
Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Pathophysiology of RP
3. Classification of RP
3.1. Mode of Inheritance
3.2. Non-Syndromic and Syndromic Forms of RP
4. Clinical Symptoms
5. Disease Onset and Prognosis
6. Diagnostic Testing in RP
7. Clinical Testing and Evaluation
7.1. Fundus Findings
7.2. Differential Diagnosis
7.3. Electrophysiological Testing
7.4. Perimetry Testing
7.5. Full-Field Stimulus Threshold Testing
7.6. Multimodal Imaging
8. Genetic Testing
8.1. Sanger Sequencing
8.2. Next-Generation Sequencing
8.3. Targeted Gene Sequencing
8.4. Whole-Exome Sequencing
8.5. Whole-Genome Sequencing
8.6. Recommendations for Genetic Testing
9. Genetic Counseling
9.1. Preconception Counseling
9.2. Pre-Implantation Genetic Testing
10. Management of RP-Associated Complications
10.1. Cataract
10.2. Cystoid Macular Edema
10.3. Other Macular Abnormalities and Retinal Detachments
10.4. Uveitis
10.5. Glaucoma
11. Rehabilitative and Psychological Management
12. Investigational Treatment Modalities
12.1. Gene-Dependent Strategies
12.2. Gene Augmentation Therapy
12.3. CRISPR/CAS9-Based Therapy
12.4. Antisense Oligonucleotide Therapy
13. Gene-Independent Strategies
13.1. Optogenetics
13.2. Stem Cell Therapy
13.3. Retinal Prostheses
13.4. Neurotrophic Factors
13.5. Neuroprotective Agents
13.6. Nutritional Therapies
14. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Haim, M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol. Scand. Suppl. 2002, 233, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Na, K.-H.; Kim, H.J.; Kim, K.H.; Han, S.; Kim, P.; Hann, H.J.; Ahn, H.S. Prevalence, Age at Diagnosis, Mortality, and Cause of Death in Retinitis Pigmentosa in Korea—A Nationwide Population-based Study. Am. J. Ophthalmol. 2017, 176, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bunker, C.H.; Berson, E.L.; Bromley, W.C.; Hayes, R.P.; Roderick, T.H. Prevalence of Retinitis Pigmentosa in Maine. Am. J. Ophthalmol. 1984, 97, 357–365. [Google Scholar] [CrossRef]
- Xu, L.; Hu, L.; Ma, K.; Li, J.; Jonas, J. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: The Beijing Eye Study. Eur. J. Ophthalmol. 2006, 16, 865–866. [Google Scholar] [CrossRef]
- Sen, P.; Bhargava, A.; George, R.; Ramesh, S.V.; Hemamalini, A.; Prema, R.; Ve, R.S.; Vijaya, L. Prevalence of Retinitis Pigmentosa in South Indian Population Aged Above 40 Years. Ophthalmic Epidemiol. 2008, 15, 279–281. [Google Scholar] [CrossRef]
- Nangia, V.; Jonas, J.B.; Khare, A.; Sinha, A. Prevalence of retinitis pigmentosa in India: The Central India Eye and Medical Study. Acta Ophthalmol. 2012, 90, e649–e650. [Google Scholar] [CrossRef]
- Sharon, D.; Banin, E. Nonsyndromic retinitis pigmentosa is highly prevalent in the Jerusalem region with a high frequency of founder mutations. Mol. Vis. 2015, 21, 783–792. [Google Scholar]
- Von Ammon, F.A. Klinische Darstellungen der Krankheiten und Bildungsfehler des Menschlichen Auges, der Augenlider, und der Thränewerkzeug; G. Reimer: Berlin, Germany, 1838. [Google Scholar]
- van Trigt, A.C. De Speculo Oculi. In Nederlandsch Lancet; Gyan Books Pvt. Ltd.: Delhi, India, 1853. [Google Scholar]
- Langenbeck, B.C.R. Observations Anatomico-Pathologica; SUB Göttingen: Göttingen, Germany, 1836. [Google Scholar]
- Donders, F.C. Beiträge zur pathologischen Anatomie des Auges. Graefe’s Arch. Clin. Exp. Ophthalmol. 1857, 3, 139–165. [Google Scholar] [CrossRef]
- Musarella, M.A.; Macdonald, I.M. Current Concepts in the Treatment of Retinitis Pigmentosa. J. Ophthalmol. 2011, 2011, 753547. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F. The Electroretinogram in Retinitis Pigmentosa. Arch. Ophthalmol. 1979, 97, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Aguirre, G.; Arden, G.; Berson, E.; Birch, D.G.; Boughman, J.A.; Carr, R.; Chatrian, G.E.; Del Monte, M.; Dowling, J.; et al. Retinitis Pigmentosa: A Symposium on Terminology and Methods of Examination. Ophthalmology 1983, 90, 126–131. [Google Scholar] [CrossRef]
- Aguirre, G.D. Concepts and Strategies in Retinal Gene Therapy. Investig. Opthalmol. Vis. Sci. 2017, 58, 5399–5411. [Google Scholar] [CrossRef]
- Chiu, W.; Lin, T.-Y.; Chang, Y.-C.; Lai, H.I.-A.M.; Lin, S.-C.; Ma, C.; Yarmishyn, A.; Lin, S.-C.; Chang, K.-J.; Chou, Y.-B.; et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int. J. Mol. Sci. 2021, 22, 4534. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.-S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Sodi, A.; Banfi, S.; Testa, F.; Della Corte, M.; Passerini, I.; Pelo, E.; Rossi, S.; Simonelli, F.; Italian IRD Working Group. RPE65-associated inherited retinal diseases: Consensus recommendations for eligibility to gene therapy. Orphanet J. Rare Dis. 2021, 16, 257. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Bennett, J.; Aleman, T.S.; Cideciyan, A.V.; Bennicelli, J.; Dejneka, N.S.; Pearce-Kelling, S.E.; Maguire, A.M.; Palczewski, K.; et al. Long-Term Restoration of Rod and Cone Vision by Single Dose rAAV-Mediated Gene Transfer to the Retina in a Canine Model of Childhood Blindness. Mol. Ther. 2005, 12, 1072–1082. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Ameri, H. Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation. J. Curr. Ophthalmol. 2018, 30, 1–2. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation–Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef]
- Kapetanovic, J.C.; E McClements, M.; de la Camara, C.M.-F.; E MacLaren, R. Molecular Strategies for RPGR Gene Therapy. Genes 2019, 10, 674. [Google Scholar] [CrossRef]
- Fischer, M.D.; McClements, M.E.; de la Camara, C.M.-F.; Bellingrath, J.-S.; Dauletbekov, D.; Ramsden, S.C.; Hickey, D.G.; Barnard, A.R.; MacLaren, R.E. Codon-Optimized RPGR Improves Stability and Efficacy of AAV8 Gene Therapy in Two Mouse Models of X-Linked Retinitis Pigmentosa. Mol. Ther. 2017, 25, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Ashtari, M.; Wellman, J.; Marshall, K.A.; Cyckowski, L.L.; Chung, D.C.; McCague, S.; Pierce, E.A.; Chen, Y.; Bennicelli, J.L.; et al. AAV2 Gene Therapy Readministration in Three Adults with Congenital Blindness. Sci. Transl. Med. 2012, 4, 120ra15. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef]
- Ku, C.A.; Pennesi, M.E. The new landscape of retinal gene therapy. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 846–859. [Google Scholar] [CrossRef]
- Chivers, M.; Li, N.; Pan, F.; Wieffer, H.; Slowik, R.; Leartsakulpanitch, J. The Burden of X-Linked Retinitis Pigmentosa on Patients and Society: A Narrative Literature Review. Clin. Outcomes Res. 2021, 13, 565–572. [Google Scholar] [CrossRef]
- Antonio-Aguirre, B.; Swenor, B.; Canner, J.K.; Singh, M.S. Risk of cystoid macular edema following cataract surgery in retinitis pigmentosa: An analysis of United States claims from 2010 to 2018. Ophthalmol. Retin. 2022, 6, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Bastek, J.V.; Heckenlively, J.R.; Straatsma, B.R. Cataract Surgery in Retinitis Pigmentosa Patients. Ophthalmology 1982, 89, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Chaumet-Riffaud, A.E.; Chaumet-Riffaud, P.; Cariou, A.; Devisme, C.; Audo, I.; Sahel, J.-A.; Mohand-Said, S. Impact of Retinitis Pigmentosa on Quality of Life, Mental Health, and Employment Among Young Adults. Am. J. Ophthalmol. 2017, 177, 169–174. [Google Scholar] [CrossRef] [PubMed]
- De Nadai, K.; Romano, M.R.; Binotto, A.; Costagliola, C.; Sato, G.; Parmeggiani, F. Clinical and Rehabilitative Management of Retinitis Pigmentosa:Up-to-Date. Curr. Genom. 2011, 12, 250–259. [Google Scholar] [CrossRef]
- Dryja, T.P.; Hahn, L.B.; Kajiwara, K.; Berson, E.L. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Investig. Opthalmol. Vis. Sci. 1997, 38, 1972–1982. [Google Scholar]
- Daiger, S.; Rossiter BJ, F.; Greenberg, J.; Christoffels, A.; Hide, W. Data services and software for identifying genes and mutations causing retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1998, 39, S295. [Google Scholar]
- Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Genetic testing for inherited retinal degenerations: Triumphs and tribulations. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 571–577. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.; Cunha, A.D.S.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Jay, M. On the heredity of retinitis pigmentosa. Br. J. Ophthalmol. 1982, 66, 405–416. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 406–420. [Google Scholar] [CrossRef]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Blankman, J.L.; Long, J.Z.; Trauger, S.A.; Siuzdak, G.; Cravatt, B.F. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad. Sci. USA 2013, 110, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-T.; Almushattat, H.; Strubbe, I.; Georgiou, M.; Li, C.H.Z.; van Schooneveld, M.J.; Joniau, I.; De Baere, E.; Florijn, R.J.; Bergen, A.A.; et al. The Phenotypic Spectrum of Patients with PHARC Syndrome Due to Variants in ABHD12: An Ophthalmic Perspective. Genes 2021, 12, 1404. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, H.; Hashimoto, T.; Murata, T.; Fukushima, K.; Sugaya, A.; Nishio, S.-Y.; Usami, S.-I. Novel ABHD12 Mutations in PHARC Patients. Ann. Otol. Rhinol. Laryngol. 2015, 124, 77S–83S. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.A.; Oftnan, R.; Ferdinandusse, S.; Ijlst, L.; Muijsers, A.O.; Skjeldal, O.H.; Stokke, O.; Jakobs, C.; Besley, G.T.; Wraith, J.E.; et al. Refsum disease is caused by mutations in the phytanoyl–CoA hydroxylase gene. Nat. Genet. 1997, 17, 190–193. [Google Scholar] [CrossRef]
- Schwartz, J.F.; Rowland, L.P.; Eder, H.; Marks, P.A.; Osserman, E.F.; Hirschberg, E.; Anderson, H. Bassen-Kornzweig Syndrome: Deficiency of Serum β-Lipoprotein: A Neuromuscular Disorder Resembling Friedreich’s Ataxia, Associated with Steatorrhea, Acanthocytosis, Retinitis Pigmentosa, and a Disorder of Lipid Metabolism. Arch. Neurol. 1963, 8, 438–454. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; Brahim, D.H.-B.; Johansson, S.; M’Zahem, A.; Haukanes, B.I.; Drouot, N.; Zimmermann, J.; Cole, A.J.; Vedeler, C.; Bredrup, C.; et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am. J. Hum. Genet. 2010, 87, 410–417. [Google Scholar] [CrossRef]
- Waterham, H.R.; Wanders, R.J.A.; Leroy, B.P. Adult Refsum Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chen, H.Y.; Welby, E.; Li, T.; Swaroop, A. Retinal disease in ciliopathies: Recent advances with a focus on stem cell-based therapies. Transl. Sci. Rare Dis. 2019, 4, 97–115. [Google Scholar] [CrossRef]
- Weleber, R.G.; Gregory-Evans, K. Chapter 17-Retinitis Pigmentosa and Allied Disorders. In Retina, 4th ed.; Ryan, S.J., Hinton, D.R., Schachat, A.P., Wilkinson, C.P., Eds.; Mosby: Edinburgh, UK, 2006; pp. 395–498. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Brown, J. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology 1998, 105, 1069–1075. [Google Scholar] [CrossRef]
- Xu, M.; Zhai, Y.; MacDonald, I.M. Visual Field Progression in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2020, 61, 56. [Google Scholar] [CrossRef]
- Timmis, M.A.; Allsop, J.; Baranian, M.; Baker, J.; Basevitch, I.; Latham, K.; Pardhan, S.; Van Paridon, K.N. Visual Search Behavior in Individuals With Retinitis Pigmentosa During Level Walking and Obstacle Crossing. Investig. Opthalmol. Vis. Sci. 2017, 58, 4737–4746. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Grewal, P.S.; Narayan, A.; Alser, M.; Ali, N.; Fujinami, K.; Webster, A.R.; Michaelides, M. Sector Retinitis Pigmentosa: Extending the Molecular Genetics Basis and Elucidating the Natural History. Am. J. Ophthalmol. 2021, 221, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Léveillard, T.; Sahel, J.-A. Rod-Derived Cone Viability Factor for Treating Blinding Diseases: From Clinic to Redox Signaling. Sci. Transl. Med. 2010, 2, 26ps16. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Classification of Diseases 11th Revision. Available online: https://icd.who.int/en (accessed on 25 December 2020).
- Vezinaw, C.M.; Fishman, G.A.; McAnany, J.J. Visual impairment in retinitis pigmentosa. Retina 2020, 40, 1630–1633. [Google Scholar] [CrossRef]
- Fishman, G.A. Retinitis Pigmentosa. Arch. Ophthalmol. 1978, 96, 1185–1188. [Google Scholar] [CrossRef]
- A Fishman, G.; Young, R.S.; Vasquez, V.; Lourenço, P. Color vision defects in retinitis pigmentosa. Ann. Ophthalmol. 1981, 13, 609–618. [Google Scholar] [PubMed]
- Bittner, A.K.; Diener-West, M.; Dagnelie, G. A survey of photopsias in self-reported retinitis pigmentosa. Retina 2009, 29, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Vaccarella, L.; Olatunji, S.; Cebulla, C.; Christoforidis, J. Diagnostic Challenges in Retinitis Pigmentosa: Genotypic Multiplicity and Phenotypic Variability. Curr. Genom. 2011, 12, 267–275. [Google Scholar] [CrossRef]
- Fishman, G.A.; Fishman, M.; Maggiano, J. Macular Lesions Associated With Retinitis Pigmentosa. Arch. Ophthalmol. 1977, 95, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Charng, J.; Roman, A.J.; Sheplock, R.; Garafalo, A.V.; Heon, E.; Jacobson, S.G. Progression in X-linked Retinitis Pigmentosa Due to ORF15-RPGR Mutations: Assessment of Localized Vision Changes Over 2 Years rod and cone sensitivity in XLRP. Investig. Opthalmol. Vis. Sci. 2018, 59, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-T.; Talib, M.; van Schooneveld, M.J.; Brinks, J.; Brink, J.T.; Florijn, R.J.; Wijnholds, J.; Verdijk, R.M.; Bergen, A.A.; Boon, C.J. RPGR-Associated Dystrophies: Clinical, Genetic, and Histopathological Features. Int. J. Mol. Sci. 2020, 21, 835. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Berson, E.L. Disease Course of Patients with X-linked Retinitis Pigmentosa due to RPGR Gene Mutations. Investig. Opthalmol. Vis. Sci. 2007, 48, 1298–1304. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. Clinical and Genetic characteristics of Male patients with RPGR-associated Retinal Dystrophies: A long-Term Follow-up Study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Van Cauwenbergh, C.; Wijnholds, J.; Brink, J.B.T.; Florijn, R.J.; Schalij-Delfos, N.E.; Dagnelie, G.; van Genderen, M.M.; De Baere, E.; et al. The Spectrum of Structural and Functional Abnormalities in Female Carriers of Pathogenic Variants in the RPGR Gene. Investig. Opthalmol. Vis. Sci. 2018, 59, 4123–4133. [Google Scholar] [CrossRef]
- Nguyen, X.-T.-A.; Talib, M.; van Cauwenbergh, C.; van Schooneveld, M.J.; Fiocco, M.; Wijnholds, J.; ten Brink, J.B.; Florijn, R.J.; Schalij-Delfos, N.E.; Dagnelie, G.; et al. CLINICAL CHARACTERISTICS AND NATURAL HISTORY OF RHO-ASSOCIATED RETINITIS PIGMENTOSA: A Long-Term Follow-Up Study. RETINA 2021, 41, 212–223. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; A Sandberg, M. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Investig. Opthalmol. Vis. Sci. 2002, 43, 3027–3036. [Google Scholar]
- Berson, E.L.; Sandberg, M.A.; Rosner, B.; Birch, D.G.; Hanson, A.H. Natural Course of Retinitis Pigmentosa Over a Three-Year Interval. Am. J. Ophthalmol. 1985, 99, 240–251. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Genderen, M.M.; Wijnholds, J.; Florijn, R.J.; Brink, J.B.T.; Schalij-Delfos, N.E.; Dagnelie, G.; Cremers, F.P.; Wolterbeek, R.; et al. Genotypic and Phenotypic Characteristics of CRB1-Associated Retinal Dystrophies. Ophthalmology 2017, 124, 884–895. [Google Scholar] [CrossRef]
- Talib, M.; Van Cauwenbergh, C.; De Zaeytijd, J.; Van Wynsberghe, D.; De Baere, E.; Boon, C.J.F.; Leroy, B.P. CRB1-associated retinal dystrophies in a Belgian cohort: Genetic characteristics and long-term clinical follow-up. Br. J. Ophthalmol. 2022, 106, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Tee, J.J.; Yang, Y.; Kalitzeos, A.; Webster, A.; Bainbridge, J.; Michaelides, M. Natural History Study of Retinal Structure, Progression, and Symmetry Using Ellipzoid Zone Metrics in RPGR-Associated Retinopathy. Am. J. Ophthalmol. 2019, 198, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, J.T.; Flood, S.R.; Seiff, S.R. Retinitis Pigmentosa Without Pigment. Am. J. Ophthalmol. 1976, 81, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, J.T. Letter: Nonpigmented Retinitis Pigmentosa and the Neurologist. Arch. Neurol. 1976, 33, 590. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.M.; Michelson, G.; Juenemann, A.G. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014, 2014, bcr2013202236. [Google Scholar] [CrossRef]
- Marsiglia, M.; Duncker, T.; Peiretti, E.; Brodie, S.E.; Tsang, S.H. Unilateral Retinitis Pigmentosa: A Proposal of Genetic Pathogenic Mechanisms. Eur. J. Ophthalmol. 2012, 22, 654–660. [Google Scholar] [CrossRef]
- Kranich, H.; Bartkowski, S.; Denton, M.J.; Krey, S.; Dickinson, P.; Duvigneau, C.; Gal, A. Autosomal dominant ‘sector’ retinitis pigmentosa due to a point mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum. Mol. Genet. 1993, 2, 813–814. [Google Scholar] [CrossRef]
- Ramon, E.; Cordomí, A.; Aguilà, M.; Srinivasan, S.; Dong, X.; Moore, A.T.; Webster, A.R.; Cheetham, M.; Garriga, P. Differential Light-induced Responses in Sectorial Inherited Retinal Degeneration. J. Biol. Chem. 2014, 289, 35918–35928. [Google Scholar] [CrossRef]
- Shah, S.P.; Wong, F.; Sharp, D.M.; Vincent, A.L. A Novel Rhodopsin Point Mutation, Proline-170-histidine, Associated with Sectoral Retinitis Pigmentosa. Ophthalmic Genet. 2014, 35, 241–247. [Google Scholar] [CrossRef]
- Balfoort, B.M.; Buijs, M.J.; Asbroek, A.L.T.; Bergen, A.A.; Boon, C.J.; Ferreira, E.A.; Houtkooper, R.H.; Wagenmakers, M.A.; Wanders, R.J.; Waterham, H.R.; et al. A review of treatment modalities in gyrate atrophy of the choroid and retina (GACR). Mol. Genet. Metab. 2021, 134, 96–116. [Google Scholar] [CrossRef]
- Benomar, A.; Yahyaoui, M.; Meggouh, F.; Bouhouche, A.; Boutchich, M.; Bouslam, N.; Zaim, A.; Schmitt, M.; Belaidi, H.; Ouazzani, R.; et al. Clinical comparison between AVED patients with 744 del A mutation and Friedreich ataxia with GAA expansion in 15 Moroccan families. J. Neurol. Sci. 2002, 198, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.; Downes, S.M. Midlife diagnosis of Refsum Disease in siblings with Retinitis Pigmentosa-the footprint is the clue: A case report. J. Med. Case Rep. 2008, 2, 80. [Google Scholar] [CrossRef] [PubMed]
- Gouras, P.; E Carr, R.; Gunkel, R.D. Retinitis pigmentosa in abetalipoproteinemia: Effects of vitamin A. Investig. Ophthalmol. 1971, 10, 784–793. [Google Scholar]
- Iwasa, K.; Shima, K.; Komai, K.; Nishida, Y.; Yokota, T.; Yamada, M. Retinitis pigmentosa and macular degeneration in a patient with ataxia with isolated vitamin E deficiency with a novel c.717 del C mutation in the TTPA gene. J. Neurol. Sci. 2014, 345, 228–230. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Odel, J.G.; Chen, C.S.; Winn, B.J. The Multifocal Electroretinogram. J. Neuro-Ophthalmol. 2003, 23, 225–235. [Google Scholar] [CrossRef]
- Alexander, K.R.; A Fishman, G. Prolonged rod dark adaptation in retinitis pigmentosa. Br. J. Ophthalmol. 1984, 68, 561–569. [Google Scholar] [CrossRef]
- Stavrou, P.; A Good, P.; Broadhurst, E.J.; Bundey, S.; Fielder, A.R.; Crews, S.J. ERG and EOG abnormalities in carriers of X-linked retinitis pigmentosa. Eye 1996, 10, 581–589. [Google Scholar] [CrossRef]
- Menghini, M.; Cehajic-Kapetanovic, J.; MacLaren, R.E. Monitoring progression of retinitis pigmentosa: Current recommendations and recent advances. Expert Opin. Orphan Drugs 2020, 8, 67–78. [Google Scholar] [CrossRef]
- Talib, M.; Dagnelie, G.; Boon, C.J.F. Recording and Analysis of Goldmann Kinetic Visual Fields. Methods Mol. Biol. 2018, 1715, 327–338. [Google Scholar] [CrossRef]
- Barnes, C.S.; Schuchard, R.A.; Birch, D.G.; Dagnelie, G.; Wood, L.; Koenekoop, R.K.; Bittner, A.K. Reliability of Semiautomated Kinetic Perimetry (SKP) and Goldmann Kinetic Perimetry in Children and Adults With Retinal Dystrophies. Transl. Vis. Sci. Technol. 2019, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.P.; Bittner, A.; Yang, L.; Marcus, R.; Iftikhar, M.H.; Dagnelie, G. Variability and Errors of Manually Digitized Goldmann Visual Fields. Optom. Vis. Sci. 2016, 93, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Bittner, A.K.; Iftikhar, M.H.; Dagnelie, G. Test-Retest, Within-Visit Variability of Goldmann Visual Fields in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2011, 52, 8042–8046. [Google Scholar] [CrossRef] [PubMed]
- Pfau, M.; Jolly, J.K.; Wu, Z.; Denniss, J.; Lad, E.M.; Guymer, R.H.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S. Fundus-controlled perimetry (microperimetry): Application as outcome measure in clinical trials. Prog. Retin. Eye Res. 2021, 82, 100907. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, I.S.; Tseng, C.; Macdonald, I.M. Microperimetry as an Outcome Measure in Choroideremia Trials: Reproducibility and Beyond. Investig. Opthalmol. Vis. Sci. 2016, 57, 4151–4161. [Google Scholar] [CrossRef] [PubMed]
- Schönbach, E.M.; Wolfson, Y.; Strauss, R.W.; Ibrahim, M.A.; Kong, X.; Muñoz, B.; Birch, D.G.; Cideciyan, A.V.; Hahn, G.-A.; Nittala, M.; et al. Macular Sensitivity Measured With Microperimetry in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study. JAMA Ophthalmol. 2017, 135, 696–703. [Google Scholar] [CrossRef]
- Iftikhar, M.; Kherani, S.; Kaur, R.; Lemus, M.; Nefalar, A.; Usmani, B.; Junaid, N.; Campochiaro, P.A.; Scholl, H.P.; Shah, S.M. Progression of Retinitis Pigmentosa as Measured on Microperimetry: The PREP-1 Study. Ophthalmol. Retin. 2018, 2, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-T.; Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Schalij-Delfos, N.E.; Klaver, C.C.; Talsma, H.E.; Fiocco, M.; Florijn, R.J.; et al. CRB1-Associated Retinal Dystrophies: A Prospective Natural History Study in Anticipation of Future Clinical Trials. Am. J. Ophthalmol. 2022, 234, 37–48. [Google Scholar] [CrossRef]
- Miura, G.; Baba, T.; Tatsumi, T.; Yokouchi, H.; Yamamoto, S. The Impact of Cataract Surgery on Contrast Visual Acuity and Retinal Sensitivity in Patients with Retinitis Pigmentosa. J. Ophthalmol. 2021, 2021, 2281834. [Google Scholar] [CrossRef]
- Bennett, L.D.; Klein, M.; Locke, K.G.; Kiser, K.; Birch, D.G. Dark-Adapted Chromatic Perimetry for Measuring Rod Visual Fields in Patients with Retinitis Pigmentosa. Transl. Vis. Sci. Technol. 2017, 6, 15. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Voigt, W.J.; Parel, J.-M.; Apathy, P.P.; Nghiem-Phu, L.; Myers, S.W.; Patella, V.M. Automated Light- and Dark- Adapted Perimetry for Evaluating Retinitis Pigmentosa. Ophthalmology 1986, 93, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, D.; Roman, A.J.; Cideciyan, A.V.; Matsui, R.; Gruzensky, M.L.; Sheplock, R.; Jacobson, S. Automated Light- and Dark-Adapted Perimetry for Evaluating Retinitis Pigmentosa: Filling a Need to Accommodate Multicenter Clinical Trials. Investig. Opthalmol. Vis. Sci. 2016, 57, 3118–3128. [Google Scholar] [CrossRef] [PubMed]
- Grewal, M.K.; Chandra, S.; Bird, A.; Jeffery, G.; Sivaprasad, S. Scotopic thresholds on dark-adapted chromatic perimetry in healthy aging and age-related macular degeneration. Sci. Rep. 2021, 11, 10349. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Beltran, W.A.; Sumaroka, A.; Swider, M.; Iwabe, S.; Roman, A.J.; Olivares, M.B.; Schwartz, S.B.; Komáromy, A.M.; et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA 2013, 110, E517–E525. [Google Scholar] [CrossRef]
- Roman, A.J.; Cideciyan, A.V.; Wu, V.; Garafalo, A.V.; Jacobson, S.G. Full-field stimulus testing: Role in the clinic and as an outcome measure in clinical trials of severe childhood retinal disease. Prog. Retin. Eye Res. 2022, 87, 101000. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, C.; Tzekov, R.; Zhu, Y.; Li, W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: A systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Cabral, T.; Sengillo, J.D.; Duong, J.K.; Justus, S.; Boudreault, K.; Schuerch, K.; Belfort Jr, R.B.; Mahajan, V.B.; Sparrow, J.R.; Tsang, S.H. Retrospective Analysis of Structural Disease Progression in Retinitis Pigmentosa Utilizing Multimodal Imaging. Sci. Rep. 2017, 7, 10347. [Google Scholar] [CrossRef]
- Cai, C.X.; Locke, K.G.; Ramachandran, R.; Birch, D.G.; Hood, D.C. A Comparison of Progressive Loss of the Ellipsoid Zone (EZ) Band in Autosomal Dominant and X-Linked Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2014, 55, 7417–7422. [Google Scholar] [CrossRef] [PubMed]
- Hariri, A.H.; Zhang, H.Y.; Ho, A.; Francis, P.; Weleber, R.G.; Birch, D.G.; Ferris, F.; Sadda, S.R. Trial of Oral Valproic Acid for Retinitis Pigmentosa Group Quantification of Ellipsoid Zone Changes in Retinitis Pigmentosa Using en Face Spectral Domain-Optical Coherence Tomography. JAMA Ophthalmol. 2016, 134, 628–635. [Google Scholar] [CrossRef]
- Liu, G.; Li, H.; Liu, X.; Xu, D.; Wang, F. Structural analysis of retinal photoreceptor ellipsoid zone and postreceptor retinal layer associated with visual acuity in patients with retinitis pigmentosa by ganglion cell analysis combined with OCT imaging. Medicine 2016, 95, e5785. [Google Scholar] [CrossRef]
- Phadikar, P.; Saxena, S.; Ruia, S.; Lai, T.Y.Y.; Meyer, C.H.; Eliott, D. The potential of spectral domain optical coherence tomography imaging based retinal biomarkers. Int. J. Retin. Vitr. 2017, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Schuerch, K.; Marsiglia, M.; Lee, W.; Tsang, S.H.; Sparrow, J.R. Multimodal imaging of disease-associated pigmentary changes in retinitis pigmentosa. Retina 2016, 36, S147–S158. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Oldani, M.; Jolly, J.; Edwards, T.; Groppe, M.; Downes, S.M.; MacLaren, R. Correlation of Optical Coherence Tomography and Autofluorescence in the Outer Retina and Choroid of Patients With Choroideremia. Investig. Opthalmol. Vis. Sci. 2016, 57, 3674–3684. [Google Scholar] [CrossRef]
- Méjécase, C.; Malka, S.; Guan, Z.; Slater, A.; Arno, G.; Moosajee, M. Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 2020, 12, 2515841420954592. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Motaee, J.; Farjami, M.; Alimardani, M.; Esmaeilie, A.; Pasdar, A. Next-generation sequencing and its application in diagnosis of retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger sequencing and molecular assay monitoring. J. Veter-Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef]
- Ng, P.C.; Kirkness, E.F. Whole genome sequencing. Methods Mol. Biol. 2010, 628, 215–226. [Google Scholar] [CrossRef]
- Fahim, A.T.; Daiger, S.P.; Weleber, R.G. Nonsyndromic Retinitis Pigmentosa Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Poptsova, M.S.; Il’Icheva, I.A.; Nechipurenko, D.Y.; Panchenko, L.A.; Khodikov, M.V.; Oparina, N.Y.; Polozov, R.V.; Nechipurenko, Y.D.; Grokhovsky, S.L. Non-random DNA fragmentation in next-generation sequencing. Sci. Rep. 2014, 4, 4532. [Google Scholar] [CrossRef]
- França, L.T.C.; Carrilho, E.; Kist, T.B.L. A review of DNA sequencing techniques. Q. Rev. Biophys. 2002, 35, 169–200. [Google Scholar] [CrossRef]
- Ge, Z.; Bowles, K.; Goetz, K.; Scholl, H.P.N.; Wang, F.; Wang, X.; Xu, S.; Wang, K.; Wang, H.; Chen, R. NGS-based Molecular diagnosis of 105 eyeGENE® probands with Retinitis Pigmentosa. Sci. Rep. 2015, 5, 18287. [Google Scholar] [CrossRef] [PubMed]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef]
- Dockery, A.; Whelan, L.; Humphries, P.; Farrar, G. Next-Generation Sequencing Applications for Inherited Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 5684. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Hart, M.R.; Biesecker, B.B.; Blout, C.L.; Christensen, K.D.; Amendola, L.M.; Bergstrom, K.L.; Biswas, S.; Bowling, K.M.; Brothers, K.B.; Conlin, L.K.; et al. Secondary findings from clinical genomic sequencing: Prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet Med. 2019, 21, 1100–1110. [Google Scholar] [CrossRef]
- Ozsolak, F. Third-generation sequencing techniques and applications to drug discovery. Expert Opin. Drug Discov. 2012, 7, 231–243. [Google Scholar] [CrossRef]
- Xiao, T.; Zhou, W. The third generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 2020, 9, 163–173. [Google Scholar] [CrossRef]
- Resta, R.; Biesecker, B.B.; Bennett, R.L.; Blum, S.; Hahn, S.E.; Strecker, M.N.; Williams, J.L. A New Definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force Report. J. Genet. Couns. 2006, 15, 77–83. [Google Scholar] [CrossRef]
- Ciarleglio, L.J.; Bennett, R.L.; Williamson, J.; Mandell, J.B.; Marks, J.H. Genetic counseling throughout the life cycle. J. Clin. Investig. 2003, 112, 1280–1286. [Google Scholar] [CrossRef]
- Middleton, A.; Hall, G.; Patch, C. Genetic counselors and Genomic Counseling in the United Kingdom. Mol. Genet. Genom. Med. 2015, 3, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Patch, C.; Middleton, A. Genetic counselling in the era of genomic medicine. Br. Med. Bull. 2018, 126, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Strait, S.; Loman, R.; Erickson, L.; DeBenedictis, M. Inherited retinal degeneration current genetics practices-A needs assessment. Ophthalmic Genet. 2020, 41, 533–538. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.L.; Caulfield, T.; Cho, M.K. Research ethics and the challenge of whole-genome sequencing. Nat. Rev. Genet. 2008, 9, 152–156. [Google Scholar] [CrossRef]
- Shintani, K.; Shechtman, D.L.; Gurwood, A.S. Review and update: Current treatment trends for patients with retinitis pigmentosa. Optom.-J. Am. Optom. Assoc. 2009, 80, 384–401. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Yang, M.; Kim, J.-W. Principles of Genetic Counseling in the Era of Next-Generation Sequencing. Ann. Lab. Med. 2018, 38, 291–295. [Google Scholar] [CrossRef]
- Berkman, B.E.; Hull, S.C. The “Right Not to Know” in the Genomic Era: Time to Break From Tradition? Am. J. Bioeth. 2014, 14, 28–31. [Google Scholar] [CrossRef]
- Bennett, R.L.; Hampel, H.L.; Mandell, J.B.; Marks, J.H. Genetic counselors: Translating genomic science into clinical practice. J. Clin. Investig. 2003, 112, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.P.; Emmertz, M.; Kristoffersson, U.; Borg, A.; Larsson, C.; Rehn, M.; Winter, C.; Saal, L.H.; Brandberg, Y.; Loman, N. Germline mutations in BRCA1 and BRCA2 incidentally revealed in a biobank research study: Experiences from re-contacting mutation carriers and relatives. J. Community Genet. 2018, 9, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Severijns, Y.; de Die-Smulders, C.E.M.; Gültzow, T.; de Vries, H.; van Osch, L.A.D.M. Hereditary diseases and child wish: Exploring motives, considerations, and the (joint) decision-making process of genetically at-risk couples. J. Community Genet. 2021, 12, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T.; Lauten, A.; Schneider, U.; Schleussner, E.; Weise, A. Noninvasive Prenatal Testing-When Is It Advantageous to Apply. Biomed. Hub 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Ahmed, M.; Potrata, B.; Willis, T.A.; Grant, H.L.; Allsop, M.J.; Hewison, J.; Downey, L.; Gale, R.; McKibbin, M. Patient attitudes towards prenatal diagnostic testing for inherited retinal disease. Prenat. Diagn. 2015, 35, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Fesahat, F.; Montazeri, F.; Hoseini, S.M. Preimplantation genetic testing in assisted reproduction technology. J. Gynecol. Obstet. Hum. Reprod. 2020, 49, 101723. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Y.; Yu, X.; Huang, Q.; Lin, C.; Zeng, J.; Lan, F.; Wang, Z. The clinical application of preimplantation genetic diagnosis for X-linked retinitis pigmentosa. J. Assist. Reprod. Genet. 2019, 36, 989–994. [Google Scholar] [CrossRef]
- Greco, E.; Litwicka, K.; Minasi, M.G.; Cursio, E.; Greco, P.F.; Barillari, P. Preimplantation Genetic Testing: Where We Are Today. Int. J. Mol. Sci. 2020, 21, 4381. [Google Scholar] [CrossRef]
- Murphy, N.M.; Samarasekera, T.S.; Macaskill, L.; Mullen, J.; Rombauts, L.J.F. Genome sequencing of human in vitro fertilisation embryos for pathogenic variation screening. Sci. Rep. 2020, 10, 3795. [Google Scholar] [CrossRef]
- Hlavatá, L.; Ďuďáková, Ľ.; Trková, M.; Soldátová, I.; Skalická, P.; Kousal, B.; Lišková, P. Preimplantation genetic diagnosis and monogenic inherited eye diseases. Cesk Slov Oftalmol 2016, 72, 167–171. [Google Scholar] [PubMed]
- Cooper, A.R.; Jungheim, E.S. Preimplantation Genetic Testing: Indications and Controversies. Clin. Lab. Med. 2010, 30, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Garway-Heath, D.; Rosen, P.; Bird, A.C.; Tuft, S.J. Outcome of cataract surgery in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2001, 85, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Ikeda, Y.; Murakami, Y.; Funatsu, J.; Nakatake, S.; Tachibana, T.; Yoshida, N.; Nakao, S.; Hisatomi, T.; Yoshida, S.; et al. Risk Factors for Posterior Subcapsular Cataract in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2017, 58, 2534–2537. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Murakami, Y.; Nakatake, S.; Fujiwara, K.; Notomi, S.; Hisatomi, T.; Ishibashi, T. Factors Affecting Visual Acuity after Cataract Surgery in Patients with Retinitis Pigmentosa. Ophthalmology 2015, 122, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Strong, S.; Bradley, P.; Severn, P.; Moore, A.T.; Webster, A.R.; Mitchell, P.; Kifley, A.; Michaelides, M. Prevalence of cystoid macular oedema, epiretinal membrane and cataract in retinitis pigmentosa. Br. J. Ophthalmol. 2019, 103, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef]
- Lu, B.; Yin, H.; Tang, Q.; Wang, W.; Luo, C.; Chen, X.; Zhang, X.; Lai, K.; Xu, J.; Chen, X.; et al. Multiple cytokine analyses of aqueous humor from the patients with retinitis pigmentosa. Cytokine 2020, 127, 154943. [Google Scholar] [CrossRef]
- E Chua, B.; Mitchell, P.; Cumming, R. Effects of cataract type and location on visual function: The Blue Mountains Eye Study. Eye 2004, 18, 765–772. [Google Scholar] [CrossRef]
- Allen, D.; Vasavada, A. Cataract and surgery for cataract. BMJ 2006, 333, 128–132. [Google Scholar] [CrossRef]
- Skiadaresi, E.; McAlinden, C.; Pesudovs, K.; Polizzi, S.; Khadka, J.; Ravalico, G. Subjective Quality of Vision Before and After Cataract Surgery. Arch. Ophthalmol. 2012, 130, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- van Bree, M.C.; Pierrache, L.; Zijlmans, B.L.; Reus, N.J.; Born, L.I.v.D.; Berg, T.J.v.D. Straylight as an Indicator for Cataract Extraction in Patients with Retinal Dystrophy. Ophthalmol. Retin. 2017, 1, 531–544. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, I.J.; Gjertsen, J.; Kruijt, B.; Witmer, J.P.; Rulo, A.; Schlingemann, R.O.; Berg, T.J.V.D. Straylight measurements as an indication for cataract surgery. J. Cataract. Refract. Surg. 2012, 38, 840–848. [Google Scholar] [CrossRef]
- Lundström, M.; Barry, P.; Henry, Y.; Rosen, P.; Stenevi, U. Visual outcome of cataract surgery; Study from the European Registry of Quality Outcomes for Cataract and Refractive Surgery. J. Cataract. Refract. Surg. 2013, 39, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, F.; Wang, Y.; Chen, Y.; Chen, Q.; Li, F. Intra and post-operative complications observed with femtosecond laser-assisted cataract surgery versus conventional phacoemulsification surgery: A systematic review and meta-analysis. BMC Ophthalmol. 2019, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Dikopf, M.S.; Chow, C.C.; Mieler, W.F.; Tu, E.Y. Cataract Extraction Outcomes and the Prevalence of Zonular Insufficiency in Retinitis Pigmentosa. Am. J. Ophthalmol. 2013, 156, 82–88.e2. [Google Scholar] [CrossRef]
- Davies, E.C.; Pineda, R. Cataract surgery outcomes and complications in retinal dystrophy patients. Can. J. Ophthalmol. 2017, 52, 543–547. [Google Scholar] [CrossRef]
- Bayyoud, T.; Bartz-Schmidt, K.U.; Yoeruek, E. Long-term clinical results after cataract surgery with and without capsular tension ring in patients with retinitis pigmentosa: A retrospective study. BMJ Open 2013, 3, e002616. [Google Scholar] [CrossRef]
- Shatriah, I.; Jin-Poi, T.; Khairy-Shamel, S.T.; Zunaina, E. Rapid anterior capsular contraction after phacoemulsification surgery in a patient with retinitis pigmentosa. Clin. Ophthalmol. 2013, 7, 839–842. [Google Scholar] [CrossRef]
- Hong, Y.; Li, H.; Sun, Y.; Ji, Y. A Review of Complicated Cataract in Retinitis Pigmentosa: Pathogenesis and Cataract Surgery. J. Ophthalmol. 2020, 2020, 6699103. [Google Scholar] [CrossRef]
- Nguyen, X.-T.; Thiadens, A.A.; Fiocco, M.; Tan, W.; McKibbin, M.; Klaver, C.C.; Meester-Smoor, M.A.; Van Cauwenbergh, C.; Strubbe, I.; Vergaro, A.; et al. Outcome of Cataract Surgery in Patients With Retinitis Pigmentosa. Am. J. Ophthalmol. 2023, 246, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Rojas, J.O.; Schuerch, K.; Mathews, P.M.; Cabral, T.; Hazan, A.; Sparrow, J.; Tsang, S.H.; Suh, L.H. Evaluating Structural Progression of Retinitis Pigmentosa After Cataract Surgery. Am. J. Ophthalmol. 2017, 180, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.C.Y.; Lam, S.C.; Mohamed, S.; Wong, R.L.M. Survival analysis of visual improvement after cataract surgery in advanced retinitis pigmentosa. Eye 2017, 31, 1747–1748. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mitamura, Y.; Hagiwara, A.; Kumagai, K.; Miura, G.; Sugawara, T.; Egawa, M.; Yamamoto, S. Relationship between retinal microstructures and visual acuity after cataract surgery in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2015, 99, 508–511. [Google Scholar] [CrossRef]
- Chen, C.X.; Da Wang, J.; Zhang, J.S.; Xiong, Y.; Li, J.; Chen, S.Y.; Sun, X.L.; Liu, Z.Y.; Mayinuer, Y.; Wan, X.H. Effect of lens capsular tension ring on preventing capsular contraction syndrome in the surgery of retinitis pigmentosa combined with cataract: Retrospective case series. Int. J. Clin. Pract. 2021, 75, e14272. [Google Scholar] [CrossRef]
- Garcia-Martin, E.; Rodriguez-Mena, D.; Dolz, I.; Almarcegui, C.; Gil-Arribas, L.; Bambo, M.P.; Larrosa, J.M.; Polo, V.; Pablo, L.E. Influence of Cataract Surgery on Optical Coherence Tomography and Neurophysiology Measurements in Patients With Retinitis Pigmentosa. Am. J. Ophthalmol. 2013, 156, 293–303.e292. [Google Scholar] [CrossRef]
- Mao, J.; Fang, D.; Chen, Y.; Tao, J.; Wu, M.; Wu, S.; Wang, P.; Zhang, Y.; Shen, L. Prediction of Visual Acuity After Cataract Surgery Using Optical Coherence Tomography Findings in Eyes With Retinitis Pigmentosa. Ophthalmic Surg. Lasers Imaging Retin. 2018, 49, 587–594. [Google Scholar] [CrossRef]
- Chatterjee, S.; Agrawal, D.; Agrawal, D.; Parchand, S.; Sahu, A. Cataract surgery in retinitis pigmentosa. Indian J. Ophthalmol. 2021, 69, 1753–1757. [Google Scholar] [CrossRef]
- Lu, Q.J.; Bi, J.; Dd, B.H.; Wang, D.; Liu, Q. Efficacy analysis of phacoemulsification combined with intraocular lens implantation in the treatment of retinitis pigmentosa complicated with cataract. Recent Adv. Ophthalmol. 2017, 37, 1064–1067. [Google Scholar]
- He, H.; Song, H.; Meng, X.; Cao, K.; Liu, Y.-X.; Wang, J.; Wan, X.; Jin, Z.-B. Effects and Prognosis of Cataract Surgery in Patients with Retinitis Pigmentosa. Ophthalmol. Ther. 2022, 11, 1975–1989. [Google Scholar] [CrossRef]
- Nakamura, S.; Fujiwara, K.; Yoshida, N.; Murakami, Y.; Shimokawa, S.; Koyanagi, Y.; Ikeda, Y.; Sonoda, K.-H. Long-term Outcomes of Cataract Surgery in Patients with Retinitis Pigmentosa. Ophthalmol. Retin. 2022, 6, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Takagi, S.; Hirami, Y.; Nakamura, M.; Kurimoto, Y. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye 2022, 37, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.W.; Li, R.W.-H.; Kee, C.-S. Blue-Light Filtering Spectacle Lenses: Optical and Clinical Performances. PLoS ONE 2017, 12, e0169114. [Google Scholar] [CrossRef] [PubMed]
- Masket, S.; Ceran, B.B.; Fram, N.R. Spontaneous dislocation of posterior chamber intraocular lenses (PC IOLs) in patients with retinitis pigmentosa–Case series. Saudi J. Ophthalmol. 2011, 26, 61–65. [Google Scholar] [CrossRef]
- Sudhir, R.R.; Rao, S.K. Capsulorhexis phimosis in retinitis pigmentosa despite capsular tension ring implantation. J. Cataract. Refract. Surg. 2001, 27, 1691–1694. [Google Scholar] [CrossRef]
- Najjar, D.M.; O Igbre, A.; Tsai, F.F. Late capsular bag contraction and intraocular lens subluxation in retinitis pigmentosa: A case report. J. Med. Case Rep. 2012, 5, 65. [Google Scholar] [CrossRef]
- Karahan, E.; Er, D.; Kaynak, S. An Overview of Nd:YAG Laser Capsulotomy. Med. Hypothesis Discov. Innov. Ophthalmol. 2014, 3, 45–50. [Google Scholar]
- Strong, S.; Liew, G.; Michaelides, M. Retinitis pigmentosa-associated cystoid macular oedema: Pathogenesis and avenues of intervention. Br. J. Ophthalmol. 2017, 101, 31–37. [Google Scholar] [CrossRef]
- Kim, Y.J.; Joe, S.G.; Lee, D.-H.; Lee, J.Y.; Kim, J.-G.; Yoon, Y.H. Correlations between Spectral-Domain OCT Measurements and Visual Acuity in Cystoid Macular Edema Associated with Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2013, 54, 1303–1309. [Google Scholar] [CrossRef]
- Bakthavatchalam, M.; Lai, F.H.; Rong, S.S.; Ng, D.S.; Brelen, M.E. Treatment of cystoid macular edema secondary to retinitis pigmentosa: A systematic review. Surv. Ophthalmol. 2018, 63, 329–339. [Google Scholar] [CrossRef]
- Hajali, M.; A Fishman, G.; Anderson, R.J. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br. J. Ophthalmol. 2008, 92, 1065–1068. [Google Scholar] [CrossRef]
- Vingolo, E.M.; Valente, S.; E Gerace, E.; Spadea, L.; Nebbioso, M. Macular hole in retinitis pigmentosa patients: Microincision vitrectomy with polydimethylsiloxane as possible treatment. Eye 2015, 29, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Iijima, H.; Gohdo, T.; Tsukahara, S. Optical coherence tomography of cystoid macular edema associated with retinitis pigmentosa. Am. J. Ophthalmol. 1999, 128, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Ffytche, T.J. Cystoid maculopathy in retinitis pigmentosa. Trans. Ophthalmol. Soc. United Kingd. 1972, 92, 265–283. [Google Scholar]
- Shahidi, M.; Fishman, G.; Ogura, Y.; Ambroz, K.; Zeimer, R. Foveal thickening in retinitis pigmentosa patients with cystoid macular edema. Retina 1994, 14, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Apushkin, M.A.; Fishman, G.A.; Janowicz, M.J. Monitoring cystoid macular edema by optical coherence tomography in patients with retinitis pigmentosa. Ophthalmology 2004, 111, 1899–1904. [Google Scholar] [CrossRef]
- Nussenblatt, R.B.; Kaufman, S.C.; Palestine, A.G.; Davis, M.D.; Ferris, F.L. Macular Thickening and Visual Acuity: Measurement in patients with cystoid macular edema. Ophthalmology 1987, 94, 1134–1139. [Google Scholar] [CrossRef]
- Yeo, J.H.; Kim, Y.J.; Yoon, Y.H. Optical coherence tomography angiography in patients with retinitis pigmentosa–associated cystoid macular edema. Retina 2020, 40, 2385–2395. [Google Scholar] [CrossRef]
- Makiyama, Y.; Oishi, A.; Otani, A.; Ogino, K.; Nakagawa, S.; Kurimoto, M.; Yoshimura, N. Prevalence and spatial distribution of cystoid spaces in retinitis pigmentosa: Investigation with spectral domain optical coherence tomography. Retina 2014, 34, 981–988. [Google Scholar] [CrossRef]
- Kjellström, U. Reduced macular function in ABCA4 carriers. Mol. Vis. 2015, 21, 767–782. [Google Scholar]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic Mutation of BEST1 Causes a Distinct Retinopathy in Humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Fishman, G.A.; Fiscella, R.G.; Adelman, A.E. Efficacy of dorzolamide hydrochloride in the management of chronic cystoid macular edema in patients with retinitis pigmentosa. Retina 1997, 17, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Gilbert, L.D.; Fiscella, R.G.; Kimura, A.E.; Jampol, L.M. Acetazolamide for Treatment of Chronic Macular Edema in Retinitis Pigmentosa. Arch. Ophthalmol. 1989, 107, 1445–1452. [Google Scholar] [CrossRef]
- Orzalesi, N.; Pierrottet, C.; Porta, A.; Aschero, M. Long-term treatment of retinitis pigmentosa with acetazolamide: A pilot study. Graefe’s Arch. Clin. Exp. Ophthalmol. 1993, 231, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Moore, A.T.; Webster, A.R.; Michaelides, M. Efficacy and Prognostic Factors of Response to Carbonic Anhydrase Inhibitors in Management of Cystoid Macular Edema in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2015, 56, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Wolfensberger, T.J. The role of carbonic anhydrase inhibitors in the management of macular edema. Doc. Ophthalmol. 1999, 97, 387–397. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, R.; Lin, X.; Xiang, Z. Efficacy of carbonic anhydrase inhibitors in management of cystoid macular edema in retinitis pigmentosa: A meta-analysis. PLoS ONE 2017, 12, e0186180. [Google Scholar] [CrossRef]
- Grover, S.; Apushkin, M.A.; Fishman, G.A. Topical Dorzolamide for the Treatment of Cystoid Macular Edema in Patients With Retinitis Pigmentosa. Am. J. Ophthalmol. 2006, 141, 850–858. [Google Scholar] [CrossRef]
- Moldow, B.; Sander, B.; Larsen, M.; Lund-Andersen, H. Effects of acetazolamide on passive and active transport of fluorescein across the normal BRB. Investig. Opthalmol. Vis. Sci. 1999, 40, 1770–1775. [Google Scholar]
- Lichter, P.R. Reducing Side Effects of Carbonic Anhydrase Inhibitors. Ophthalmology 1981, 88, 266–269. [Google Scholar] [CrossRef]
- Schmickl, C.N.; Owens, R.L.; E Orr, J.; A Edwards, B.; Malhotra, A. Side effects of acetazolamide: A systematic review and meta-analysis assessing overall risk and dose dependence. BMJ Open Respir. Res. 2020, 7, e000557. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrand, C.; Tiselius, H.-G. Urine Composition and Stone Formation During Treatment with Acetazolamide. Scand. J. Urol. Nephrol. 1987, 21, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Kass, M.A.; Kolker, A.E.; Gordon, M.; Goldberg, I.; Gieser, D.K.; Krupin, T.; Becker, B. Acetazolamide and Urolithiasis. Ophthalmology 1981, 88, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A. Efficacy of Sustained Topical Dorzolamide Therapy for Cystic Macular Lesions in Patients With Retinitis Pigmentosa and Usher Syndrome. Arch. Ophthalmol. 2010, 128, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Yoshida, N.; Notomi, S.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Therapeutic effect of prolonged treatment with topical dorzolamide for cystoid macular oedema in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2013, 97, 1187–1191. [Google Scholar] [CrossRef]
- Apushkin, M.A.; Fishman, G.A.; Grover, S.; Janowicz, M.J. Rebound of cystoid macular edema with continued use of acetazolamide in patients with retinitis pigmentosa. Retina 2007, 27, 1112–1118. [Google Scholar] [CrossRef]
- Fishman, G.A.; A Apushkin, M. Continued use of dorzolamide for the treatment of cystoid macular oedema in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2007, 91, 743–745. [Google Scholar] [CrossRef]
- Ozdemir, H.; Karacorlu, M.; Karacorlu, S. Intravitreal triamcinolone acetonide for treatment of cystoid macular oedema in patients with retinitis pigmentosa. Acta Ophthalmol. Scand. 2005, 83, 248–251. [Google Scholar] [CrossRef]
- Srour, M.; Querques, G.; Leveziel, N.; Zerbib, J.; Tilleul, J.; Boulanger-Scemama, E.; Souied, E.H. Intravitreal dexamethasone implant (Ozurdex) for macular edema secondary to retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 1501–1506. [Google Scholar] [CrossRef]
- Strong, S.A.; Gurbaxani, A.; Michaelides, M. Treatment of Retinitis Pigmentosa-Associated Cystoid Macular Oedema Using Intravitreal Aflibercept (Eylea) despite Minimal Response to Ranibizumab (Lucentis): A Case Report. Case Rep. Ophthalmol. 2016, 7, 389–397. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar] [PubMed]
- Veritti, D.; Sarao, V.; De Nadai, K.; Chizzolini, M.; Parmeggiani, F.; Perissin, L.; Lanzetta, P. Dexamethasone Implant Produces Better Outcomes than Oral Acetazolamide in Patients with Cystoid Macular Edema Secondary to Retinitis Pigmentosa. J. Ocul. Pharmacol. Ther. 2020, 36, 190–197. [Google Scholar] [CrossRef]
- Ahn, S.J.; Kim, K.E.; Woo, S.J.; Park, K.H. The Effect of an Intravitreal Dexamethasone Implant for Cystoid Macular Edema in Retinitis Pigmentosa: A Case Report and Literature Review. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Sudhalkar, A.; Kodjikian, L.; Borse, N. Intravitreal dexamethasone implant for recalcitrant cystoid macular edema secondary to retinitis pigmentosa: A pilot study. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Park, U.C.; Park, J.H.; Ma, D.J.; Cho, I.H.; Oh, B.-L.; Yu, H.G. A randomized paired-eye trial of intravitreal dexamethasone implant for cystoid macular edema in retinitis pigmentosa. Retina 2020, 40, 1359–1366. [Google Scholar] [CrossRef]
- Celik, N.; Khoramnia, R.; Auffarth, G.U.; Sel, S.; Mayer, C.S. Complications of dexamethasone implants: Risk factors, prevention, and clinical management. Int. J. Ophthalmol. 2020, 13, 1612–1620. [Google Scholar] [CrossRef]
- Hagiwara, A.; Yamamoto, S.; Ogata, K.; Sugawara, T.; Hiramatsu, A.; Shibata, M.; Mitamura, Y. Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011, 89, e122–e125. [Google Scholar] [CrossRef]
- Fujiwara, K.; Ikeda, Y.; Murakami, Y.; Nakatake, S.; Tachibana, T.; Yoshida, N.; Nakao, S.; Hisatomi, T.; Yoshitomi, T.; Sonoda, K.-H.; et al. Association Between Aqueous Flare and Epiretinal Membrane in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2016, 57, 4282–4286. [Google Scholar] [CrossRef]
- Ikeda, Y.; Yoshida, N.; Murakami, Y.; Nakatake, S.; Notomi, S.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Long-term Surgical Outcomes of Epiretinal Membrane in Patients with Retinitis Pigmentosa. Sci. Rep. 2015, 5, 13078. [Google Scholar] [CrossRef]
- Jin, Z.-B.; Gan, D.-K.; Xu, G.-Z.; Nao-I, N. Macular Hole Formation in Patients With Retinitis Pigmentosa and Prognosis of Pars Plana Vitrectomy. Retina 2008, 28, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Shah, G.; Blinder, K.J. Bilateral Macular Hole Formation in a Patient With Retinitis Pigmentosa. Ophthalmic Surg. Lasers Imaging Retin. 2002, 33, 152–154. [Google Scholar] [CrossRef]
- García-Fernández, M.; Castro-Navarro, J.; Bajo-Fuente, A. Unilateral recurrent macular hole in a patient with retinitis pigmentosa: A case report. J. Med. Case Rep. 2013, 7, 69. [Google Scholar] [CrossRef]
- Chan, W.O.; Brennan, N.; Webster, A.R.; Michaelides, M.; Muqit, M.M.K. Retinal detachment in retinitis pigmentosa. BMJ Open Ophthalmol. 2020, 5, e000454. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.P.; Jalali, S.; Nayaka, A.; Pappuru, R.R.; Pathengay, A.; Das, T. Clinical presentations and outcomes of rhegmatogenous retinal detachment in retinitis pigmentosa. Retina 2016, 36, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Rishi, E.; Rishi, P.; Bhende, M.; Koundanya, V.V.; Sidramayya, R.; Maitray, A.; Rao, C.; Susvar, P.; Bhende, P.; Sharma, T. Retinal Detachment in 31 Eyes with Retinitis Pigmentosa. Ophthalmol. Retin. 2018, 2, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.D.; Menia, N.; Roy, R.; Sen, P.; George, A.E.; Ganesh, S.K.; Biswas, J. Uveitis in Patients with Retinitis Pigmentosa: 30 Years’ Consecutive Data. Ocul. Immunol. Inflamm. 2017, 26, 1283–1288. [Google Scholar] [CrossRef]
- Li, A.S.; Pasricha, M.V.; Mishra, K.; Nguyen, Q.D.; Beres, S.J.; Wood, E.H. CRB1-associated retinal dystrophy presenting as self-resolving opsoclonus and posterior uveitis. Am. J. Ophthalmol. Case Rep. 2022, 26, 101444. [Google Scholar] [CrossRef]
- Murro, V.; Mucciolo, D.P.; Sodi, A.; Vannozzi, L.; De Libero, C.; Simonini, G.; Rizzo, S. Retinal capillaritis in a CRB1-associated retinal dystrophy. Ophthalmic Genet. 2017, 38, 555–558. [Google Scholar] [CrossRef]
- Verhagen, F.; Kuiper, J.; Nierkens, S.; Imhof, S.M.; Radstake, T.; De Boer, J. Systemic inflammatory immune signatures in a patient with CRB1 linked retinal dystrophy. Expert Rev. Clin. Immunol. 2016, 12, 1359–1362. [Google Scholar] [CrossRef]
- Duncker, T.; Lee, W.; Jiang, F.; Ramachandran, R.; Hood, D.C.; Tsang, S.H.; Sparrow, J.R.; Greenstein, V.C. Acute zonal occult outer retinopathy: Structural and Functional Analysis Across the Transition Zone Between Healthy and Diseased Retina. Retina 2018, 38, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Willermain, F.; Greiner, K.; Forrester, J.V. Atypical end-stage birdshot retinochoroidopathy. Ocul. Immunol. Inflamm. 2003, 11, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Zamir, E.; Banin, E.; Merin, S. Retinitis pigmentosa associated with Fuchs’ heterochromic uveitis. Arch. Ophthalmol. 2000, 118, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Díez-Cattini, G.F.; Ancona-Lezama, D.A.; Valdés-Lara, C.; Morales-Cantón, V. The unusual association of inverse retinitis pigmentosa and Fuchs’ heterochromic iridocyclitis. Int. J. Retin. Vitr. 2017, 3, 3. [Google Scholar] [CrossRef]
- Born, L.I.V.D.; van Schooneveld, M.J.; de Jong, P.T.; Bleeker-Wagemakers, E.M. Fuchs’ heterochromic uveitis associated with retinitis pigmentosa in a father and son. Br. J. Ophthalmol. 1994, 78, 504–505. [Google Scholar] [CrossRef]
- Vuorre, I.; Saari, M.; Tiilikainen, A.; Rasanen, O. Fuchs’ heterochromic cyclitis associated with retinitis pigmentosa: A family study. Can. J. Ophthalmol. 1979, 14, 10–16. [Google Scholar]
- Yalvaç, I.S.; Altintas, A.K.; Gökdere, A.; Duman, S. Fuchs’ heterochromic uveitis associated with retinitis pigmentosa. Acta Ophthalmol. Scand. 1998, 76, 243–244. [Google Scholar] [CrossRef]
- Sandinha, T. Retinitis pigmentosa associated with Fuchs’ heterochromic uveitis. Eye 2003, 17, 778–779. [Google Scholar] [CrossRef]
- Lichtinger, A.; Chowers, I.; Amer, R. Usher syndrome associated with Fuchs’ heterochromic uveitis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 1481–1485. [Google Scholar] [CrossRef]
- Sevgi, D.D.; Davoudi, S.; Comander, J.; Sobrin, L. Retinal pigmentary changes in chronic uveitis mimicking retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1801–1810. [Google Scholar] [CrossRef]
- Hettinga, Y.M.; van Genderen, M.M.; Wieringa, W.; Norel, J.O.-V.; de Boer, J.H. Retinal Dystrophy in 6 Young Patients Who Presented with Intermediate Uveitis. Ophthalmology 2016, 123, 2043–2046. [Google Scholar] [CrossRef] [PubMed]
- Szabó, E.; Brichová, M.; Lišková, P.; Svozílková, P.; Ríhová, E. Retinitis pigmentosa mimicking uveitis. A case report. Cesk Slov. Oftalmol. 2013, 69, 32–36. [Google Scholar]
- Latorre, R.H.; Fernandez-perez, S.; Garcia-martin, E.; Satue, M.; Idoipe, M.; De La Mata, G.; Torrón, C. Bilateral intermediate uveitis asociated with retinosis pigmentosa. Acta Ophthalmol. 2012, 90, S249. [Google Scholar] [CrossRef]
- Kaufman, M.; Medina-Mendez, C.; Friberg, T.; Eller, A. Evaluation of peripheral retinal vasculitis in retinitis pigmentosa using wide-field fluorescein angiography. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4018. [Google Scholar]
- Badeeb, O.; Trope, G.; Musarella, M. Primary angle closure glaucoma and retinitis pigmentosa. Acta Ophthalmol. 1993, 71, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-C.; Chen, Y.-Y. Association between retinitis pigmentosa and an increased risk of primary angle closure glaucoma: A population-based cohort study. PLoS ONE 2022, 17, e0274066. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, C.; Khadka, S.; Joshi, P. Angle Closure Glaucoma in Retinitis Pigmentosa. Case Rep. Ophthalmol. Med. 2020, 2020, 602358. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Choy, B.N.K.; Shum, J.W.H. Management of Primary Angle-Closure Glaucoma. Asia-Pacific J. Ophthalmol. 2016, 5, 59–62. [Google Scholar] [CrossRef]
- Slade, A.; Isa, F.; Kyte, D.; Pankhurst, T.; Kerecuk, L.; Ferguson, J.; Lipkin, G.; Calvert, M. Patient reported outcome measures in rare diseases: A narrative review. Orphanet J. Rare Dis. 2018, 13, 61. [Google Scholar] [CrossRef]
- Wilkinson, M.E.; Shahid, K.S. Low vision rehabilitation: An update. Saudi J. Ophthalmol. 2018, 32, 134–138. [Google Scholar] [CrossRef]
- Langelaan, M.; de Boer, M.R.; van Nispen, R.M.; Wouters, B.; Moll, A.C.; van Rens, G.H. Change in quality of life after rehabilitation: Prognostic factors for visually impaired adults. Int. J. Rehabil. Res. 2009, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- van Nispen, R.M.; Virgili, G.; Hoeben, M.; Langelaan, M.; Klevering, J.; Keunen, J.E.; van Rens, G.H. Low vision rehabilitation for better quality of life in visually impaired adults. Cochrane Database Syst. Rev. 2020, 1, CD006543. [Google Scholar] [CrossRef] [PubMed]
- Owsley, C. Characteristics of Low-Vision Rehabilitation Services in the United States. Arch. Ophthalmol. 2009, 127, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, E.L.; Pallant, J.F.; Pesudovs, K.; Rees, G.; Hassell, J.B.; Keeffe, J.E. The Effectiveness of Low-Vision Rehabilitation on Participation in Daily Living and Quality of Life. Investig. Opthalmol. Vis. Sci. 2007, 48, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Stelmack, J.A.; Tang, X.C.; Wei, Y.; Massof, R.W.; for the Low-Vision Intervention Trial Study Group. The Effectiveness of Low-Vision Rehabilitation in 2 Cohorts Derived From the Veterans Affairs Low-Vision Intervention Trial. JAMA Ophthalmol. 2012, 130, 1162–1168. [Google Scholar] [CrossRef]
- Massof, R.W.; Ahmadian, L.; Grover, L.L.; Deremeik, J.T.; Goldstein, J.E.; Rainey, C.; Epstein, C.; Barnett, G.D. The Activity Inventory: An Adaptive Visual Function Questionnaire. Optom. Vis. Sci. 2007, 84, 763–774. [Google Scholar] [CrossRef]
- Mangione, C.M.; Lee, P.; Gutierrez, P.R.; Spritzer, K.; Berry, S.; Hays, R.D. Development of the 25-list-item National Eye Institute Visual Function Questionnaire. Arch. Ophthalmol. 2001, 119, 1050–1058. [Google Scholar] [CrossRef]
- Bruijning, J.; Van Nispen, R.; Verstraten, P.; Van Rens, G. A Dutch ICF Version of the Activity Inventory: Results from Focus Groups with Visually Impaired Persons and Experts. Ophthalmic Epidemiol. 2010, 17, 366–377. [Google Scholar] [CrossRef]
- Lacy, G.D.; Abalem, M.F.; Andrews, C.A.; Popova, L.T.; Santos, E.P.; Yu, G.; Rakine, H.Y.; Baig, N.; Ehrlich, J.R.; Fahim, A.T.; et al. The Michigan Retinal Degeneration Questionnaire: A Patient-Reported Outcome Instrument for Inherited Retinal Degenerations. Am. J. Ophthalmol. 2020, 222, 60–68. [Google Scholar] [CrossRef]
- Virgili, G.; Acosta, R.; Bentley, S.A.; Giacomelli, G.; Allcock, C.; Evans, J.R. Reading aids for adults with low vision. Cochrane Database Syst. Rev. 2018, 4, CD003303. [Google Scholar] [CrossRef]
- Nguyen, X.; Koopman, J.; Genderen, M.M.; Stam, H.L.; Boon, C.J. Artificial vision: The effectiveness of the OrCam in patients with advanced inherited retinal dystrophies. Acta Ophthalmol. 2021, 100, e986–e993. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, M.-C.; Wittich, W. Factors related to the use of magnifying low vision aids: A scoping review. Disabil. Rehabil. 2020, 42, 3525–3537. [Google Scholar] [CrossRef] [PubMed]
- Wittich, W.; Lorenzini, M.-C.; Markowitz, S.N.; Tolentino, M.; Gartner, S.A.; Goldstein, J.E.; Dagnelie, G. The Effect of a Head-mounted Low Vision Device on Visual Function. Optom. Vis. Sci. 2018, 95, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M. Occupational therapy interventions in low vision rehabilitation. Can. J. Ophthalmol. 2006, 41, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.W.; Bressler, N.M.; Ffolkes, S.; Wittenborn, J.S.; Jorkasky, J. Public Attitudes About Eye and Vision Health. JAMA Ophthalmol. 2016, 134, 1111–1118. [Google Scholar] [CrossRef]
- Demmin, D.L.; Silverstein, S.M. Visual Impairment and Mental Health: Unmet Needs and Treatment Options. Clin. Ophthalmol. 2020, 14, 4229–4251. [Google Scholar] [CrossRef]
- Munaw, M.B.; Tegegn, M.T. Visual impairment and psychological distress among adults attending the University of Gondar tertiary eye care and training center, Northwest Ethiopia: A comparative cross-sectional study. PLoS ONE 2022, 17, e0264113. [Google Scholar] [CrossRef]
- Horowitz, A.; Leonard, R.; Reinhardt, J.P. Measuring Psychosocial and Functional Outcomes of a Group Model of Vision Rehabilitation Services for Older Adults. J. Vis. Impair. Blind. 2000, 94, 328–337. [Google Scholar] [CrossRef]
- Rees, G.; Ponczek, E.; Hassell, J.; E Keeffe, J.; Lamoureux, E.L. Psychological outcomes following interventions for people with low vision: A systematic review. Expert Rev. Ophthalmol. 2010, 5, 385–403. [Google Scholar] [CrossRef]
- Veldman, M.H.J.; van der Aa, H.P.A.; Bode, C.; Knoop, H.; Hulshof, C.T.J.; Koopmanschap, M.; Stavleu, E.; van Rens, G.H.M.B.; van Nispen, R.M.A. E-nergEYEze, a vision-specific eHealth intervention based on cognitive behavioral therapy and self-management to reduce fatigue in adults with visual impairment: Study protocol for a randomized controlled trial. Trials 2021, 22, 966. [Google Scholar] [CrossRef]
- Virgili, G.; Rubin, G. Orientation and mobility training for adults with low vision. Cochrane Database Syst. Rev. 2010, 2010, CD003925. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, G.R.; Ballemans, J.; Kempen, G.I. Orientation and mobility training for adults with low vision: A new standardized approach. Clin. Rehabil. 2013, 27, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.E.; Mahajan, V.B.; Tsang, S.H. Gene therapy and genome surgery in the retina. J. Clin. Investig. 2018, 128, 2177–2188. [Google Scholar] [CrossRef]
- Botto, C.; Rucli, M.; Tekinsoy, M.D.; Pulman, J.; Sahel, J.-A.; Dalkara, D. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog. Retin. Eye Res. 2022, 86, 100975. [Google Scholar] [CrossRef]
- Leroy, B.P.; Fischer, M.D.; Flannery, J.G.; MacLaren, R.E.; Dalkara, D.; Scholl, H.P.; Chung, D.C.; Spera, C.; Viriato, D.; Banhazi, J. Gene therapy for inherited retinal disease: Long-term durability of effect. Ophthalmic Res. 2022, 66, 179–196. [Google Scholar] [CrossRef]
- Gange, W.S.; Sisk, R.A.; Besirli, C.G.; Lee, T.C.; Havunjian, M.; Schwartz, H.; Borchert, M.; Sengillo, J.D.; Mendoza, C.; Berrocal, A.M.; et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmol. Retin. 2022, 6, 58–64. [Google Scholar] [CrossRef]
- Reichel, F.F.; Seitz, I.; Wozar, F.; Dimopoulos, S.; Jung, R.; Kempf, M.; Kohl, S.; Kortüm, F.C.; Ott, S.; Pohl, L.; et al. Development of retinal atrophy after subretinal gene therapy with voretigene neparvovec. Br. J. Ophthalmol. 2022. [Google Scholar] [CrossRef]
- Peng, Y.; Tang, L.; Zhou, Y. Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic Res. 2017, 58, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Ghoraba, H.H.; Akhavanrezayat, A.; Karaca, I.; Yavari, N.; Lajevardi, S.; Hwang, J.; Regenold, J.; Matsumiya, W.; Pham, B.; Zaidi, M.; et al. Ocular Gene Therapy: A Literature Review with Special Focus on Immune and Inflammatory Responses. Clin. Ophthalmol. 2022, 16, 1753–1771. [Google Scholar] [CrossRef] [PubMed]
- Boon, N.; Wijnholds, J.; Pellissier, L.P. Research Models and Gene Augmentation Therapy for CRB1 Retinal Dystrophies. Front. Neurosci. 2020, 14, 860. [Google Scholar] [CrossRef]
- Bansal, M.; Acharya, S.; Sharma, S.; Phutela, R.; Rauthan, R.; Maiti, S.; Chakraborty, D. CRISPR Cas9 based genome editing in inherited retinal dystrophies. Ophthalmic Genet. 2021, 42, 365–374. [Google Scholar] [CrossRef]
- Burnight, E.R.; Giacalone, J.C.; Cooke, J.A.; Thompson, J.R.; Bohrer, L.R.; Chirco, K.R.; Drack, A.V.; Fingert, J.H.; Worthington, K.S.; Wiley, L.A.; et al. CRISPR-Cas9 genome engineering: Treating inherited retinal degeneration. Prog. Retin. Eye Res. 2018, 65, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Pulman, J.; Sahel, J.-A.; Dalkara, D. New Editing Tools for Gene Therapy in Inherited Retinal Dystrophies. CRISPR J. 2022, 5, 377–388. [Google Scholar] [CrossRef]
- Rasul, M.F.; Hussen, B.M.; Salihi, A.; Ismael, B.S.; Jalal, P.J.; Zanichelli, A.; Jamali, E.; Baniahmad, A.; Ghafouri-Fard, S.; Basiri, A.; et al. Strategies to overcome the main challenges of the use of CRISPR/Cas9 as a replacement for cancer therapy. Mol. Cancer 2022, 21, 64. [Google Scholar] [CrossRef]
- Collin, R.W.; Garanto, A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr. Opin. Ophthalmol. 2017, 28, 260–266. [Google Scholar] [CrossRef]
- Girach, A.; Audo, I.; Birch, D.G.; Huckfeldt, R.M.; Lam, B.L.; Leroy, B.P.; Michaelides, M.; Russell, S.R.; Sallum, J.M.; Stingl, K.; et al. RNA-based therapies in inherited retinal diseases. Ther. Adv. Ophthalmol. 2022, 14, 25158414221134602. [Google Scholar] [CrossRef]
- Collin, R.W.; Den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther.-Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Duebel, J.; Marazova, K.; Sahel, J.-A. Optogenetics. Curr. Opin. Ophthalmol. 2015, 26, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Tomita, H.; Maeda, A. Optogenetic Therapy for Visual Restoration. Int. J. Mol. Sci. 2022, 23, 15041. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.-A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Degli Esposti, S.; Delaux, A.; de Saint Aubert, J.-B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y. Stem cell therapies for retinal diseases: From bench to bedside. J. Mol. Med. 2020, 98, 1347–1368. [Google Scholar] [CrossRef]
- Bacakova, L.; Zarubova, J.; Travnickova, M.; Musilkova, J.; Pajorova, J.; Slepicka, P.; Kasalkova, N.S.; Svorcik, V.; Kolska, Z.; Motarjemi, H.; et al. Stem cells: Their source, potency and use in regenerative therapies with focus on adipose-derived stem cells–a review. Biotechnol. Adv. 2018, 36, 1111–1126. [Google Scholar] [CrossRef]
- Sharma, A.; Jaganathan, B.G. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biol. Targets Ther. 2021, 15, 299–306. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Sangkitporn, S.; Trinavarat, A.; Pawestri, A.R.; Vamvanij, V.; Ruangchainikom, M.; Luksanapruksa, P.; Pongpaksupasin, P.; Khorchai, A.; Dambua, A.; et al. Intravitreal autologous mesenchymal stem cell transplantation: A non-randomized phase I clinical trial in patients with retinitis pigmentosa. Stem Cell Res. Ther. 2021, 12, 52. [Google Scholar] [CrossRef]
- Gagliardi, G.; Ben M’Barek, K.; Goureau, O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: A pluripotent stem cell-based approach. Prog. Retin. Eye Res. 2019, 71, 1–25. [Google Scholar] [CrossRef]
- Siqueira, R.C. Stem cell therapy in retinal diseases? Rev. Bras. Hematol. Hemoter. 2012, 34, 222–226. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, Y.; Wang, Y.; Zhang, D.; Shen, B.; Luo, M.; Gu, P. Progress of stem/progenitor cell-based therapy for retinal degeneration. J. Transl. Med. 2017, 15, 99. [Google Scholar] [CrossRef]
- Blum, B.; Benvenisty, N. The Tumorigenicity of Human Embryonic Stem Cells. Adv. Cancer Res. 2008, 100, 133–158. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Agosta, P.; McKee, C.; Walker, K.; Mazzella, M.; Alamri, A.; Svinarich, D.; Chaudhry, G.R. Human primitive mesenchymal stem cell-derived retinal progenitor cells improved neuroprotection, neurogenesis, and vision in rd12 mouse model of retinitis pigmentosa. Stem Cell Res. Ther. 2022, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.J.; Li, S.Y.; Qu, L.H.; Meng, X.H.; Wang, Y.; Xu, H.W.; Liang, Z.Q.; Yin, Z.Q. Long-term safety of human retinal progenitor cell transplantation in retinitis pigmentosa patients. Stem Cell Res. Ther. 2017, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Park, S.S.; Albini, T.A.; Canto-Soler, M.V.; Klassen, H.; MacLaren, R.E.; Takahashi, M.; Nagiel, A.; Schwartz, S.D.; Bharti, K. Retinal stem cell transplantation: Balancing safety and potential. Prog. Retin. Eye Res. 2020, 75, 100779. [Google Scholar] [CrossRef] [PubMed]
- Musiał-Wysocka, A.; Kot, M.; Majka, M. The Pros and Cons of Mesenchymal Stem Cell-Based Therapies. Cell Transplant. 2019, 28, 801–812. [Google Scholar] [CrossRef]
- Ayton, L.N.; Barnes, N.; Dagnelie, G.; Fujikado, T.; Goetz, G.; Hornig, R.; Jones, B.W.; Muqit, M.M.; Rathbun, D.L.; Stingl, K.; et al. An update on retinal prostheses. Clin. Neurophysiol. 2020, 131, 1383–1398. [Google Scholar] [CrossRef]
- Miyoshi, T.; Morimoto, T.; Sawai, H.; Fujikado, T. Spatial Resolution of Suprachoroidal–Transretinal Stimulation Estimated by Recording Single-Unit Activity From the Cat Lateral Geniculate Nucleus. Front. Neurosci. 2021, 15, 717429. [Google Scholar] [CrossRef]
- Shim, S.; Eom, K.; Jeong, J.; Kim, S.J. Retinal Prosthetic Approaches to Enhance Visual Perception for Blind Patients. Micromachines 2020, 11, 535. [Google Scholar] [CrossRef]
- Ayton, L.N.; Blamey, P.J.; Guymer, R.H.; Luu, C.D.; Nayagam, D.A.X.; Sinclair, N.C.; Shivdasani, M.N.; Yeoh, J.; McCombe, M.F.; Briggs, R.J.; et al. First-in-Human Trial of a Novel Suprachoroidal Retinal Prosthesis. PLoS ONE 2014, 9, e115239. [Google Scholar] [CrossRef]
- Fujikado, T.; Kamei, M.; Sakaguchi, H.; Kanda, H.; Morimoto, T.; Ikuno, Y.; Nishida, K.; Kishima, H.; Maruo, T.; Konoma, K.; et al. Testing of Semichronically Implanted Retinal Prosthesis by Suprachoroidal-Transretinal Stimulation in Patients with Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2011, 52, 4726–4733. [Google Scholar] [CrossRef]
- White, J.; Knight, L.; da Cruz, L.; E Stanga, P.; Patrick, H.; Powell, H.; Berry, L.; Withers, K.; Carolan-Rees, G.; Jackson, T.L. Effects of the Argus II Retinal Prosthesis System on the Quality of Life of Patients With Ultra-Low Vision Due to Retinitis Pigmentosa: Protocol for a Single-Arm, Mixed Methods Study. JMIR Res. Protoc. 2021, 10, e17436. [Google Scholar] [CrossRef] [PubMed]
- Chuang, A.T.; E Margo, C.; Greenberg, P.B. Retinal implants: A systematic review. Br. J. Ophthalmol. 2014, 98, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Stronks, H.C.; Dagnelie, G. The functional performance of the Argus II retinal prosthesis. Expert Rev. Med. Devices 2014, 11, 23–30. [Google Scholar] [CrossRef]
- Sieving, P.A.; Caruso, R.C.; Tao, W.; Coleman, H.R.; Thompson, D.J.S.; Fullmer, K.R.; Bush, R.A. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc. Natl. Acad. Sci. USA 2006, 103, 3896–3901. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G.; Weleber, R.G.; Duncan, J.L.; Jaffe, G.J.; Tao, W. Randomized Trial of Ciliary Neurotrophic Factor Delivered by Encapsulated Cell Intraocular Implants for Retinitis Pigmentosa. Am. J. Ophthalmol. 2013, 156, 283–292.e281. [Google Scholar] [CrossRef]
- Birch, D.G.; Bennett, L.D.; Duncan, J.L.; Weleber, R.G.; Pennesi, M.E. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am. J. Ophthalmol. 2016, 170, 10–14. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investig. 2020, 130, 1527–1541. [Google Scholar] [CrossRef]
- Kong, X.; Hafiz, G.; Wehling, D.; Akhlaq, A.; Campochiaro, P.A. Locus-Level Changes in Macular Sensitivity in Patients with Retinitis Pigmentosa Treated with Oral N-acetylcysteine. Am. J. Ophthalmol. 2021, 221, 105–114. [Google Scholar] [CrossRef]
- Maccarone, R.; Tisi, A.; Passacantando, M.; Ciancaglini, M. Ophthalmic Applications of Cerium Oxide Nanoparticles. J. Ocul. Pharmacol. Ther. 2020, 36, 376–383. [Google Scholar] [CrossRef]
- Wong, L.L.; Pye, Q.N.; Chen, L.; Seal, S.; McGinnis, J.F. Defining the Catalytic Activity of Nanoceria in the P23H-1 Rat, a Photoreceptor Degeneration Model. PLoS ONE 2015, 10, e0121977. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.; Anderson, E.J.; Gaudio, A.R.; et al. Further Evaluation of Docosahexaenoic Acid in Patients With RetinitisPigmentosa Receiving Vitamin A Treatment: Subgroup analyses. Arch. Ophthalmol. 2004, 122, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Weigel-DiFranco, C.; Rosner, B.; Gaudio, A.R.; Sandberg, M.A. Association of Vitamin A Supplementation With Disease Course in Children With Retinitis Pigmentosa. JAMA Ophthalmol. 2018, 136, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Klaver, C.C.W.; Thiadens, A.A.H.J. Vitamin A for Children With Retinitis Pigmentosa: An Unresolved Mystery. JAMA Ophthalmol. 2018, 136, 496–497. [Google Scholar] [CrossRef]
- Sajovic, J.; Meglič, A.; Glavač, D.; Markelj, Š.; Hawlina, M.; Fakin, A. The Role of Vitamin A in Retinal Diseases. Int. J. Mol. Sci. 2022, 23, 1014. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Pearson, N.S.; Fish, G.E.; Spencer, R.; Takacs, A.; Klein, M.; Locke, K.G.; Birch, D. Four-Year Placebo-Controlled Trial of Docosahexaenoic Acid in X-Linked Retinitis Pigmentosa (DHAX Trial): A randomized clinical trial. JAMA Ophthalmol. 2014, 132, 866–873. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Spencer, R.; Fish, G.E.; Pearson, N.S.; Wang, Y.-Z.; Klein, M.; Takacs, A.; Locke, K.G.; Birch, D.G. Docosahexaenoic Acid Slows Visual Field Progression in X-Linked Retinitis Pigmentosa: Ancillary Outcomes of the DHAX Trial. Investig. Opthalmol. Vis. Sci. 2015, 56, 6646–6653. [Google Scholar] [CrossRef]
- Rayapudi, S.; Schwartz, S.G.; Wang, X.; Chavis, P. Vitamin A and fish oils for retinitis pigmentosa. Cochrane Database Syst. Rev. 2013, 2013, CD008428. [Google Scholar] [CrossRef]
- Schwartz, S.G.; Wang, X.; Chavis, P.; E Kuriyan, A.; A Abariga, S. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst. Rev. 2020, 6, CD008428. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, K.; Liu, R.; Pan, J.; Zhang, L.; Lu, X. Vitamins and Mineral Supplements for Retinitis Pigmentosa. J. Ophthalmol. 2019, 2019, 8524607. [Google Scholar] [CrossRef]
- Massof, R.W.; A Fishman, G. How Strong Is the Evidence That Nutritional Supplements Slow the Progression of Retinitis Pigmentosa? Arch. Ophthalmol. 2010, 128, 493–495. [Google Scholar] [CrossRef]
- Sofi, F.; Sodi, A.; Franco, F.; Murro, V.; Biagini, D.; Miele, A.; Abbruzzese, G.; Mucciolo, D.P.; Virgili, G.; Menchini, U.; et al. Dietary profile of patients with Stargardt’s disease and Retinitis Pigmentosa: Is there a role for a nutritional approach? BMC Ophthalmol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated Accumulation of Lipofuscin Pigments in the RPE of a Mouse Model for ABCA4-Mediated Retinal Dystrophies following Vitamin A Supplementation. Investig. Opthalmol. Vis. Sci. 2008, 49, 3821–3829. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, C.A.; Bertelsen, M.; Kessel, L. Vitamin A in Stargardt disease—An evidence-based update. Ophthalmic Genet. 2018, 39, 555–559. [Google Scholar] [CrossRef] [PubMed]






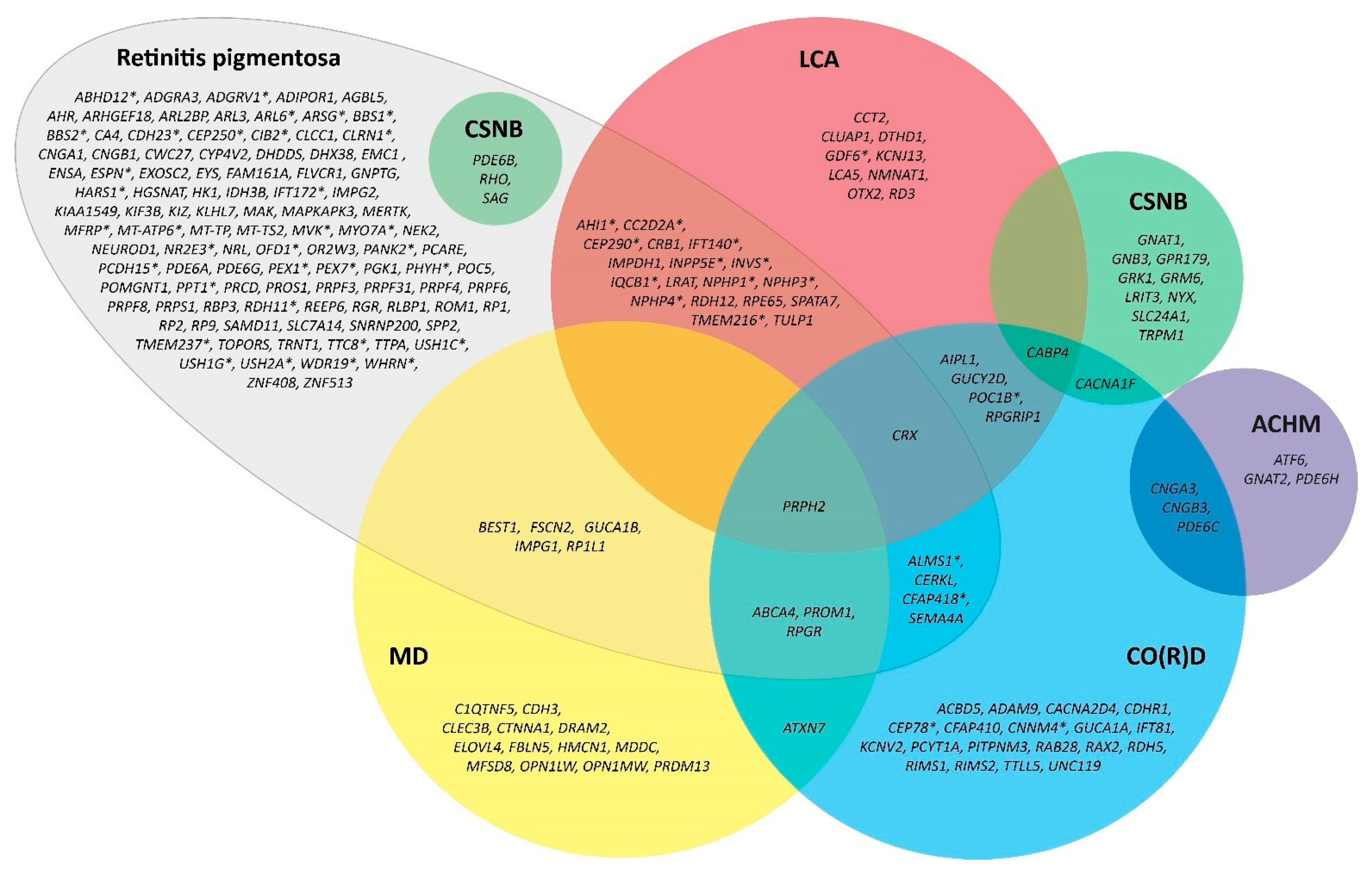{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Pts | Eyes | Follow-Up | Baseline BCVA | Postoperative BCVA | BCVA Change | Complications |
|---|---|---|---|---|---|---|---|
| Jackson et al., 2001 [160] | 89 | 142 | 32.7 months | 1.05 ± 0.38 logMAR | 0.63 ± 0.49 logMAR | −0.42 logMAR | PCO (63%), CME (14%), CCS (10%) |
| Dikopf et al., 2013 [173] | 47 | 80 | 23.3 months | 1.23 ± 0.99 logMAR | 0.81 ± 0.87 logMAR | −0.42 logMAR | PCO (83%), IOL dislocation (3%) |
| Bayyoud et al., 2013 [175] | 52 | 46 | 26.0 months | 1.45 ± 0.85 logMAR | 1.32 ± 0.95 logMAR | −0.13 logMAR | PCO (44%), CME (4%), CCS (4%) |
| Garcia-Martin et al., 2013 [183] | 35 | 35 | 1.0 month | 0.10 ± 0.23 Snellen | 0.48 ± 0.21 Snellen | 0.38 Snellen | N/A |
| Nakamura et al., 2015 [188] | 43 | 58 | 3.0 months | 0.81 ± 0.51 logMAR | 0.34 ± 0.43 logMAR | −0.47 logMAR | None |
| Yoshida et al., 2015 [162] | 40 | 56 | 37.5 ± 22.6 months | 0.76 ± 0.65 logMAR | 0.45 ± 0.53 logMAR | −0.31 logMAR | PCO (84%), CCS (23%) |
| Davies et al., 2017 [174] | 18 | 30 | 3.7 ± 3.3 months | 1.09 ± 0.69 logMAR | 0.61 ± 0.45 logMAR | −0.47 logMAR | CME (13.3%), PCO (66.7%) |
| Chan et al., 2017 [180] | 42 | 67 | 6.9 ± 4.4 years | 1.27 ± 0.42 logMAR | 1.18 ± 0.49 logMAR | −0.09 logMAR | N/A |
| De Rojas et al., 2017 [179] | 19 | 19 | 259 days | 0.33 ± 0.20 logMAR | 0.19 ± 0.17 logMAR | −0.14 logMAR | CME (32%), PCO (95%) |
| Lu et al., 2017 [186] | 52 | 101 | 5.09 ± 2.2 months | 0.12 ± 0.09 Snellen | 0.21 ± 0.16 Snellen | 0.09 Snellen | CCS (2%), increased IOP (2%) |
| Mao et al., 2018 [184] | 70 | 109 | 3 months | 0.80 ± 0.59 logMAR | 0.45 ± 0.41 logMAR | −0.35 logMAR | N/A |
| Chatterjee et al., 2021 [185] | 103 | 132 | 13.5 ± 25.1 months | 1.21 ± 0.87 logMAR | 0.66 ± 0.64 logMAR | −0.55 logMAR | PCO (17%), CME (5%), zonulolysis (3%). PCR (2%), uveitis (4%) |
| Chen et al., 2021 182] | 63 | 84 | 6 months | 1.3 ± 0.7 logMAR | 0.91 ± 0.88 logMAR | −0.39 logMAR | CCS (5%) |
| Miura et al., 2021 [105] | 62 | 62 | 3 months | 0.45 ± 0.25 logMAR | 0.11 ± 0.19 logMAR | −0.33 logMAR | None |
| Nakamura et al., 2022 [181] | 64 | 96 | 5.8 ± 2.4 years | 0.64 ± 0.52 logMAR | 0.61 ± 0.52 logMAR | −0.03 logMAR | PCO (53%), CME (3%), ERM (2%), macular hole (1%), VMT (1%) |
| Nguyen et al., 2022 [178] | 225 | 295 | 0.8 ± 1.6 years | 1.03 ± 0.79 logMAR | 0.81 ± 0.87 logMAR | −0.22 logMAR | PCO (38%), CME (5%), zonulolysis (5%), CCS (2%), IOL dislocation (1%), PCR (<1%), endophthalmitis (<1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, X.-T.-A.; Moekotte, L.; Plomp, A.S.; Bergen, A.A.; van Genderen, M.M.; Boon, C.J.F. Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Int. J. Mol. Sci. 2023, 24, 7481. https://doi.org/10.3390/ijms24087481
Nguyen X-T-A, Moekotte L, Plomp AS, Bergen AA, van Genderen MM, Boon CJF. Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. International Journal of Molecular Sciences. 2023; 24(8):7481. https://doi.org/10.3390/ijms24087481
Chicago/Turabian StyleNguyen, Xuan-Thanh-An, Lude Moekotte, Astrid S. Plomp, Arthur A. Bergen, Maria M. van Genderen, and Camiel J. F. Boon. 2023. "Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies" International Journal of Molecular Sciences 24, no. 8: 7481. https://doi.org/10.3390/ijms24087481
APA StyleNguyen, X.-T.-A., Moekotte, L., Plomp, A. S., Bergen, A. A., van Genderen, M. M., & Boon, C. J. F. (2023). Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. International Journal of Molecular Sciences, 24(8), 7481. https://doi.org/10.3390/ijms24087481