
Structure-Based GC Investigation Sheds New Light on ITS2 Evolution in Corydalis Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
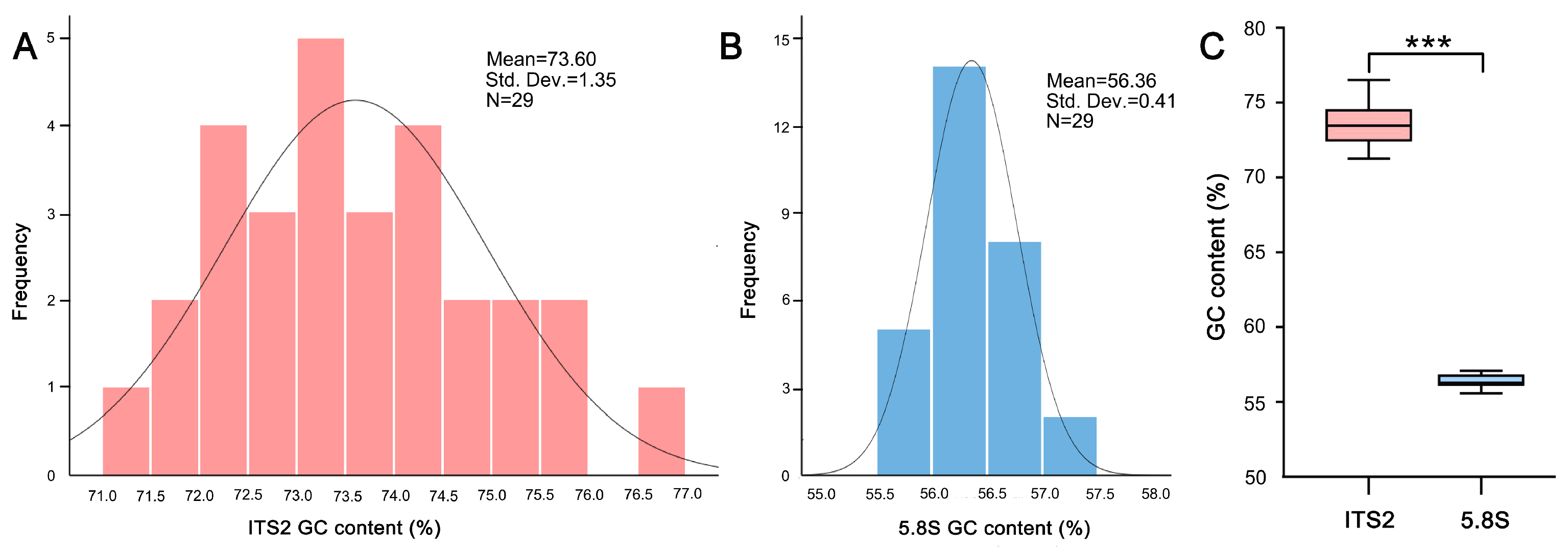
2.1. GC Content Differs Significantly between ITS2 and 5.8S Region
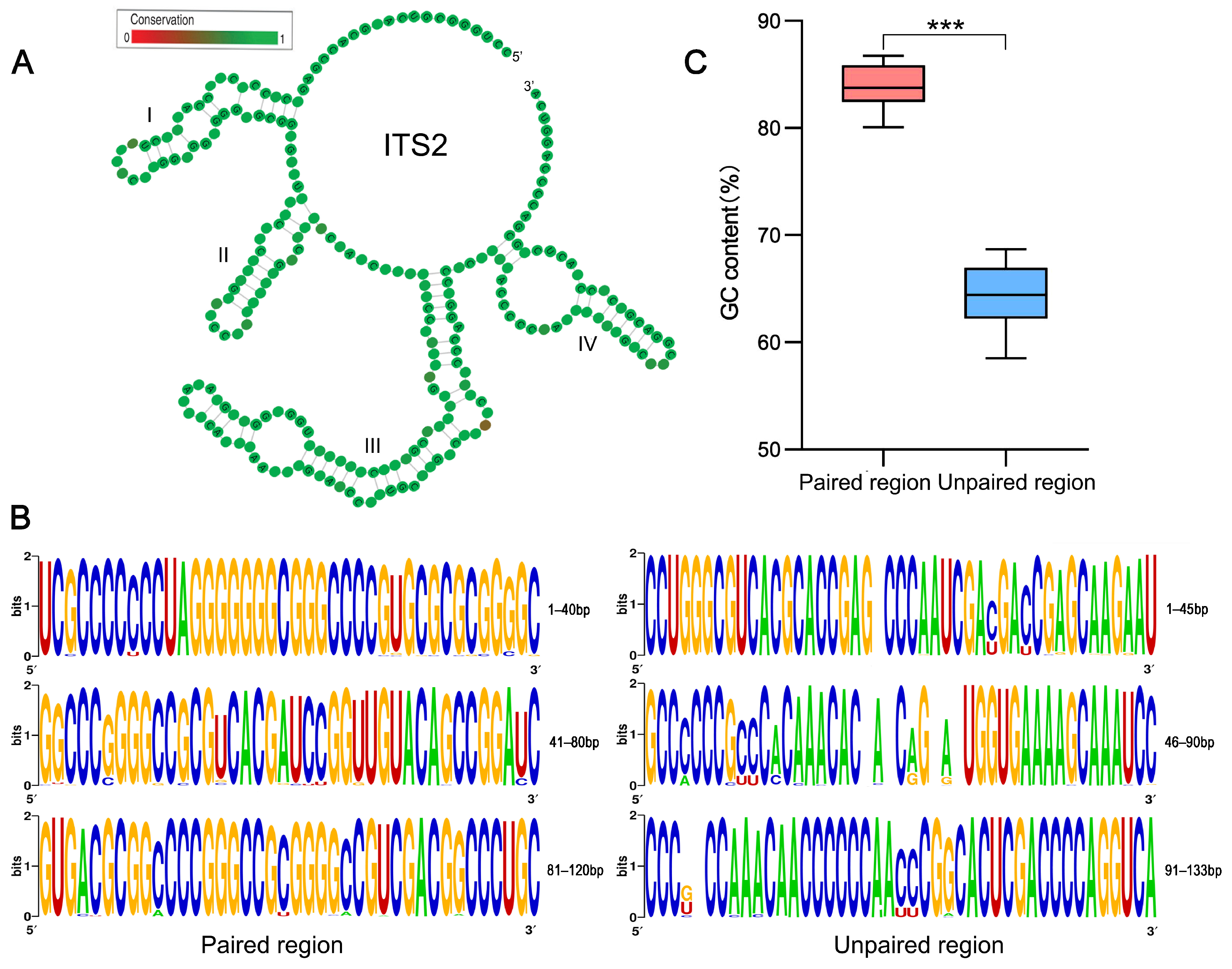
2.2. Comparison of GC and GC* Content between ITS2 Paired and Unpaired Regions
2.3. Correlation between GC Content and Sequence Homogeneity
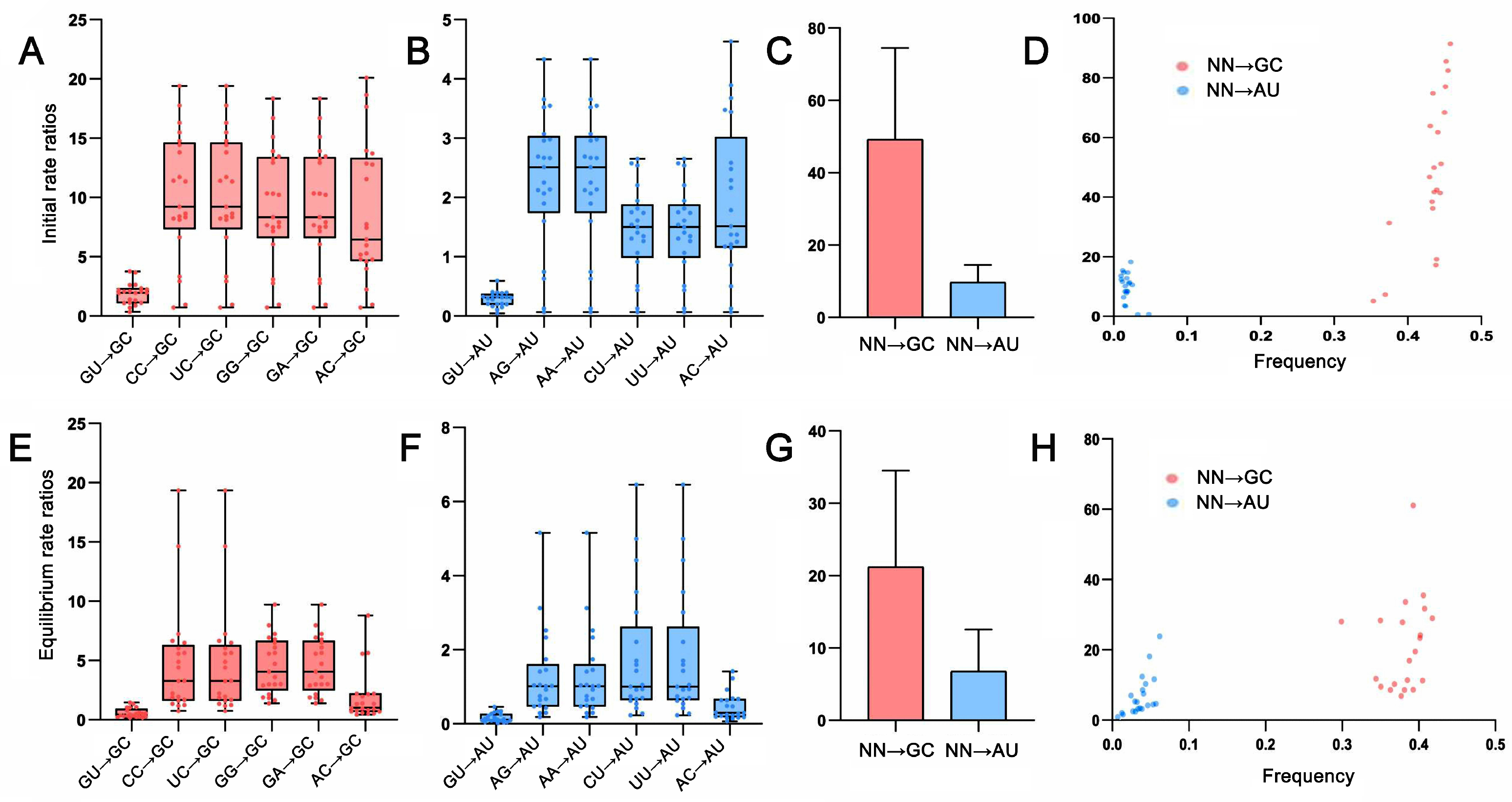
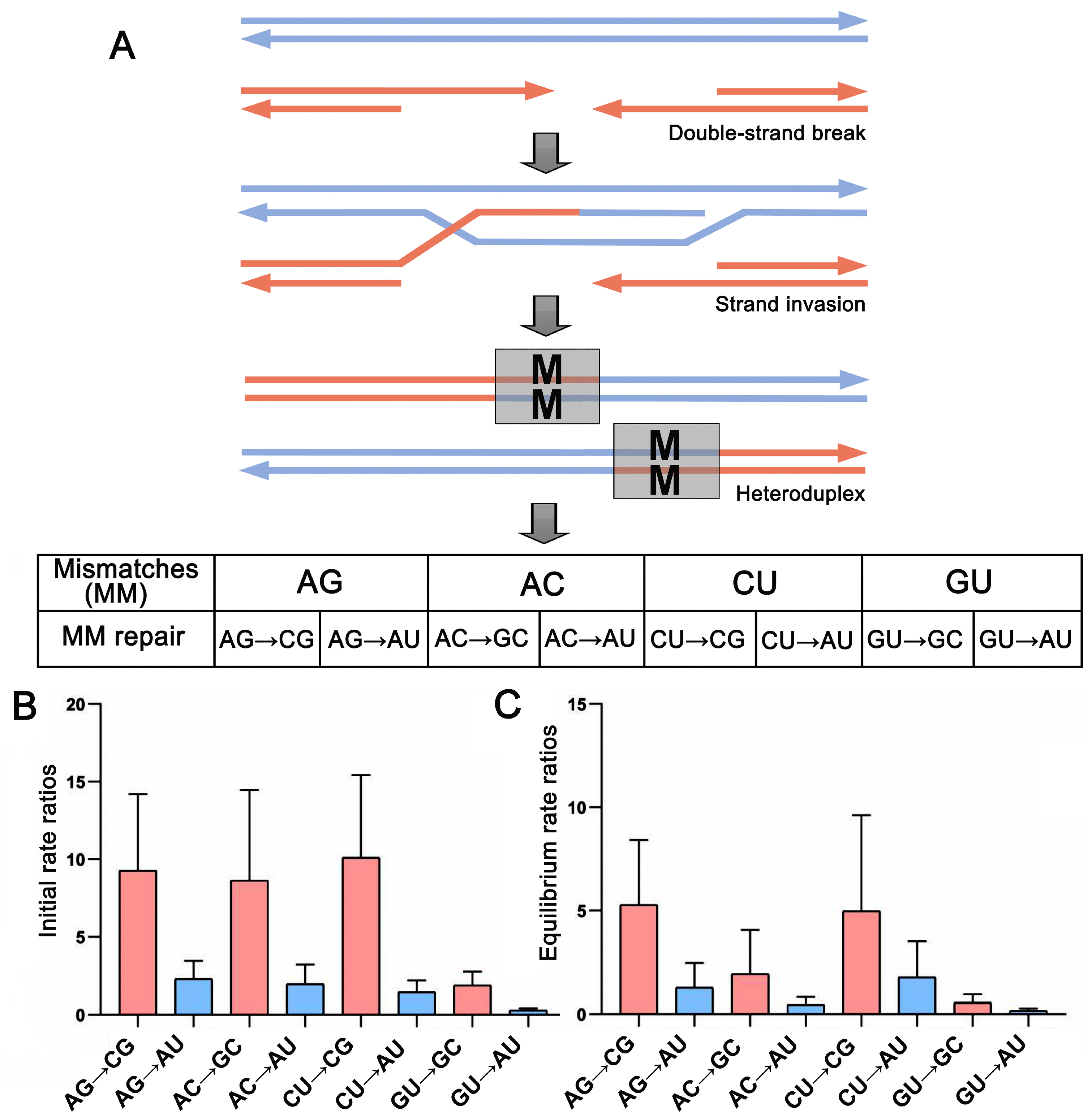
2.4. Base Pair Transformations in ITS2 Paired Regions
3. Discussion
4. Materials and Methods
4.1. Taxon Sampling and Sequence Acquisition
4.2. ITS2 Secondary Structure Prediction and Partition
4.3. Inferring Substitution Parameters of ITS2 Sequence Structure
4.4. Calculation of GC Content and Sequences Homogenization
4.5. Phylogenetic and Statistic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.Q.; Du, D.L. Variation, evolution, and correlation analysis of C plus G content and genome or chromosome size in different kingdoms and phyla. PLoS ONE 2014, 9, e88339. [Google Scholar]
- Hershberg, R.; Petrov, D.A. Evidence That Mutation Is Universally Biased towards AT in Bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012, 10, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Smarda, P.; Knapek, O.; Brezinova, A.; Horova, L.; Grulich, V.; Danihelka, J.; Vesely, P.; Smerda, J.; Rotreklova, O.; Bures, P. Ge-nome sizes and genomic guanine plus cytosine (GC) contents of the Czech vascular flora with new estimates for 1700 species. Preslia 2019, 91, 117–142. [Google Scholar] [CrossRef]
- Serres-Giardi, L.; Belkhir, K.; David, J.; Glémin, S. Patterns and Evolution of Nucleotide Landscapes in Seed Plants. Plant Cell 2012, 24, 1379–1397. [Google Scholar] [CrossRef]
- Singh, R.; Ming, R.; Yu, Q. Comparative Analysis of GC Content Variations in Plant Genomes. Trop. Plant Biol. 2016, 9, 136–149. [Google Scholar] [CrossRef]
- Eyre-Walker, A.; Hurst, L.D. The evolution of isochores. Nat. Rev. Genet. 2001, 2, 549–555. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Muyle, A.; Serres-Giardi, L.; Ressayre, A.; Escobar, J.; Glémin, S. GC-Biased Gene Conversion and Selection Affect GC Content in the Oryza Genus (rice). Mol. Biol. Evol. 2011, 28, 2695–2706. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Sharp, P.M.; Li, W.-H. Mutation rates differ among regions of the mammalian genome. Nature 1989, 337, 283–285. [Google Scholar] [CrossRef]
- Duret, L.; Galtier, N. Biased Gene Conversion and the Evolution of Mammalian Genomic Landscapes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A. Recombination and mammalian genome evolution. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1993, 252, 237–243. [Google Scholar] [CrossRef]
- Lassalle, F.; Périan, S.; Bataillon, T.; Nesme, X.; Duret, L.; Daubin, V. GC-Content Evolution in Bacterial Genomes: The Biased Gene Conversion Hypothesis Expands. PLoS Genet. 2015, 11, e1004941. [Google Scholar] [CrossRef] [PubMed]
- Marais, G. Biased gene conversion: Implications for genome and sex evolution. Trends Genet. 2003, 19, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.G. Modeling Compositional Heterogeneity. Syst. Biol. 2004, 53, 485–495. [Google Scholar] [CrossRef]
- Gruber, K.F.; Voss, R.S.; Jansa, S.A. Base-compositional heterogeneity in the RAG1 locus among didelphid marsupials: Implications for phylogenetic inference and the evolution of GC content. Syst. Biol. 2007, 56, 83–96. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.Z.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenetics Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Mooers, A.Ø.; Holmes, E.C. The evolution of base composition and phylogenetic inference. Trends Ecol. Evol. 2000, 15, 365–369. [Google Scholar] [CrossRef]
- Álvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenetics Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef]
- Chen, S.L.; Yao, H.; Han, J.P.; Liu, C.; Song, J.Y.; Shi, L.C.; Zhu, Y.J.; Ma, X.Y.; Gao, T.; Pang, X.H.; et al. Validation of the ITS2 Region as a Novel DNA Barcode for Identifying Medicinal Plant Species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef]
- Li, D.-Z.; Gao, L.-M.; Li, H.-T.; Wang, H.; Ge, X.-J.; Liu, J.-Q.; Chen, Z.-D.; Zhou, S.-L.; Chen, S.-L.; Yang, J.-B.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [CrossRef]
- Qin, Y.; Li, M.; Cao, Y.; Gao, Y.; Zhang, W. Molecular thresholds of ITS2 and their implications for molecular evolution and species identification in seed plants. Sci. Rep. 2017, 7, 17316. [Google Scholar] [CrossRef]
- Zhang, W.; Tian, W.; Gao, Z.P.; Wang, G.L.; Zhao, H. Phylogenetic Utility of rRNA ITS2 Sequence-Structure under Functional Constraint. Int. J. Mol. Sci. 2020, 21, 6395. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Wang, D. Comparative Chloroplast Genomics of Corydalis Species (Papaveraceae): Evolutionary Perspectives on Their Unusual Large Scale Rearrangements. Front. Plant Sci. 2021, 11, 600354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, M.H.; Zhao, F.x.; Chu, S.S.; Zha, L.P.; Xu, T.; Peng, H.S.; Zhang, W. Molecular Identification and Taxonomic Implication of Herbal Species in Genus Corydalis (Papaveraceae). Molecules 2018, 23, 1393. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zhao, H.; Zhao, F.X.; Jiang, L.; Peng, H.S.; Zhang, W.; Simmons, M.P. Alternative analyses of compensatory base changes in an ITS2 phylogeny of Corydalis (Papaveraceae). Ann. Bot. 2019, 124, 233–243. [Google Scholar] [CrossRef]
- Ren, F.-M.; Wang, Y.-W.; Xu, Z.-C.; Li, Y.; Xin, T.-Y.; Zhou, J.-G.; Qi, Y.-D.; Wei, X.-P.; Yao, H.; Song, J.-Y. DNA barcoding of Corydalis, the most taxonomically complicated genus of Papaveraceae. Ecol. Evol. 2019, 9, 1934–1945. [Google Scholar] [CrossRef]
- Coleman, A.W. ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet. 2003, 19, 370–375. [Google Scholar] [CrossRef]
- Hershkovitz, M.A.; Zimmer, E.A. Conservation patterns in angiosperm rDNA ITS2 sequences. Nucleic Acids Res. 1996, 24, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Maisel, S.; Gerlach, D.; Müller, T.; Wolf, M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 2005, 11, 361–364. [Google Scholar] [CrossRef]
- Myers, S.; Bottolo, L.; Freeman, C.; McVean, G.; Donnelly, P. A Fine-Scale Map of Recombination Rates and Hotspots Across the Human Genome. Science 2005, 310, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Coop, G.; Wen, X.; Ober, C.; Pritchard, J.K.; Przeworski, M. High-Resolution Mapping of Crossovers Reveals Extensive Variation in Fine-Scale Recombination Patterns Among Humans. Science 2008, 319, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Variation in recombination frequency and distribution across eukaryotes: Patterns and processes. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160455. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Steenkamp, E.T.; Coetzee, M.P.A.; Wingfield, M.J.; Wingfield, B.D. Concerted Evolution in the Ribosomal RNA Cistron. PLoS ONE 2013, 8, e59355. [Google Scholar] [CrossRef]
- Galtier, N. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 2003, 19, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Mugal, C.F.; Weber, C.C.; Ellegren, H. GC-biased gene conversion links the recombination landscape and demography to genomic base composition GC-biased gene conversion drives genomic base composition across a wide range of species. BioEssays 2015, 37, 1317–1326. [Google Scholar] [CrossRef]
- Fromm, L.; Falk, S.; Flemming, D.; Schuller, J.M.; Thoms, M.; Conti, E.; Hurt, E. Reconstitution of the complete pathway of ITS2 processing at the pre-ribosome. Nat. Commun. 2017, 8, 1787. [Google Scholar] [CrossRef]
- Higgs, P.G. RNA secondary structure: Physical and computational aspects. Q. Rev. Biophys. 2000, 33, 199–253. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.; Murphy, R.W. Recent trends in molecular phylogenetic analysis: Where to next? J. Hered. 2011, 102, 130–138. [Google Scholar] [CrossRef]
- Kainer, D.; Lanfear, R. The Effects of Partitioning on Phylogenetic Inference. Mol. Biol. Evol. 2015, 32, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.M.; Holland, B.R. Comparing partitioned models to mixture models: Do information criteria apply? Syst. Biol. 2022, 71, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Rota, J.; Malm, T.; Chazot, N.; Peña, C.; Wahlberg, N. A simple method for data partitioning based on relative evolutionary rates. PeerJ 2018, 6, e5498. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Fan, Y.; Wu, R.; Chen, M.-H.; Kuo, L.; Lewis, P.O. Choosing among Partition Models in Bayesian Phylogenetics. Mol. Biol. Evol. 2011, 28, 523–532. [Google Scholar] [CrossRef]
- Cao, R.X.; Tong, S.Y.; Luan, T.J.; Zheng, H.Y.; Zhang, W. Compensatory base changes and varying phylogenetic effects on an-giosperm ITS2 genetic distances. Plants 2022, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Li, X.X.; Wang, D. New Insights into the Backbone Phylogeny and Character Evolution of Corydalis (Papaveraceae) Based on Plastome Data. Front. Plant Sci. 2022, 13, 926574. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccha-ride and polyphenol components. Plant Mol. Biol. Report. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F.Q. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Selig, C.; Wolf, M.; Müller, T.; Dandekar, T.; Schultz, J. The ITS2 Database II: Homology modelling RNA structure for molecular systematics. Nucleic Acids Res. 2008, 36, D377–D380. [Google Scholar] [CrossRef]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. 4SALE—A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 2006, 7, 498. [Google Scholar] [CrossRef]
- Allen, J.E.; Whelan, S. Assessing the State of Substitution Models Describing Noncoding RNA Evolution. Genome Biol. Evol. 2014, 6, 65–75. [Google Scholar] [CrossRef]
- Savill, N.J.; Hoyle, D.C.; Higgs, P.G. RNA sequence evolution with secondary structure constraints: Comparison of substitu-tion rate models using maximum-likelihood methods. Genetics 2001, 157, 399–411. [Google Scholar] [CrossRef]
- Sueoka, N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc. Natl. Acad. Sci. USA 1962, 48, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11 molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xian, Q.; Wang, S.; Liu, Y.; Kan, S.; Zhang, W. Structure-Based GC Investigation Sheds New Light on ITS2 Evolution in Corydalis Species. Int. J. Mol. Sci. 2023, 24, 7716. https://doi.org/10.3390/ijms24097716
Xian Q, Wang S, Liu Y, Kan S, Zhang W. Structure-Based GC Investigation Sheds New Light on ITS2 Evolution in Corydalis Species. International Journal of Molecular Sciences. 2023; 24(9):7716. https://doi.org/10.3390/ijms24097716
Chicago/Turabian StyleXian, Qing, Suyin Wang, Yanyan Liu, Shenglong Kan, and Wei Zhang. 2023. "Structure-Based GC Investigation Sheds New Light on ITS2 Evolution in Corydalis Species" International Journal of Molecular Sciences 24, no. 9: 7716. https://doi.org/10.3390/ijms24097716