
Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies


Abstract
1. Introduction
2. Results and Discussion
2.1. QSAR Modelling to Identify Potential Hit Compounds
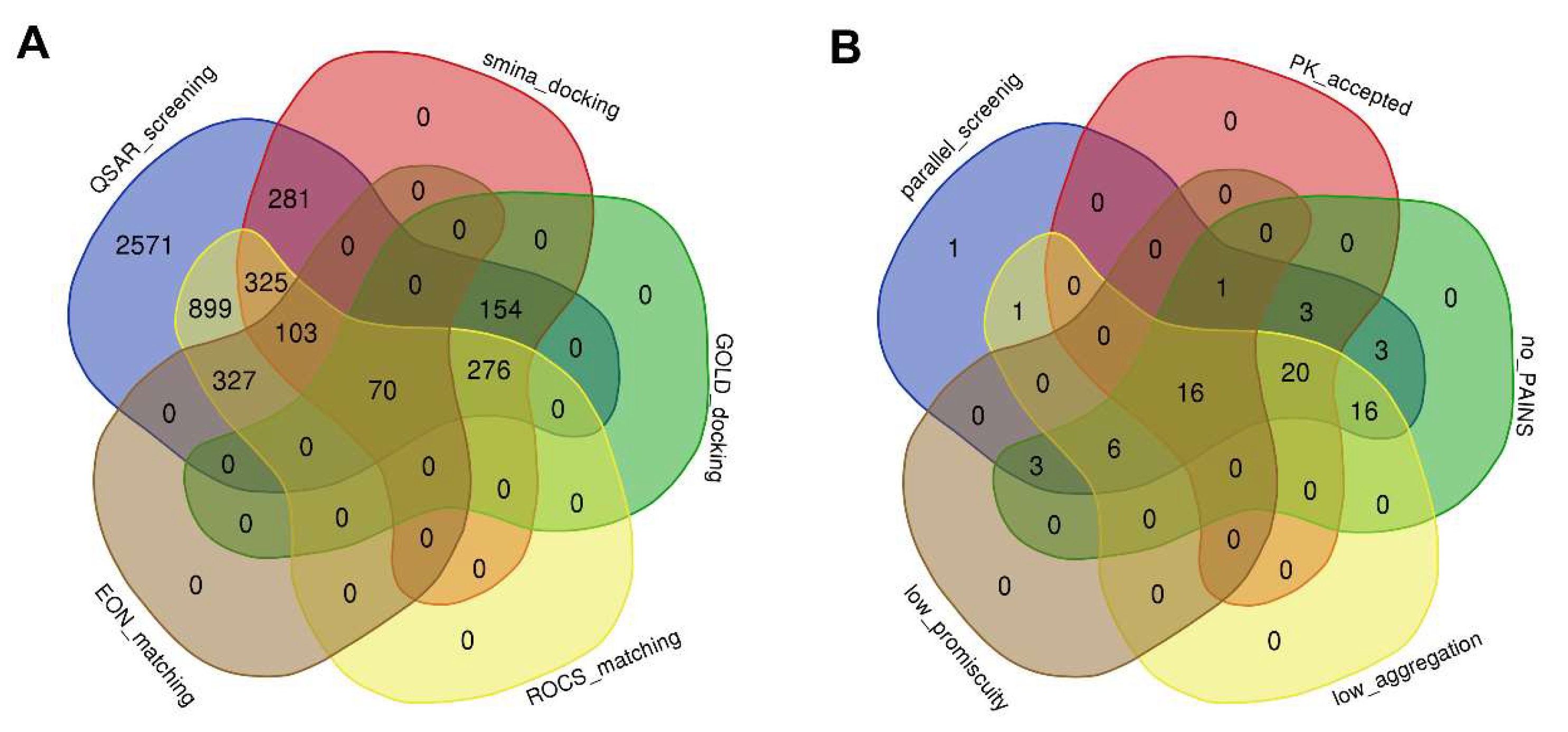
2.2. Secondary Screening Using Molecular Docking and Ligand-Based Methods, and Follow-Up Negative Design to Shortlist Hit Compounds
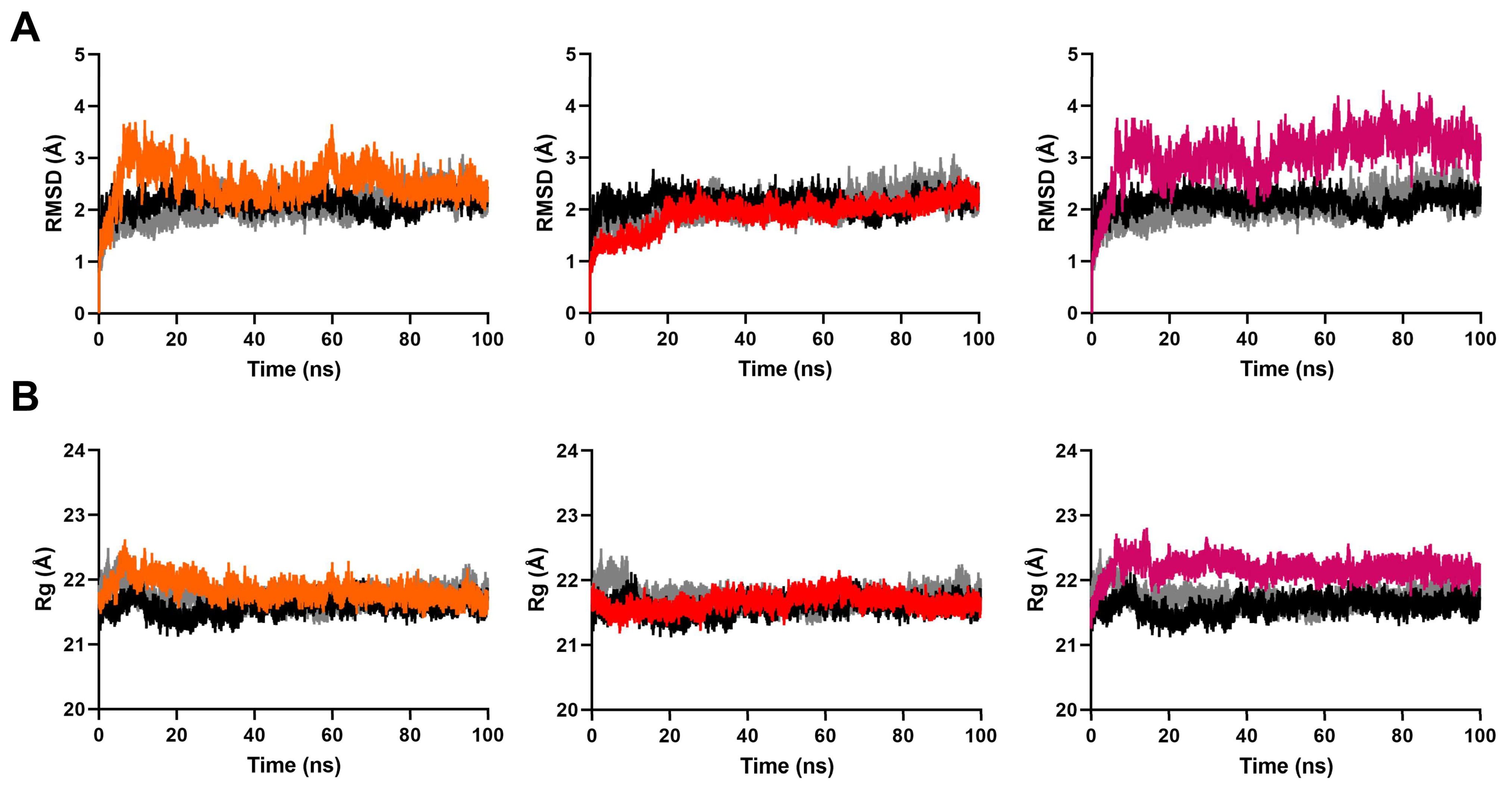
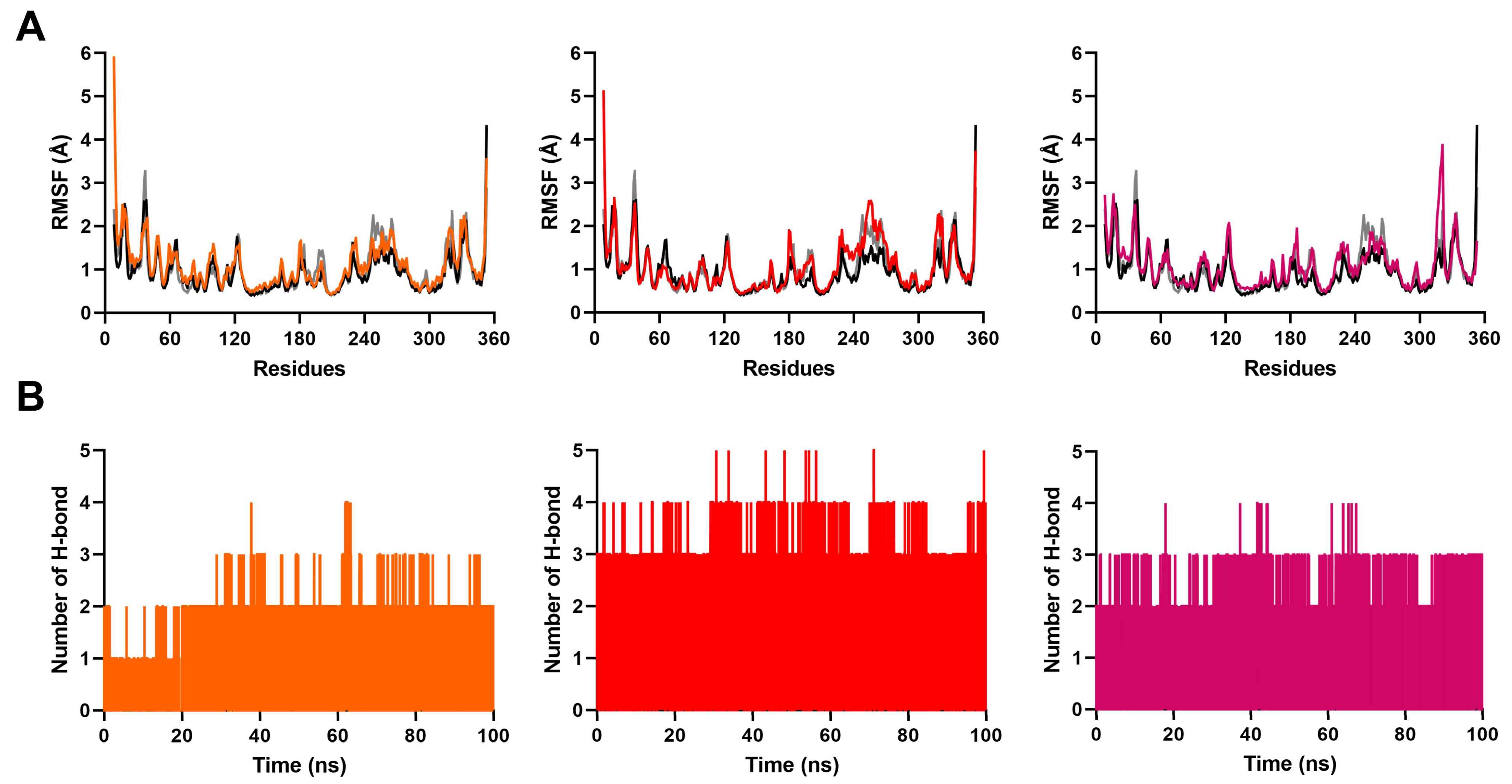
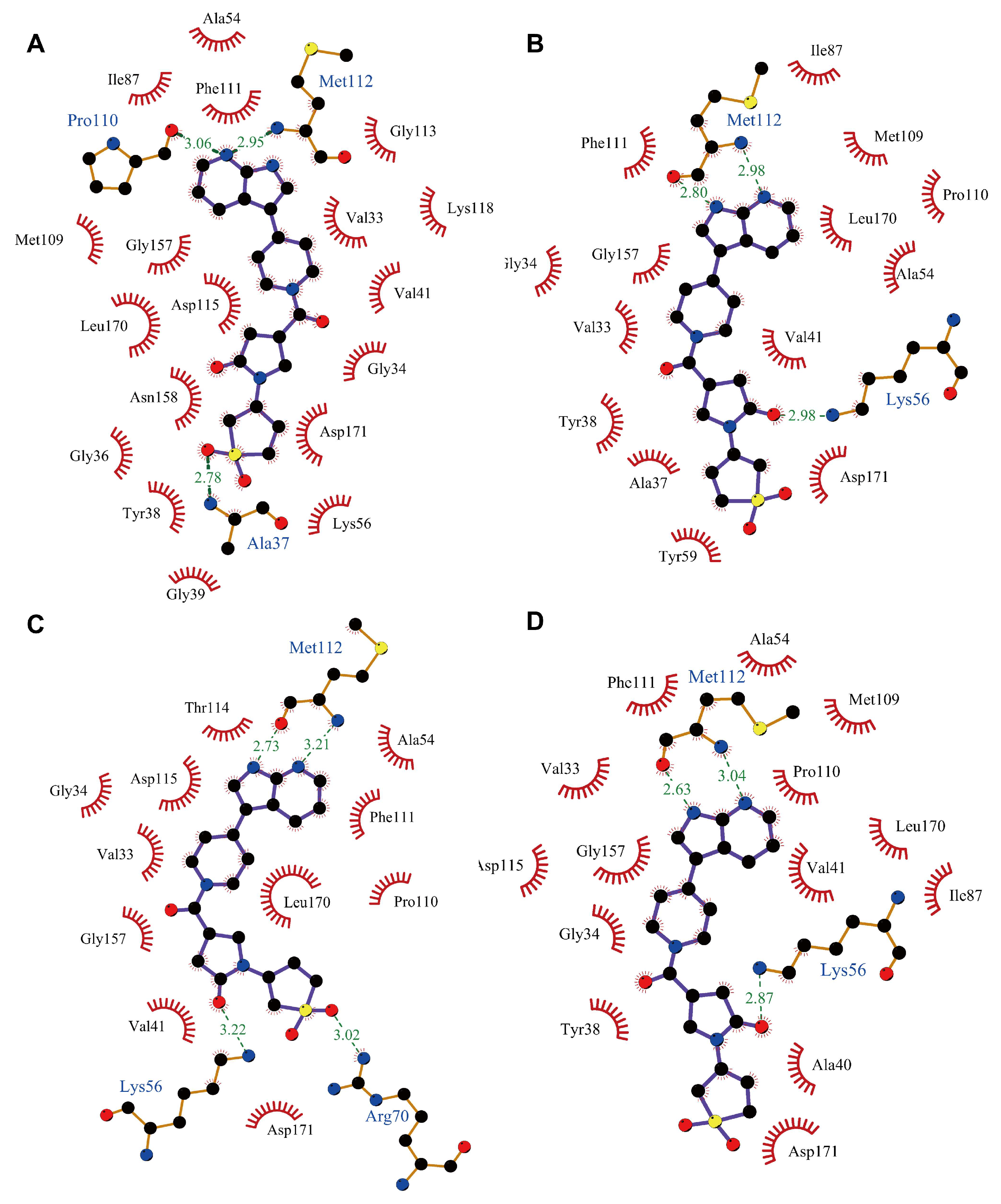
2.3. MD Simulation to Study Binding Stability of Hit Compounds
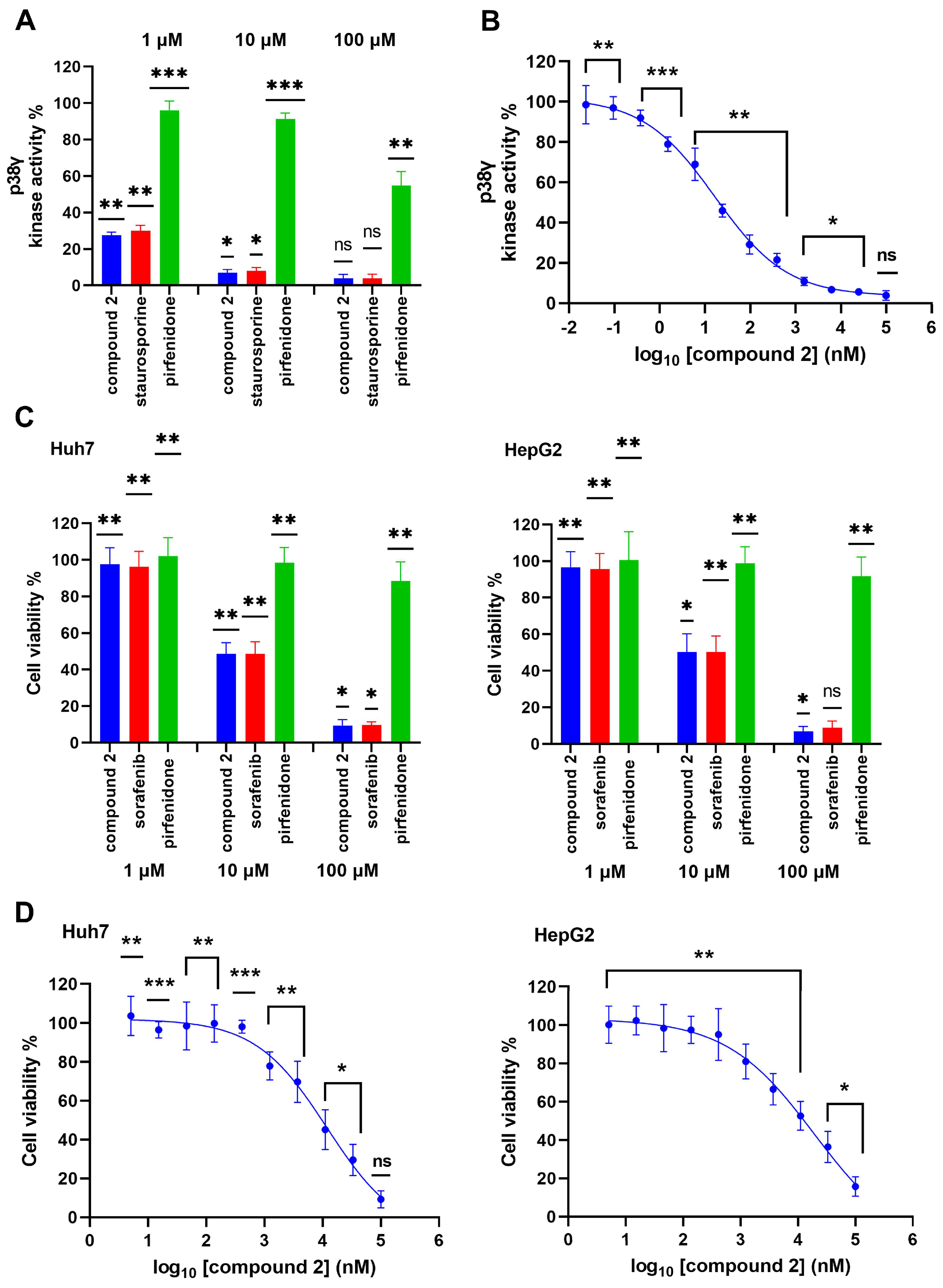
2.4. In Vitro Evaluation for p38γ Inhibitory Activity of Compound 2
2.5. Binding Analysis of Compound 2 to p38γ
3. Materials and Methods
3.1. QSAR Modelling
3.2. Consensus Docking
3.3. Shape/electrostatic Matching
3.4. Negative Design Approaches
3.5. Molecular Dynamics Simulations
3.6. MD Simulations Analysis
3.7. Kinase Assays of p38γ
3.8. Cytotoxicity Assays
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten Things You Should Know about Protein Kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Xu, W.; Liu, R.; Dai, Y.; Hong, S.; Dong, H.; Wang, H. The Role of P38γ in Cancer: From Review to Outlook. Int. J. Biol. Sci. 2021, 17, 4036–4046. [Google Scholar] [CrossRef]
- Yin, D.-P.; Zheng, Y.-F.; Sun, P.; Yao, M.-Y.; Xie, L.-X.; Dou, X.-W.; Tian, Y.; Liu, J.-S. The Pro-Tumorigenic Activity of P38γ Overexpression in Nasopharyngeal Carcinoma. Cell Death Dis. 2022, 13, 210. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhang, H.; Liu, G.; Kreike, B.; Chen, W.; Sethi, S.; Miller, F.R.; Wu, G. P38γ Mitogen-Activated Protein Kinase Contributes to Oncogenic Properties Maintenance and Resistance to Poly (ADP-Ribose)-Polymerase-1 Inhibition in Breast Cancer. Neoplasia 2011, 13, 472–482. [Google Scholar] [CrossRef][Green Version]
- Shi, C.; Cheng, W.-N.; Wang, Y.; Li, D.-Z.; Zhou, L.-N.; Zhu, Y.-C.; Zhou, X.-Z. P38γ Overexpression Promotes Osteosarcoma Cell Progression. Aging 2020, 12, 18384–18395. [Google Scholar] [CrossRef]
- Tomás-Loba, A.; Manieri, E.; González-Terán, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodríguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. P38γ Is Essential for Cell Cycle Progression and Liver Tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef]
- Su, C.; Sun, Q.; Liu, S.; Wang, H.; Feng, L.; Cao, Y. Targeting P38γ to Inhibit Human Colorectal Cancer Cell Progression. Biochem. Biophys. Res. Commun. 2019, 517, 172–179. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Ren, Z.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Ke, Z.-J.; Shi, X.; Luo, J. Chronic Ethanol Exposure Enhances the Aggressiveness of Breast Cancer: The Role of P38γ. Oncotarget 2016, 7, 3489–3505. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Qi, X.; Tsai, S.; Lu, Y.; Basir, Z.; Oshima, K.; Thomas, J.P.; Myers, C.R.; Stoner, G.; Chen, G. P38γ MAPK Is Required for Inflammation-Associated Colon Tumorigenesis. Oncogene 2016, 35, 1039–1048. [Google Scholar] [CrossRef]
- Moran, N. P38 Kinase Inhibitor Approved for Idiopathic Pulmonary Fibrosis. Nat. Biotechnol. 2011, 29, 301. [Google Scholar] [CrossRef] [PubMed]
- Krämer, M.; Markart, P.; Drakopanagiotakis, F.; Mamazhakypov, A.; Schaefer, L.; Didiasova, M.; Wygrecka, M. Pirfenidone Inhibits Motility of NSCLC Cells by Interfering with the Urokinase System. Cell. Signal. 2020, 65, 109432. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Watanabe, M.; Sugimura, Y. Pirfenidone, an Anti-Fibrotic Drug, Suppresses the Growth of Human Prostate Cancer Cells by Inducing G1 Cell Cycle Arrest. JCM 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Lepp, A.; Ji, Y.; Mortensen, M.; Hou, S.; Qi, X.-M.; Myers, C.R.; Chen, G. The K-Ras Effector P38γ MAPK Confers Intrinsic Resistance to Tyrosine Kinase Inhibitors by Stimulating EGFR Transcription and EGFR Dephosphorylation. J. Biol. Chem. 2017, 292, 15070–15079. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yin, N.; Ma, S.; Lepp, A.; Tang, J.; Jing, W.; Johnson, B.; Dwinell, M.B.; Chitambar, C.R.; Chen, G. P38γ MAPK Is a Therapeutic Target for Triple-Negative Breast Cancer by Stimulation of Cancer Stem-Like Cell Expansion: P38γ MAPK Stimulates CSC Expansion. Stem Cells 2015, 33, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Amran, S.I.; Thompson, P.E.; Jennings, I.G. Isoform-Selective Inhibition of Phosphoinositide 3-Kinase: Identification of a New Region of Nonconserved Amino Acids Critical for P110α Inhibition. Mol. Pharmacol. 2011, 80, 657–664. [Google Scholar] [CrossRef]
- Zhang, X.H.; Nam, S.; Wu, J.; Chen, C.-H.; Liu, X.; Li, H.; McKeithan, T.; Gong, Q.; Chan, W.C.; Yin, H.H.; et al. Multi-Kinase Inhibitor with Anti-P38γ Activity in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 2377–2387. [Google Scholar] [CrossRef]
- Tesch, R.; Becker, C.; Müller, M.P.; Beck, M.E.; Quambusch, L.; Getlik, M.; Lategahn, J.; Uhlenbrock, N.; Costa, F.N.; Polêto, M.D.; et al. An Unusual Intramolecular Halogen Bond Guides Conformational Selection. Angew. Chem. Int. Ed. Engl. 2018, 57, 9970–9975. [Google Scholar] [CrossRef]
- Vyas, V. Virtual Screening: A Fast Tool for Drug Design. Sci. Pharm. 2008, 76, 333–360. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Wang, T.; Wu, M.-B.; Lin, J.-P.; Yang, L.-R. Quantitative Structure–Activity Relationship: Promising Advances in Drug Discovery Platforms. Expert Opin. Drug Discov. 2015, 10, 1283–1300. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An Overview of Molecular Fingerprint Similarity Search in Virtual Screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom Typing, Depiction, Molecular Formulas, and Substructure Searching. J. Cheminf. 2017, 9, 33. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A Public Information System for Analyzing Bioactivities of Small Molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Klekota, J.; Roth, F.P. Chemical Substructures That Enrich for Biological Activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature Selection: A Data Perspective. ACM Comput. Surv. 2018, 50, 1–45. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA Data Mining Software: An Update. SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Gramatica, P. External Evaluation of QSAR Models, in Addition to Cross-Validation: Verification of Predictive Capability on Totally New Chemicals. Mol. Inf. 2014, 33, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Models Validation: Internal and External. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Gramatica, P.; Sangion, A. A Historical Excursus on the Statistical Validation Parameters for QSAR Models: A Clarification Concerning Metrics and Terminology. J. Chem. Inf. Model. 2016, 56, 1127–1131. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of Q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models. Part 2. New Intercomparable Thresholds for Different Validation Criteria and the Need for Scatter Plot Inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef]
- Gramatica, P. On the Development and Validation of QSAR Models. In Computational Toxicology; Reisfeld, B., Mayeno, A.N., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 930, pp. 499–526. ISBN 978-1-62703-058-8. [Google Scholar]
- Zhang, Y.; Wang, Y.; Zhou, W.; Fan, Y.; Zhao, J.; Zhu, L.; Lu, S.; Lu, T.; Chen, Y.; Liu, H. A Combined Drug Discovery Strategy Based on Machine Learning and Molecular Docking. Chem. Biol. Drug Des. 2019, 93, 685–699. [Google Scholar] [CrossRef]
- Liñares-Blanco, J.; Munteanu, C.R.; Pazos, A.; Fernandez-Lozano, C. Molecular Docking and Machine Learning Analysis of Abemaciclib in Colon Cancer. BMC Mol. Cell Biol. 2020, 21, 52. [Google Scholar] [CrossRef]
- Agrawal, P.; Singh, H.; Srivastava, H.K.; Singh, S.; Kishore, G.; Raghava, G.P.S. Benchmarking of Different Molecular Docking Methods for Protein-Peptide Docking. BMC Bioinform. 2019, 19, 426. [Google Scholar] [CrossRef]
- Ogrizek, M.; Turk, S.; Lešnik, S.; Sosič, I.; Hodošček, M.; Mirković, B.; Kos, J.; Janežič, D.; Gobec, S.; Konc, J. Molecular Dynamics to Enhance Structure-Based Virtual Screening on Cathepsin B. J. Comput. Aided Mol. Des. 2015, 29, 707–712. [Google Scholar] [CrossRef]
- Svensson, F.; Karlén, A.; Sköld, C. Virtual Screening Data Fusion Using Both Structure- and Ligand-Based Methods. J. Chem. Inf. Model. 2012, 52, 225–232. [Google Scholar] [CrossRef]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Hung, T.-C.; Lee, W.-Y.; Chen, K.-B.; Huang, H.-J.; Chan, Y.-C.; Lee, C.-C.; Chen, C.Y.-C. Possible Inhibitor from Traditional Chinese Medicine for the β Form of Calcium-Dependent Protein Kinase Type II in the Treatment of Major Depressive Disorder. Biomed. Res. Int. 2014, 2014, 761849. [Google Scholar] [CrossRef]
- Wentsch, H.K.; Walter, N.M.; Bührmann, M.; Mayer-Wrangowski, S.; Rauh, D.; Zaman, G.J.R.; Willemsen-Seegers, N.; Buijsman, R.C.; Henning, M.; Dauch, D.; et al. Optimized Target Residence Time: Type I1/2 Inhibitors for P38α MAP Kinase with Improved Binding Kinetics through Direct Interaction with the R-Spine. Angew. Chem. Int. Ed. Engl. 2017, 56, 5363–5367. [Google Scholar] [CrossRef]
- Bellon, S.; Fitzgibbon, M.J.; Fox, T.; Hsiao, H.-M.; Wilson, K.P. The Structure of Phosphorylated P38γ Is Monomeric and Reveals a Conserved Activation-Loop Conformation. Structure 1999, 7, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Beenstock, J.; Ben-Yehuda, S.; Melamed, D.; Admon, A.; Livnah, O.; Ahn, N.G.; Engelberg, D. The P38β Mitogen-Activated Protein Kinase Possesses an Intrinsic Autophosphorylation Activity, Generated by a Short Region Composed of the α-G Helix and MAPK Insert. J. Biol. Chem. 2014, 289, 23546–23556. [Google Scholar] [CrossRef] [PubMed]
- Getlik, M.; Simard, J.R.; Termathe, M.; Grütter, C.; Rabiller, M.; van Otterlo, W.A.L.; Rauh, D. Fluorophore Labeled Kinase Detects Ligands That Bind within the MAPK Insert of P38α Kinase. PLoS ONE 2012, 7, e39713. [Google Scholar] [CrossRef] [PubMed]
- Aoto, P.C.; Martin, B.T.; Wright, P.E. NMR Characterization of Information Flow and Allosteric Communities in the MAP Kinase P38γ. Sci. Rep. 2016, 6, 28655. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Romanova, M.A.; Zadorozhny, A.D.; Kurilenko, N.S.; Shilov, B.V.; Pogodin, P.V.; Ivanov, S.M.; Filimonov, D.A.; Poroikov, V.V. Comparison of Quantitative and Qualitative (Q)SAR Models Created for the Prediction of Ki and IC50 Values of Antitarget Inhibitors. Front. Pharmacol. 2018, 9, 1136. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. Support Vector Machines for Drug Discovery. Expert Opin. Drug Discov. 2014, 9, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, Y.; Luk, B.L. Combined Genetic Algorithm Optimization and Regularized Orthogonal Least Squares Learning for Radial Basis Function Networks. IEEE Trans. Neural Netw. 1999, 10, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H. Stochastic Gradient Boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Huang, S.-C.; Huang, Y.-F. Bounds on the Number of Hidden Neurons in Multilayer Perceptrons. IEEE Trans. Neural Netw. 1991, 2, 47–55. [Google Scholar] [CrossRef]
- Sahigara, F.; Ballabio, D.; Todeschini, R.; Consonni, V. Defining a Novel K-Nearest Neighbours Approach to Assess the Applicability Domain of a QSAR Model for Reliable Predictions. J. Cheminf. 2013, 5, 27. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Equation | Proposed Threshold | Best Model Value | References |
|---|---|---|---|---|
| Cross validation | ||||
| Determination coefficient | > 0.5 | = 0.7491 = 0.7600 | [33] | |
| External validation | ||||
| Determination coefficient | > 0.6 | = 0.7475 | [33] | |
| Slope of lines for RTO | 0.85 ≤ ≤ 1.15 or 0.85 ≤ ≤ 1.15 | = 0.9919 = 1.0006 | [33] | |
| Determination coefficient for RTO | (−)/ < 0.1 or (R2−)/ < 0.1 and − < 0.3 | (−)/ = 0.0026 (−)/ = 0.0719 − = 0.0518 | [33,38] | |
| Concordance correlation coefficient | ≥ 0.85 | = 0.8604 | [37] | |
| functions | ≥ 0.70 | = 0.7435 = 0.7431 = 0.7150 | [37] | |
| Regression function | ≥ 0.65 | = 0.6585 | [37] | |
| RMSD (Å) | AUC | EF (5%) | |
|---|---|---|---|
| AutoDock SMINA | |||
| smina | 0.474 | 0.7372 | 0.351 |
| dkoes | 2.540 | 0.7712 | 0.000 |
| vinardo | 0.214 | 0.6850 | 1.053 |
| ad4 | 1.477 | 0.6895 | 0.702 |
| GOLD | |||
| CHEMPLP | 1.342 | 0.6794 | 1.754 |
| GoldScore | 0.983 | 0.7018 | 1.053 |
| ChemScore | 1.012 | 0.6100 | 1.053 |
| ASP | 1.393 | 0.7248 | 1.404 |
| GOLD consensus scoring | |||
| ASP+CHEMPLP | 0.7067 | 1.053 | |
| ASP+GoldScore | 0.7243 | 1.053 | |
| ASP+CHEMPLP+GoldScore | 0.7052 | 1.053 | |
| Compound | Compound 1 | Compound 2 | Compound 3 | Pirfenidone | PIK75 |
|---|---|---|---|---|---|
| VDWAALS 1 | −34.12 | −49.82 | −39.24 | −24.08 | −49.64 |
| EEL 2 | −30.33 | −34.69 | −86.51 | −12.25 | −18.95 |
| EGB 3 | 41.72 | 51.55 | 98.85 | 15.10 | 33.90 |
| ESURF 4 | −4.02 | −6.03 | −5.81 | −3.41 | −5.71 |
| DELTA TOTAL | −26.76 | −38.99 | −32.71 | −24.64 | −40.39 |
 | Acceptor | Donor | Average Distance Å | Occupancy % |
| compound 2_N2 | p38γ_MET112_N | 3.0976 | 76.456 | |
| compound 2_O1 | p38γ_LYS56_N | 2.8444 | 57.222 | |
| p38γ_MET112_O | compound 2_N1 | 2.8687 | 84.702 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Bhave, M.; Hwang, S.S.; Rahman, T.; Chee, X.W. Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies. Int. J. Mol. Sci. 2023, 24, 7360. https://doi.org/10.3390/ijms24087360
Cheng Z, Bhave M, Hwang SS, Rahman T, Chee XW. Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences. 2023; 24(8):7360. https://doi.org/10.3390/ijms24087360
Chicago/Turabian StyleCheng, Zixuan, Mrinal Bhave, Siaw San Hwang, Taufiq Rahman, and Xavier Wezen Chee. 2023. "Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies" International Journal of Molecular Sciences 24, no. 8: 7360. https://doi.org/10.3390/ijms24087360
APA StyleCheng, Z., Bhave, M., Hwang, S. S., Rahman, T., & Chee, X. W. (2023). Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences, 24(8), 7360. https://doi.org/10.3390/ijms24087360