
Spin-Forbidden Addition of Molecular Oxygen to Stable Enol Intermediates—Decarboxylation of 2-Methyl-1-tetralone-2-carboxylic Acid

 , ,
, ,  , and
, and
Abstract
:1. Introduction
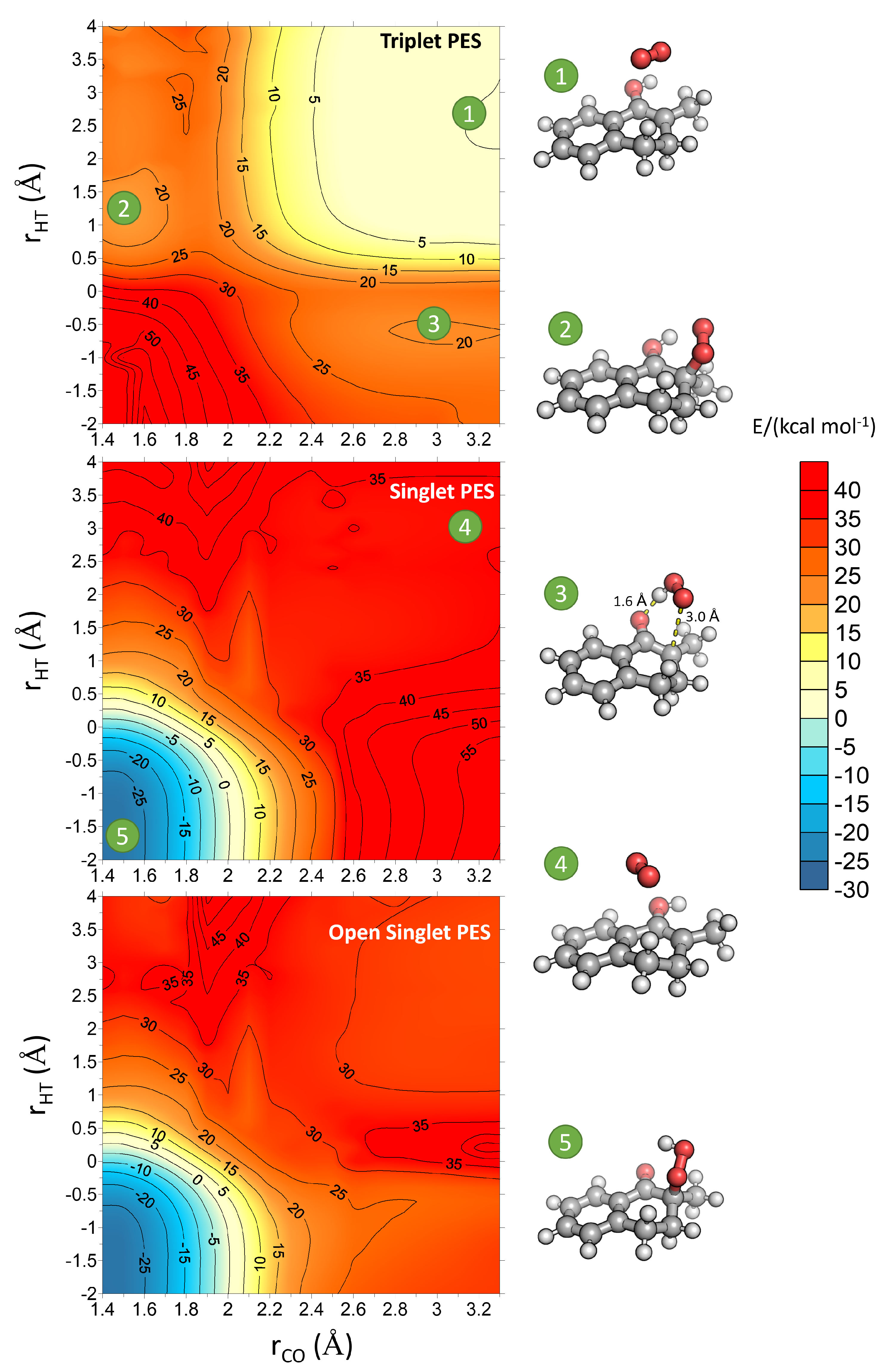
2. Results and Discussion
Phosphite Reduction Mechanism
3. Materials and Methods
3.1. Calculations for the Peroxidation of 2-Methyl-3,4-dihydro-1-naphtol (2)
3.2. Spin-Orbit Coupling and Hopping Probabilities
3.3. Calculations for Reduction with Triethylphosphine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, V.N.; Yan, Y.; Zhao, J.; Yoon, J. Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer. Acc. Chem. Res. 2020, 54, 207–220. [Google Scholar] [CrossRef]
- Gunaydin, G.; Gedik, M.E.; Ayan, S. Photodynamic Therapy—Current Limitations and Novel Approaches. Front. Chem. 2021, 9, 691697. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, K.R.; Gomer, C.J.; Dougherty, T.J. Identification of singlet oxygen as the cytotoxic agent in photoinactivation of a murine tumor. Cancer Res. 1976, 36, 2326–2329. [Google Scholar]
- Schiff, L.J.; Eisenberg, W.C.; Dziuba, J.; Taylor, K.; Moore, S.J. Cytotoxic effects of singlet oxygen. Environ. Health Perspect. 1987, 76, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Edge, R.; Truscott, T.G. The Reactive Oxygen Species Singlet Oxygen, Hydroxy Radicals, and the Superoxide Radical Anion—Examples of Their Roles in Biology and Medicine. Oxygen 2021, 1, 77–95. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Thorning, F.; Jensen, F.; Ogilby, P.R. Modeling the Effect of Solvents on Nonradiative Singlet Oxygen Deactivation: Going beyond Weak Coupling in Intermolecular Electronic-to-Vibrational Energy Transfer. J. Phys. Chem. B 2020, 124, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1758. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D. Dioxygenase enzymes: Catalytic mechanisms and chemical models. Tetrahedron 2003, 59, 7075–7101. [Google Scholar] [CrossRef]
- Sahu, S.; Goldberg, D.P. Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc. 2016, 138, 11410–11428. [Google Scholar] [CrossRef]
- Jasniewski, A.J.; Que, L. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen Activation and Energy Conservation by Cytochrome Oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [PubMed]
- Kisgeropoulos, E.C.; Griese, J.J.; Smith, Z.R.; Branca, R.M.M.; Schneider, C.R.; Högbom, M.; Shafaat, H.S. Key Structural Motifs Balance Metal Binding and Oxidative Reactivity in a Heterobimetallic Mn/Fe Protein. J. Am. Chem. Soc. 2020, 142, 5338–5354. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.S.; Ramanan, R.; Lehnert, N.; Schofield, C.J.; Karabencheva-Christova, T.G.; Christov, C.Z. Catalysis by the Non-Heme Iron(II) Histone Demethylase PHF8 Involves Iron Center Rearrangement and Conformational Modulation of Substrate Orientation. ACS Catal. 2019, 10, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Suardíaz, R.; Jambrina, P.G.; Masgrau, L.; González-Lafont, À.; Rosta, E.; Lluch, J.M. Understanding the Mechanism of the Hydrogen Abstraction from Arachidonic Acid Catalyzed by the Human Enzyme 15-Lipoxygenase-2. A Quantum Mechanics/Molecular Mechanics Free Energy Simulation. J. Chem. Theory Comput. 2016, 12, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Ryde, U. How O2 Binds to Heme. J. Biol. Chem. 2004, 279, 14561–14569. [Google Scholar] [CrossRef]
- Romero, E.; Castellanos, J.R.G.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef]
- Thierbach, S.; Bui, N.; Zapp, J.; Chhabra, S.R.; Kappl, R.; Fetzner, S. Substrate-Assisted O2 Activation in a Cofactor-Independent Dioxygenase. Chem. Biol 2014, 21, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Bui, S.; Steiner, R.A. New insight into cofactor-free oxygenation from combined experimental and computational approaches. Curr. Opin. Struct. Biol. 2016, 41, 109–118. [Google Scholar] [CrossRef]
- Fetzner, S.; Steiner, R.A. Cofactor-independent oxidases and oxygenases. Appl. Microbiol. Biotechnol 2010, 86, 791–804. [Google Scholar] [CrossRef]
- Chaiyen, P.; Fraaije, M.W.; Mattevi, A. The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 2012, 37, 373–380. [Google Scholar] [CrossRef]
- Mattevi, A. To be or not to be an oxidase: Challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci. 2006, 31, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Bui, S.; von Stetten, D.; Jambrina, P.G.; Prangé, T.; Colloc’h, N.; de Sanctis, D.; Royant, A.; Rosta, E.; Steiner, R.A. Direct Evidence for a Peroxide Intermediate and a Reactive Enzyme-Substrate-Dioxygen Configuration in a Cofactor-free Oxidase. Angew. Chem. Int. Ed. 2014, 53, 13710–13714. [Google Scholar] [CrossRef]
- Matthews, A.; Saleem-Batcha, R.; Sanders, J.N.; Stull, F.; Houk, K.N.; Teufel, R. Aminoperoxide adducts expand the catalytic repertoire of flavin monooxygenases. Nat. Chem. Biol. 2020, 16, 556–563. [Google Scholar] [CrossRef]
- Kiss, D.J.; Ferenczy, G.G. A detailed mechanism of the oxidative half-reaction ofd-amino acid oxidase: Another route for flavin oxidation. Org. Biomol. Chem. 2019, 17, 7973–7984. [Google Scholar] [CrossRef]
- Ortega, P.; Zanchet, A.; Sanz-Sanz, C.; Gómez-Carrasco, S.; González-Sánchez, L.; Jambrina, P.G. DpgC-Catalyzed Peroxidation of 3, 5-Dihydroxyphenylacetyl-CoA (DPA-CoA): Insights into the Spin-Forbidden Transition and Charge Transfer Mechanisms. Chem. Eur. J. 2020, 27, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Quan, J.M. Intermediate-Assisted Multifunctional Catalysis in the Conversion of Flavin to 5, 6-Dimethylbenzimidazole by BluB: A Density Functional Theory Study. J. Am. Chem. Soc. 2011, 133, 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heyes, D.J.; Steiner, R.A.; Scrutton, N.S.; de Visser, S.P. Catalytic Mechanism of Cofactor-Free Dioxygenases and How They Circumvent Spin-Forbidden Oxygenation of Their Substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. [Google Scholar] [CrossRef]
- Silva, P.J. Refining the reaction mechanism of O2 towards its co-substrate in cofactor-free dioxygenases. PeerJ 2016, 4, e2805. [Google Scholar] [CrossRef]
- Prabhakar, R.; Siegbahn, P.E.M.; Minaev, B.F.; Ågren, H. Activation of Triplet Dioxygen by Glucose Oxidase: Spin-Orbit Coupling in the Superoxide Ion. J. Phys. Chem. B 2002, 106, 3742–3750. [Google Scholar] [CrossRef]
- Minaev, B. How cofactor-free oxygenases can overcome spin prohibition in substrates oxygenation by dioxygen. Chem. Phys. 2019, 521, 61–68. [Google Scholar] [CrossRef]
- Wei, D.; Huang, X.; Qiao, Y.; Rao, J.; Wang, L.; Liao, F.; Zhan, C.G. Catalytic Mechanisms for Cofactor-Free Oxidase-Catalyzed Reactions: Reaction Pathways of Uricase-Catalyzed Oxidation and Hydration of Uric Acid. ACS Catal. 2017, 7, 4623–4636. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, F.G.C.; DuBois, J.L.; de Visser, S.P. Catalytic Mechanism of Nogalamycin Monoxygenase: How Does Nature Synthesize Antibiotics without a Metal Cofactor? J. Phys. Chem. B 2018, 122, 10841–10854. [Google Scholar] [CrossRef]
- Visitsatthawong, S.; Chenprakhon, P.; Chaiyen, P.; Surawatanawong, P. Mechanism of Oxygen Activation in a Flavin-Dependent Monooxygenase: A Nearly Barrierless Formation of C4a-Hydroperoxyflavin via Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 9363–9374. [Google Scholar] [CrossRef]
- Ortega, P.; Gil-Guerrero, S.; Veselinova, A.; Zanchet, A.; González-Sánchez, L.; Jambrina, P.G.; Sanz-Sanz, C. Multi- and single-reference methods for the analysis of multi-state peroxidation of enolates. J. Chem. Phys. 2021, 154, 144303. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Understanding the kinetics of spin-forbidden chemical reactions. Phys. Chem. Chem. Phys. 2007, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Spin-forbidden reactions: Computational insight into mechanisms and kinetics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 4, 1–14. [Google Scholar] [CrossRef]
- Lykhin, A.O.; Kaliakin, D.S.; dePolo, G.E.; Kuzubov, A.A.; Varganov, S.A. Nonadiabatic transition state theory: Application to intersystem crossings in the active sites of metal-sulfur proteins. Int. J. Quant. Chem. 2016, 116, 750–761. [Google Scholar] [CrossRef]
- Marian, C.M. Spin-orbit coupling and intersystem crossing in molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 187–203. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Spin—Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Thorning, F.; Henke, P.; Ogilby, P.R. Perturbed and Activated Decay: The Lifetime of Singlet Oxygen in Liquid Organic Solvents. J. Am. Chem. Soc. 2022, 144, 10902–10911. [Google Scholar] [CrossRef] [PubMed]
- Thorning, F.; Jensen, F.; Ogilby, P.R. Geometry Dependence of Spin-Orbit Coupling in Complexes of Molecular Oxygen with Atoms, H2, or Organic Molecules. J. Phys. Chem. A 2022, 126, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Zener, C. Non-adiabatic crossing of energy levels. Proc. R. Soc. Lond. 1932, 137, 696–702. [Google Scholar] [CrossRef]
- Rooein, M.; Varganov, S.A. How to calculate the rate constants for nonradiative transitions between the M components of spin multiplets? Mol. Phys. 2022, e2116364. [Google Scholar] [CrossRef]
- Liu, X.; Ryabenkova, Y.; Conte, M. Catalytic oxygen activation versus autoxidation for industrial applications: A physicochemical approach. Phys. Chem. Chem. Phys. 2015, 17, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Singh, A.K.; Niu, L.; Yi, H.; Lei, A. Visible-Light-Mediated Oxygenation Reactions using Molecular Oxygen. Asian J. Org. Chem. 2017, 6, 386–396. [Google Scholar] [CrossRef]
- Tang, C.; Qiu, X.; Cheng, Z.; Jiao, N. Molecular oxygen-mediated oxygenation reactions involving radicals. Chem. Soc. Rev. 2021, 50, 8067–8101. [Google Scholar] [CrossRef]
- Taniguchi, T. Strategy for the Use of Molecular Oxygen in Organic Synthesis. Synlett 2020, 32, 573–581. [Google Scholar] [CrossRef]
- Piera, J.; Bäckvall, J.E. Catalytic Oxidation of Organic Substrates by Molecular Oxygen and Hydrogen Peroxide by Multistep Electron Transfer—A Biomimetic Approach. Angew. Chem. Int. Ed. 2008, 47, 3506–3523. [Google Scholar] [CrossRef]
- Riahi, A.; Muzart, J.; Abe, M.; Hoffmann, N. On the decarboxylation of 2-methyl-1-tetralone-2-carboxylic acid-oxidation of the enol intermediate by triplet oxygen. New J. Chem. 2013, 37, 2245. [Google Scholar] [CrossRef]
- Chen, B.C.; Zhou, P.; Davis, F.A.; Ciganek, E. α-Hydroxylation of Enolates and Silyl Enol Ethers. Org. React. 2004, 64, 1–356. [Google Scholar]
- Lévesque, F.; Seeberger, P.H. Continuous-Flow Synthesis of the Anti-Malaria Drug Artemisinin. Angew. Chem. Int. Ed. 2012, 51, 1706–1709. [Google Scholar] [CrossRef]
- Sim, S.B.D.; Wang, M.; Zhao, Y. Phase-Transfer-Catalyzed Enantioselective α-Hydroxylation of Acyclic and Cyclic Ketones with Oxygen. ACS Catal. 2015, 5, 3609–3612. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, L.; Bao, H.; Zhou, B.; Wu, H.; Liu, Y. Reactivity Umpolung of the C-N Bond in Quinoxaline Scaffold Enabling Direct Nucleophilic Attack of Alkyl Grignard Reagents at the N-Terminus. Org. Lett. 2022, 24, 3982–3986. [Google Scholar] [CrossRef]
- Inukai, T.; Kano, T.; Maruoka, K. Asymmetric α-Hydroxylation of α-Aryl-δ-lactams with Molecular Oxygen under Phase-Transfer Conditions. Org. Lett. 2021, 23, 792–796. [Google Scholar] [CrossRef]
- Anderson, T.; Andia, A.A.; Woerpel, K. Chemiluminescence-promoted oxidation of alkyl enol ethers by NHPI under mild conditions and in the dark. Tetrahedron 2021, 82, 131874. [Google Scholar] [CrossRef]
- Herzberg, G. Spectra of Diatomic Molecules; Van Nostrand Reinhold: New York, NY, USA, 1950. [Google Scholar]
- Rienstra-Kiracofe, J.C.; Allen, W.D.; Schaefer, H.F. The C2H5 + O2 Reaction Mechanism: High-Level ab Initio Characterizations. J. Phys. Chem. A 2000, 104, 9823–9840. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quant. Chem. 2009, 36, 199–207. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Celani, P.; Györffy, W.; Hesselmann, A.; Kats, D.; Knizia, G.; Köhn, A.; Korona, T.; Kreplin, D.; et al. MOLPRO; Version 2020.1, a Package of ab Initio Programs. 2020. Available online: https://www.molpro.net (accessed on 16 February 2023).
- Ryu, H.; Park, J.; Kim, H.K.; Park, J.Y.; Kim, S.T.; Baik, M.H. Pitfalls in Computational Modeling of Chemical Reactions and How To Avoid Them. Organometallics 2018, 37, 3228–3239. [Google Scholar] [CrossRef]
- Denney, D.B.; Goodyear, W.F.; Goldstein, B. Concerning the Mechanism of the Reduction of Hydroperoxides by Trisubstituted Phosphines and Trisubstituted Phosphites. J. Am. Chem. Soc. 1960, 82, 1393–1395. [Google Scholar] [CrossRef]
- Tweedy, S.E.; Rodríguez Benítez, A.; Narayan, A.R.H.; Zimmerman, P.M.; Brooks, C.L.; Wymore, T. Hydroxyl Radical-Coupled Electron-Transfer Mechanism of Flavin-Dependent Hydroxylases. J. Phys. Chem. B 2019, 123, 8065–8073. [Google Scholar] [CrossRef] [PubMed]
- Papajak, E.; Leverentz, H.R.; Zheng, J.; Truhlar, D.G. Efficient Diffuse Basis Sets: Cc-pVxZ+ and maug-cc-pVxZ. J. Chem. Theory Comput. 2009, 5, 3330. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian-16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yamanaka, S.; Nishino, M.; Takano, Y.; Kitagawa, Y.; Nagao, H.; Yoshioka, Y. Symmetry and broken symmetries in molecular orbital descriptions of unstable molecules II. Alignment, flustration and tunneling of spins in mesoscopic molecular magnets. Theor. Chim. Acta 1999, 102, 328–345. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Nakanishi, Y.; Kataoka, Y.; Matsui, T.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Spin Contamination Error in Optimized Geometry of Singlet Carbene by Broken-Symmetry Method. J. Phys. Chem. A 2009, 113, 15041–15046. [Google Scholar] [CrossRef]
- Saito, T.; Nishihara, S.; Kataoka, Y.; Nakanishi, Y.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M.; Yamaguchi, K. Reinvestigation of the Reaction of Ethylene and Singlet Oxygen by the Approximate Spin Projection Method. Comparison with Multireference Coupled-Cluster Calculations. J. Phys. Chem. A 2010, 114, 7967–7974. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chim. Acta 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Siegbahn, P.E. A new direct CI method for large CI expansions in a small orbital space. Chem. Phys. Lett. 1984, 109, 417–423. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Olsen, J. The CASSCF method: A perspective and commentary. Int. J. Quant. Chem. 2011, 111, 3267–3272. [Google Scholar] [CrossRef]
- Mai, S.; González, L. Molecular Photochemistry: Recent Developments in Theory. Angew. Chem. Int. Ed. 2020, 59, 16832–16846. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chim. Acta 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Ma, Q.; Werner, H.J. Explicitly correlated local coupled-cluster methods using pair natural orbitals. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, c1371. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; Vico, L.D.; Galván, I.F.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comp. Chem. 2015, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Giambiagi, M.; de Giambiagi, M.S.; Mundim, K.C. Definition of a multicenter bond index. Struct. Chem. 1990, 1, 423–427. [Google Scholar] [CrossRef]
- Bultinck, P.; Ponec, R.; Damme, S.V. Multicenter bond indices as a new measure of aromaticity in polycyclic aromatic hydrocarbons. J. Phys. Org. Chem. 2005, 18, 706–718. [Google Scholar] [CrossRef]
- Mundim, K.C.; Giambiagi, M.; de Giambiagi, M.S. Multicenter Bond Index: Grassmann Algebra and N-Order Density Functional. J. Phys. Chem. 1994, 98, 6118–6119. [Google Scholar] [CrossRef]
- Mandado, M.; González-Moa, M.J.; Mosquera, R.A. QTAIMn-center delocalization indices as descriptors of aromaticity in mono and poly heterocycles. J. Comp. Chem. 2006, 28, 127–136. [Google Scholar] [CrossRef]
- Mulliken, R.S. Criteria for the Construction of Good Self-Consistent-Field Molecular Orbital Wave Functions, and the Significance of LCAO-MO Population Analysis. J. Chem. Phys. 1962, 36, 3428–3439. [Google Scholar] [CrossRef]
- Gannon, K.L.; Blitz, M.A.; Liang, C.H.; Pilling, M.J.; Seakins, P.W.; Glowacki, D.R.; Harvey, J.N. An experimental and theoretical investigation of the competition between chemical reaction and relaxation for the reactions of 1CH2 with acetylene and ethene: Implications for the chemistry of the giant planets. Faraday Discuss. 2010, 147, 173. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO-pathway | M062X-D3 | DLPNO-CCSD(T) | CASPT2(8,5) |
| Reactant complex (triplet) | 0.00 | 0.00 | 0.00 |
| O | 36.9 (27.9) | 31.8 | 23.2 |
| TS (triplet) | 22.5 | 25.7 | 28.6 |
| Intermediate (triplet) | 17.1 | 20.8 | 26.1 |
| MECP | 22.6 (17.2) | 26.2 | 27.0 |
| Product (singlet) | −27.8 | −25.5 | −20.5 |
| OOH-pathway | M062X-D3 | DLPNO-CCSD(T) | CASPT2(8,5) |
| Reactant complex (triplet) | 0.00 | 0.00 | 0.00 |
| O | 37.0 (27.9) | 31.8 | 23.2 |
| TS (triplet) | 22.7 | 30.4 | 32.9 |
| Intermediate (triplet) | 19.6 | 22.5 | 26.6 |
| MECP | 30.5 (20.0) | 26.9 | 27.7 |
| Product (singlet) | −27.8 | −25.5 | −20.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, P.; Gil-Guerrero, S.; González-Sánchez, L.; Sanz-Sanz, C.; Jambrina, P.G. Spin-Forbidden Addition of Molecular Oxygen to Stable Enol Intermediates—Decarboxylation of 2-Methyl-1-tetralone-2-carboxylic Acid. Int. J. Mol. Sci. 2023, 24, 7424. https://doi.org/10.3390/ijms24087424
Ortega P, Gil-Guerrero S, González-Sánchez L, Sanz-Sanz C, Jambrina PG. Spin-Forbidden Addition of Molecular Oxygen to Stable Enol Intermediates—Decarboxylation of 2-Methyl-1-tetralone-2-carboxylic Acid. International Journal of Molecular Sciences. 2023; 24(8):7424. https://doi.org/10.3390/ijms24087424
Chicago/Turabian StyleOrtega, Pablo, Sara Gil-Guerrero, Lola González-Sánchez, Cristina Sanz-Sanz, and Pablo G. Jambrina. 2023. "Spin-Forbidden Addition of Molecular Oxygen to Stable Enol Intermediates—Decarboxylation of 2-Methyl-1-tetralone-2-carboxylic Acid" International Journal of Molecular Sciences 24, no. 8: 7424. https://doi.org/10.3390/ijms24087424
APA StyleOrtega, P., Gil-Guerrero, S., González-Sánchez, L., Sanz-Sanz, C., & Jambrina, P. G. (2023). Spin-Forbidden Addition of Molecular Oxygen to Stable Enol Intermediates—Decarboxylation of 2-Methyl-1-tetralone-2-carboxylic Acid. International Journal of Molecular Sciences, 24(8), 7424. https://doi.org/10.3390/ijms24087424