo: greatest significance for IP-10, MCP-1, RANTES, and IL-6; lower concentrations of Evans blue dye, IP-10, RANTES, TNF-α, MCP-1, IL-1β, IL-6, and IL-8 | In vitro and in vivo (mice) | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yudhawati, R.; Shimizu, K. PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 7299. https://doi.org/10.3390/ijms24087299
Yudhawati R, Shimizu K. PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway. International Journal of Molecular Sciences. 2023; 24(8):7299. https://doi.org/10.3390/ijms24087299
Chicago/Turabian StyleYudhawati, Resti, and Kazufumi Shimizu. 2023. "PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway" International Journal of Molecular Sciences 24, no. 8: 7299. https://doi.org/10.3390/ijms24087299
APA StyleYudhawati, R., & Shimizu, K. (2023). PGE2 Produced by Exogenous MSCs Promotes Immunoregulation in ARDS Induced by Highly Pathogenic Influenza A through Activation of the Wnt-β-Catenin Signaling Pathway. International Journal of Molecular Sciences, 24(8), 7299. https://doi.org/10.3390/ijms24087299