
The Contribution of the Nrf2/ARE System to Mechanotransduction in Musculoskeletal and Periodontal Tissues

 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanobiology and the Nrf2/ARE System in Striated Muscles
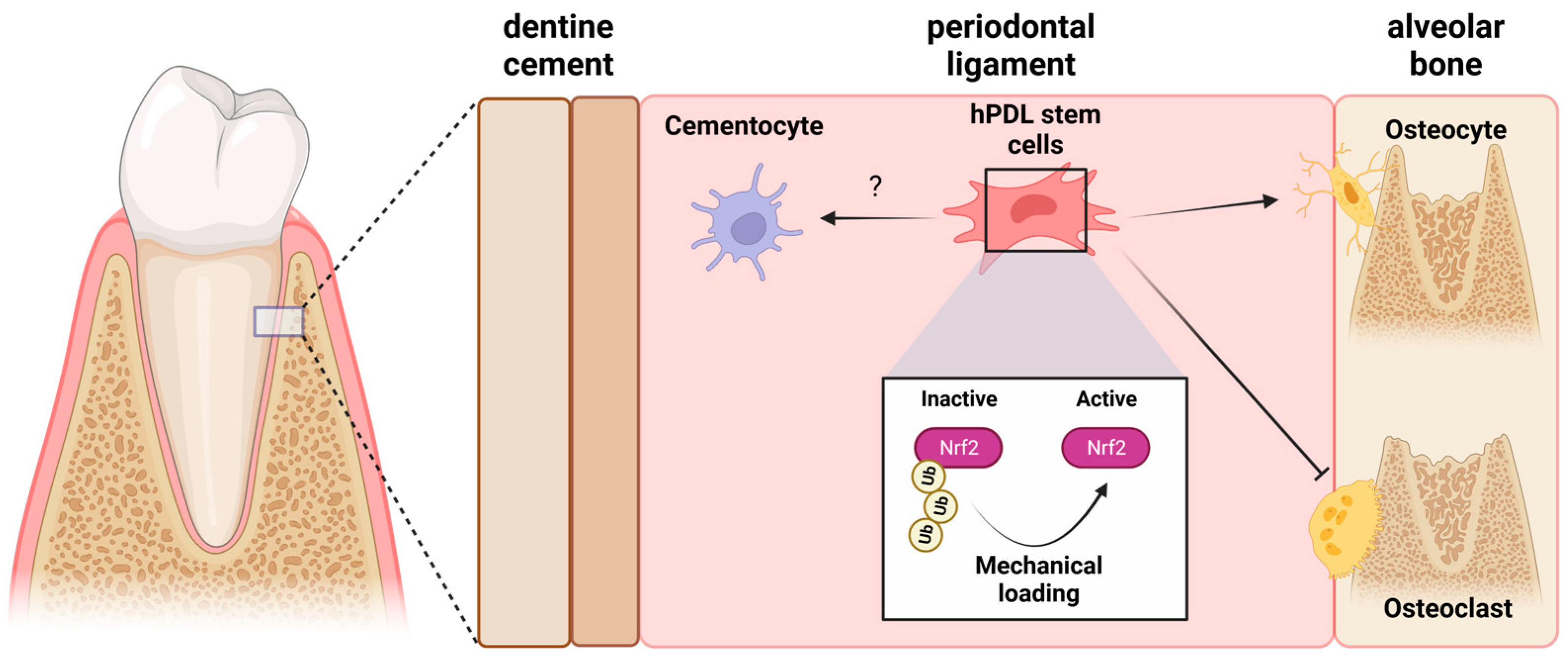
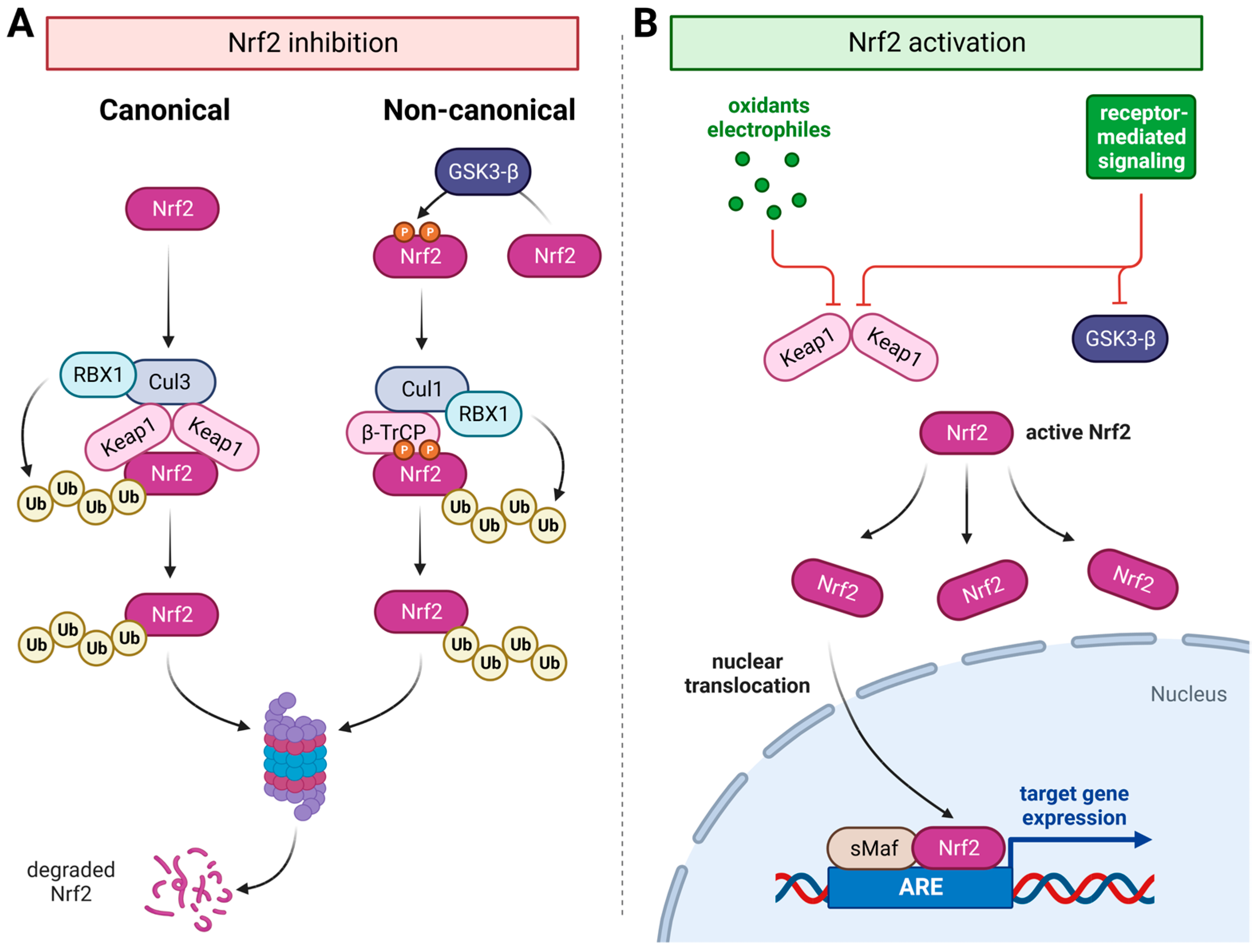
3. Mechanobiology and the Nrf2/ARE System in Periodontal Alveolar Remodeling
4. Mechanobiology and the Nrf2/ARE System in Bones
5. Mechanobiology and the Nrf2/ARE System in Tendons
6. Mechanobiology and the Nrf2-Keap1 System in Cartilage
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Legate, K.R.; Montanez, E.; Kudlacek, O.; Fassler, R. Ilk, pinch and parvin: The tipp of integrin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol. Rev. 2004, 84, 649–698. [Google Scholar] [CrossRef] [PubMed]
- Pingel, J.; Suhr, F. Are mechanically sensitive regulators involved in the function and (patho) physiology of cerebral palsy-related contractures? J. Muscle Res. Cell Motil. 2017, 38, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Mathes, S.; Vanmunster, M.; Bloch, W.; Suhr, F. Evidence for skeletal muscle fiber type-specific expressions of mechanosensors. Cell. Mol. Life Sci. CMLS 2019, 76, 2987–3004. [Google Scholar] [CrossRef]
- Vanmunster, M.; Rojo Garcia, A.V.; Pacolet, A.; Dalle, S.; Koppo, K.; Jonkers, I.; Lories, R.; Suhr, F. Mechanosensors control skeletal muscle mass, molecular clocks, and metabolism. Cell. Mol. Life Sci. CMLS 2022, 79, 321. [Google Scholar] [CrossRef]
- Andresen, B.; de Marees, M.; Schiffer, T.; Bloch, W.; Suhr, F. Skeletal muscle fiber type-specific expressions of mechanosensors integrin-linked kinase, talin, and vinculin and their modulation by loading and environmental conditions in humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22458. [Google Scholar] [CrossRef]
- Langberg, H.; Skovgaard, D.; Petersen, L.J.; Bulow, J.; Kjaer, M. Type i collagen synthesis and degradation in peritendinous tissue after exercise determined by microdialysis in humans. J. Physiol. 1999, 521 Pt 1, 299–306. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H.; Ursini, F. Homeostatic control of redox status and health. IUBMB Life 2022, 74, 24–28. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The golden mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Concept and some practical aspects. Antioxidants 2020, 9, 852. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ros) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, S.; Pinkus, R.; Daniel, V. Induction of ap-1 (fos/jun) by chemical agents mediates activation of glutathione s-transferase and quinone reductase gene expression. Oncogene 1994, 9, 565–571. [Google Scholar]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione s-transferase ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef]
- Rushmore, T.H.; King, R.G.; Paulson, K.E.; Pickett, C.B. Regulation of glutathione s-transferase ya subunit gene expression: Identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc. Natl. Acad. Sci. USA 1990, 87, 3826–3830. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by nrf2 through binding to the amino-terminal neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. Scf/beta-trcp promotes glycogen synthase kinase 3-dependent degradation of the nrf2 transcription factor in a keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Kweider, N.; Fragoulis, A.; Rosen, C.; Pecks, U.; Rath, W.; Pufe, T.; Wruck, C.J. Interplay between vascular endothelial growth factor (vegf) and nuclear factor erythroid 2-related factor-2 (nrf2): Implications for preeclampsia. J. Biol. Chem. 2011, 286, 42863–42872. [Google Scholar] [CrossRef]
- Reiss, L.K.; Fragoulis, A.; Siegl, S.; Platen, C.; Kan, Y.W.; Nautiyal, J.; Parker, M.; Pufe, T.; Uhlig, U.; Martin, C.; et al. Interplay between nuclear factor erythroid 2-related factor 2 and amphiregulin during mechanical ventilation. Am. J. Respir. Cell Mol. Biol. 2014, 51, 668–677. [Google Scholar] [CrossRef]
- Furusawa, Y.; Uruno, A.; Yagishita, Y.; Higashi, C.; Yamamoto, M. Nrf2 induces fibroblast growth factor 21 in diabetic mice. Genes Cells Devoted Mol. Cell. Mech. 2014, 19, 864–878. [Google Scholar] [CrossRef]
- Lastra, D.; Escoll, M.; Cuadrado, A. Transcription factor nrf2 participates in cell cycle progression at the level of g1/s and mitotic checkpoints. Antioxidants 2022, 11, 946. [Google Scholar] [CrossRef]
- Zou, Y.; Hu, M.; Lee, J.; Nambiar, S.M.; Garcia, V.; Bao, Q.; Chan, J.Y.; Dai, G. Nrf2 is essential for timely m phase entry of replicating hepatocytes during liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G262–G268. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, R.; Vogel, A.; Schuett, J.; Lupke, M.; Spieker, S.M.; Kettern, N.; Hildt, E.; Melter, M.; Weiss, T.S. Nrf2 activates augmenter of liver regeneration (alr) via antioxidant response element and links oxidative stress to liver regeneration. Mol. Med. 2013, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, A.; Schenkel, J.; Herzog, M.; Schellenberg, T.; Jahr, H.; Pufe, T.; Trautwein, C.; Kensler, T.W.; Streetz, K.L.; Wruck, C.J. Nrf2 ameliorates ddc-induced sclerosing cholangitis and biliary fibrosis and improves the regenerative capacity of the liver. Toxicol. Sci. Off. J. Soc. Toxicol. 2019, 169, 485–498. [Google Scholar] [CrossRef]
- Fragoulis, A.; Schenkel, J.; Schroder, N.; Brandt, E.F.; Weiand, M.; Neu, T.; Ramadori, P.; Caspers, T.; Kant, S.; Pufe, T.; et al. Nrf2 induces malignant transformation of hepatic progenitor cells by inducing beta-catenin expression. Redox Biol. 2022, 57, 102453. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Traustadottir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. Bone 2014, 63, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, O.; Diermeier, S.; Larsson, L. Weak by the machines: Muscle motor protein dysfunction-a side effect of intensive care unit treatment. Acta Physiol. 2018, 222, e12885. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Murgia, M.; Ciciliot, S. Signatures of muscle disuse in spaceflight and bed rest revealed by single muscle fiber proteomics. PNAS Nexus 2022, 1, pgac086. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Samarel, A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2291–H2301. [Google Scholar] [CrossRef]
- Greiwe, L.; Vinck, M.; Suhr, F. The muscle contraction mode determines lymphangiogenesis differentially in rat skeletal and cardiac muscles by modifying local lymphatic extracellular matrix microenvironments. Acta Physiol. 2016, 217, 61–79. [Google Scholar] [CrossRef]
- Thievessen, I.; Suhr, F.; Vergarajauregui, S.; Bottcher, R.T.; Brixius, K.; Rosenberger, G.; Dewald, O.; Fleischmann, B.K.; Ghanem, A.; Kruger, M.; et al. The focal adhesion protein beta-parvin controls cardiomyocyte shape and sarcomere assembly in response to mechanical load. Curr. Biol. CB 2022, 32, 3033–3047.e9. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Huang, D.D.; Fan, S.D.; Chen, X.Y.; Yan, X.L.; Zhang, X.Z.; Ma, B.W.; Yu, D.Y.; Xiao, W.Y.; Zhuang, C.L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef]
- Narasimhan, M.; Hong, J.; Atieno, N.; Muthusamy, V.R.; Davidson, C.J.; Abu-Rmaileh, N.; Richardson, R.S.; Gomes, A.V.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency promotes apoptosis and impairs pax7/myod expression in aging skeletal muscle cells. Free Radic. Biol. Med. 2014, 71, 402–414. [Google Scholar] [CrossRef]
- Al-Sawaf, O.; Fragoulis, A.; Rosen, C.; Keimes, N.; Liehn, E.A.; Holzle, F.; Kan, Y.W.; Pufe, T.; Sonmez, T.T.; Wruck, C.J. Nrf2 augments skeletal muscle regeneration after ischaemia-reperfusion injury. J. Pathol. 2014, 234, 538–547. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The two faces of reactive oxygen species in cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Davies, K.J. Adaptive homeostasis. Mol. Asp. Med. 2016, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic nrf2 activators: Can the clinician’s expectation be matched by the reality? Oxidative Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef]
- Li, W.; Trieu, J.; Blazev, R.; Parker, B.L.; Murphy, K.T.; Swiderski, K.; Lynch, G.S. Sulforaphane attenuates cancer cell-induced atrophy of c2c12 myotubes. Am. J. Physiol. Cell Physiol. 2023, 324, C205–C221. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Kudo, T.; Fujita, R.; Fujita, S.I.; Tsubouchi, H.; Fuseya, S.; Suzuki, R.; Hamada, M.; Okada, R.; Muratani, M.; et al. Nuclear factor e2-related factor 2 (nrf2) deficiency accelerates fast fibre type transition in soleus muscle during space flight. Commun. Biol. 2021, 4, 787. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Seymour, E.M.; Bennink, M.R.; Bolling, S.F. Diet-relevant phytochemical intake affects the cardiac ahr and nrf2 transcriptome and reduces heart failure in hypertensive rats. J. Nutr. Biochem. 2013, 24, 1580–1586. [Google Scholar] [CrossRef]
- Xue, M.; Momiji, H.; Rabbani, N.; Barker, G.; Bretschneider, T.; Shmygol, A.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of nrf2 mediate the antioxidant response element cytoprotective transcriptional response. Antioxid. Redox Signal. 2015, 23, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of nf-e2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Silvério, K.G.; Rodrigues, T.L.; Coletta, R.D.; Benevides, L.; Da Silva, J.S.; Casati, M.Z.; Sallum, E.A.; Nociti, F.H., Jr. Mesenchymal stem cell properties of periodontal ligament cells from deciduous and permanent teeth. J. Periodontol. 2010, 81, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Chu, A.K.Y.; Ono, N.; Welch, J.D.; Ono, W. Single-cell transcriptomic analysis reveals developmental relationships and specific markers of mouse periodontium cellular subsets. Front. Dent. Med. 2021, 2, 679937. [Google Scholar] [CrossRef]
- Maeda, H.; Wada, N.; Tomokiyo, A.; Monnouchi, S.; Akamine, A. Prospective potency of tgf-β1 on maintenance and regeneration of periodontal tissue. Int. Rev. Cell Mol. Biol. 2013, 304, 283–367. [Google Scholar] [PubMed]
- Meikle, M.C. The tissue, cellular, and molecular regulation of orthodontic tooth movement: 100 years after carl sandstedt. Eur. J. Orthod. 2006, 28, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Davidovitch, Z. Cellular, molecular, and tissue-level reactions to orthodontic force. American journal of orthodontics and dentofacial orthopedics: Official publication of the American Association of Orthodontists, its constituent societies, and the American Board of Orthodontics. Am. J. Orthod. Dentofac. Orthop. 2006, 129, 469.e1–469.e32. [Google Scholar]
- Aveic, S.; Craveiro, R.B.; Wolf, M.; Fischer, H. Current trends in in vitro modeling to mimic cellular crosstalk in periodontal tissue. Adv. Healthc. Mater. 2021, 10, 2001269. [Google Scholar] [CrossRef]
- Murray, D.; Whyte, A. Dental panoramic tomography: What the general radiologist needs to know. Clin. Radiol. 2002, 57, 1–7. [Google Scholar] [CrossRef]
- Craveiro, R.B.; Florea, A.; Niederau, C.; Brenji, S.; Kiessling, F.; Sahnoun, S.E.M.; Morgenroth, A.; Mottaghy, F.M.; Wolf, M. [(68)ga]ga-pentixafor and sodium [(18)f]fluoride pet can non-invasively identify and monitor the dynamics of orthodontic tooth movement in mouse model. Cells 2022, 11, 2949. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.E.; Craveiro, R.B.; Niederau, C.; Malyaran, H.; Neuss, S.; Jankowski, J.; Wolf, M. Mechanical compression by simulating orthodontic tooth movement in an in vitro model modulates phosphorylation of akt and mapks via tlr4 in human periodontal ligament cells. Int. J. Mol. Sci. 2022, 23, 8062. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Davidovitch, Z. The effect of drugs on orthodontic tooth movement. Orthod. Craniofacial Res. 2006, 9, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Davidovitch, Z. On a path to unfolding the biological mechanisms of orthodontic tooth movement. J. Dent. Res. 2009, 88, 597–608. [Google Scholar] [CrossRef]
- Sokos, D.; Everts, V.; de Vries, T.J. Role of periodontal ligament fibroblasts in osteoclastogenesis: A review. J. Periodontal Res. 2015, 50, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, C.; Yang, Y. Effects of mechanical forces on osteogenesis and osteoclastogenesis in human periodontal ligament fibroblasts: A systematic review of in vitro studies. Bone Jt. Res. 2019, 8, 19–31. [Google Scholar] [CrossRef]
- Yamaguchi, M. Rank/rankl/opg during orthodontic tooth movement. Orthod Craniofac Res 2009, 12, 113–119. [Google Scholar] [CrossRef]
- Garlet, T.P.; Coelho, U.; Silva, J.S.; Garlet, G.P. Cytokine expression pattern in compression and tension sides of the periodontal ligament during orthodontic tooth movement in humans. Eur. J. Oral Sci. 2007, 115, 355–362. [Google Scholar] [CrossRef]
- Xing, Y.; Zhang, Y.; Wu, X.; Zhao, B.; Ji, Y.; Xu, X. A comprehensive study on donor-matched comparisons of three types of mesenchymal stem cells-containing cells from human dental tissue. J. Periodontal Res. 2019, 54, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.M.; Miura, M.; Gronthos, S.; Bartold, P.M.; Batouli, S.; Brahim, J.; Young, M.; Robey, P.G.; Wang, C.Y.; Shi, S. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 2004, 364, 149–155. [Google Scholar] [CrossRef]
- Li, Y.; Jacox, L.A.; Little, S.H.; Ko, C.C. Orthodontic tooth movement: The biology and clinical implications. Kaohsiung J. Med. Sci. 2018, 34, 207–214. [Google Scholar] [CrossRef]
- Linkous, E.R.; Trojan, T.M.; Harris, E.F. External apical root resorption and vectors of orthodontic tooth movement. American journal of orthodontics and dentofacial orthopedics: Official publication of the American Association of Orthodontists, its constituent societies, and the American Board of Orthodontics. Am. J. Orthod. Dentofac. Orthop. 2020, 158, 700–709. [Google Scholar]
- Jager, F.; Mah, J.K.; Bumann, A. Peridental bone changes after orthodontic tooth movement with fixed appliances: A cone-beam computed tomographic study. Angle Orthod. 2017, 87, 672–680. [Google Scholar] [CrossRef]
- Artun, J.; Van ’t Hullenaar, R.; Doppel, D.; Kuijpers-Jagtman, A.M. Identification of orthodontic patients at risk of severe apical root resorption. Am. J. Orthod. Dentofac. Orthop. 2009, 135, 448–455. [Google Scholar] [CrossRef]
- Li, S.; Zhang, H.; Li, S.; Yang, Y.; Huo, B.; Zhang, D. Connexin 43 and erk regulate tension-induced signal transduction in human periodontal ligament fibroblasts. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2015, 33, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Zhao, Y.; Liu, H.; Li, Z.; Chen, S.; Liu, D. Nrf2 activation is involved in osteogenic differentiation of periodontal ligament stem cells under cyclic mechanical stretch. Exp. Cell Res. 2021, 403, 112598. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Li, Z.; Liu, H.; Chen, S.; Liu, D. Nrf2 activation is involved in cyclic mechanical stress-stimulated osteogenic differentiation in periodontal ligament stem cells via pi3k/akt signaling and ho1-sod2 interaction. Front. Cell Dev. Biol. 2021, 9, 816000. [Google Scholar] [CrossRef]
- Xi, X.; Li, Z.X.; Zhao, Y.; Liu, H.; Chen, S.; Liu, D.X. N-acetylcysteine promotes cyclic mechanical stress-induced osteogenic differentiation of periodontal ligament stem cells by down-regulating nrf2 expression. J. Dent. Sci. 2022, 17, 750–762. [Google Scholar] [CrossRef]
- Grabowski, P. Physiology of bone. Endocr. Dev. 2015, 28, 33–55. [Google Scholar]
- Malone, A.M.; Anderson, C.T.; Tummala, P.; Kwon, R.Y.; Johnston, T.R.; Stearns, T.; Jacobs, C.R. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 13325–13330. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Bakker, A.D.; Bacabac, R.G.; Vatsa, A.; Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 2013, 54, 182–190. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell. And more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- Feller, L.; Khammissa, R.A.; Schechter, I.; Moodley, A.; Thomadakis, G.; Lemmer, J. Periodontal biological events associated with orthodontic tooth movement: The biomechanics of the cytoskeleton and the extracellular matrix. Sci. World J. 2015, 2015, 894123. [Google Scholar] [CrossRef]
- Qin, L.; Liu, W.; Cao, H.; Xiao, G. Molecular mechanosensors in osteocytes. Bone Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Sanchez-de-Diego, C.; Pedrazza, L.; Pimenta-Lopes, C.; Martinez-Martinez, A.; Dahdah, N.; Valer, J.A.; Garcia-Roves, P.; Rosa, J.L.; Ventura, F. Nrf2 function in osteocytes is required for bone homeostasis and drives osteocytic gene expression. Redox Biol. 2021, 40, 101845. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E.; Suzuki, T.; Morita, M.; Taguchi, K.; Tsuchida, K.; Motohashi, H.; Doita, M.; Yamamoto, M. Hyperactivation of nrf2 leads to hypoplasia of bone in vivo. Genes Cells Devoted Mol. Cell. Mech. 2018, 23, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.G.; Cregor, M.; McAndrews, K.; Morales, C.C.; McCabe, L.D.; McCabe, G.P.; Peacock, M.; Burr, D.; Weaver, C.; Bellido, T. Nrf2 regulates mass accrual and the antioxidant endogenous response in bone differently depending on the sex and age. PLoS ONE 2017, 12, e0171161. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Ferrandiz, M.L.; Brines, R.; Guede, D.; Cuadrado, A.; Alcaraz, M.J. Effects of nrf2 deficiency on bone microarchitecture in an experimental model of osteoporosis. Oxidative Med. Cell. Longev. 2014, 2014, 726590. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Gonzalez, J.A.H.; Beckmann, R.; Weiler, M.; Pahlavani, H.; Saldivar, M.C.; Szymanski, K.; Rosenhain, S.; Fragoulis, A.; Leeflang, S.; et al. Nuclear factor erythroid 2-related factor 2 (nrf2) deficiency causes age-dependent progression of female osteoporosis. BMC Musculoskelet. Disord. 2022, 23, 1015. [Google Scholar] [CrossRef]
- Kubo, Y.; Wruck, C.J.; Fragoulis, A.; Drescher, W.; Pape, H.C.; Lichte, P.; Fischer, H.; Tohidnezhad, M.; Hildebrand, F.; Pufe, T.; et al. Role of nrf2 in fracture healing: Clinical aspects of oxidative stress. Calcif. Tissue Int. 2019, 105, 341–352. [Google Scholar] [CrossRef]
- Lippross, S.; Beckmann, R.; Streubesand, N.; Ayub, F.; Tohidnezhad, M.; Campbell, G.; Kan, Y.W.; Horst, F.; Sonmez, T.T.; Varoga, D.; et al. Nrf2 deficiency impairs fracture healing in mice. Calcif. Tissue Int. 2014, 95, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhu, X.; Wei, A.; Chen, F.; Gao, Q.; Lu, K.; Jiang, Q.; Cao, W. Nrf2 epigenetic derepression induced by running exercise protects against osteoporosis. Bone Res. 2021, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Yelin, E.; Weinstein, S.; King, T. The burden of musculoskeletal diseases in the united states. Semin. Arthritis Rheum. 2016, 46, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Murrell, G.A. The basic science of tendinopathy. Clin. Orthop. Relat. Res. 2008, 466, 1528–1538. [Google Scholar] [CrossRef]
- Gardner, K.; Arnoczky, S.P.; Caballero, O.; Lavagnino, M. The effect of stress-deprivation and cyclic loading on the timp/mmp ratio in tendon cells: An in vitro experimental study. Disabil. Rehabil. 2008, 30, 1523–1529. [Google Scholar] [CrossRef]
- Koch, S.; Tillmann, B. The distal tendon of the biceps brachii. Structure and clinical correlations. Ann. Anat. Anat. Anz. Off. Organ Anat. Ges. 1995, 177, 467–474. [Google Scholar]
- Ralphs, J.R.; Benjamin, M.; Waggett, A.D.; Russell, D.C.; Messner, K.; Gao, J. Regional differences in cell shape and gap junction expression in rat achilles tendon: Relation to fibrocartilage differentiation. J. Anat. 1998, 193 Pt 2, 215–222. [Google Scholar] [CrossRef]
- Buckley, M.R.; Evans, E.B.; Matuszewski, P.E.; Chen, Y.L.; Satchel, L.N.; Elliott, D.M.; Soslowsky, L.J.; Dodge, G.R. Distributions of types i, ii and iii collagen by region in the human supraspinatus tendon. Connect. Tissue Res. 2013, 54, 374–379. [Google Scholar] [CrossRef]
- Petersen, W.; Hohmann, G.; Pufe, T.; Tsokos, M.; Zantop, T.; Paulsen, F.; Tillmann, B. Structure of the human tibialis posterior tendon. Arch. Orthop. Trauma Surg. 2004, 124, 237–242. [Google Scholar] [CrossRef]
- Pufe, T.; Petersen, W.J.; Mentlein, R.; Tillmann, B.N. The role of vasculature and angiogenesis for the pathogenesis of degenerative tendons disease. Scand. J. Med. Sci. Sport. 2005, 15, 211–222. [Google Scholar] [CrossRef]
- Benjamin, M.; Ralphs, J.R. Biology of fibrocartilage cells. Int. Rev. Cytol. 2004, 233, 1–45. [Google Scholar]
- Abrahamsson, S.O.; Lundborg, G.; Lohmander, L.S. Segmental variation in microstructure, matrix synthesis and cell proliferation in rabbit flexor tendon. Scand. J. Plast. Reconstr. Surg. Hand Surg. 1989, 23, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Takimoto, A.; Akiyama, H.; Kist, R.; Scherer, G.; Nakamura, T.; Hiraki, Y.; Shukunami, C. Scx+/sox9+ progenitors contribute to the establishment of the junction between cartilage and tendon/ligament. Development 2013, 140, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into tgf-beta/smad signaling in tissue fibrosis. Chem.-Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, M.; Suzuki, K.; Yamaguchi, A. Effect of mechanical tension on fibroblast transcriptome profile and regulatory mechanisms of myocardial collagen turnover. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2023, 37, e22841. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.R.; Evanko, S.P.; Vogel, K.G. Mechanical loading and tgf-beta regulate proteoglycan synthesis in tendon. Arch. Biochem. Biophys. 1997, 342, 203–211. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Wang, D.; Pun, C.C.M.; Huang, S.; Tang, T.C.M.; Ho, K.K.W.; Rothrauff, B.B.; Yung, P.S.H.; Blocki, A.M.; Ker, E.D.F.; Tuan, R.S. Tendon-derived extracellular matrix induces mesenchymal stem cell tenogenesis via an integrin/transforming growth factor-beta crosstalk-mediated mechanism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 8172–8186. [Google Scholar]
- Wang, X.; Liu, S.; Yu, T.; An, S.; Deng, R.; Tan, X.; Crane, J.; Zhang, W.; Pan, D.; Wan, M.; et al. Inhibition of integrin alphavbeta6 activation of tgf-beta attenuates tendinopathy. Adv. Sci. 2022, 9, e2104469. [Google Scholar] [CrossRef]
- de Mos, M.; Koevoet, W.; van Schie, H.T.; Kops, N.; Jahr, H.; Verhaar, J.A.; van Osch, G.J. In vitro model to study chondrogenic differentiation in tendinopathy. Am. J. Sport. Med. 2009, 37, 1214–1222. [Google Scholar] [CrossRef]
- Scott, A.; Lian, O.; Roberts, C.R.; Cook, J.L.; Handley, C.J.; Bahr, R.; Samiric, T.; Ilic, M.Z.; Parkinson, J.; Hart, D.A.; et al. Increased versican content is associated with tendinosis pathology in the patellar tendon of athletes with jumper’s knee. Scand. J. Med. Sci. Sport. 2008, 18, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Tohidnezhad, M.; Zander, J.; Slowik, A.; Kubo, Y.; Dursun, G.; Willenberg, W.; Zendedel, A.; Kweider, N.; Stoffel, M.; Pufe, T. Impact of uniaxial stretching on both gliding and traction areas of tendon explants in a novel bioreactor. Int. J. Mol. Sci. 2020, 21, 2925. [Google Scholar] [CrossRef] [PubMed]
- Docheva, D.; Popov, C.; Alberton, P.; Aszodi, A. Integrin signaling in skeletal development and function. Birth Defects Res. Part C Embryo Today Rev. 2014, 102, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, R.; Hojabrpour, P.; Eltit, F.; McDonald, P.C.; Dedhar, S.; McCormack, R.G.; Duronio, V.; Jafarnejad, S.M.; Scott, A. Beta1 integrin, ilk and mtor regulate collagen synthesis in mechanically loaded tendon cells. Sci. Rep. 2020, 10, 12644. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hasan, H.; Anderson, D.E.; Lee, W. The role of mechanically-activated ion channels piezo1, piezo2, and trpv4 in chondrocyte mechanotransduction and mechano-therapeutics for osteoarthritis. Front. Cell Dev. Biol. 2022, 10, 885224. [Google Scholar] [CrossRef]
- Lewis, A.H.; Grandl, J. Stretch and poke stimulation for characterizing mechanically activated ion channels. Methods Enzymol. 2021, 654, 225–253. [Google Scholar]
- Poole, K. The diverse physiological functions of mechanically activated ion channels in mammals. Annu. Rev. Physiol. 2022, 84, 307–329. [Google Scholar] [CrossRef]
- Coste, B. piezo proteins form a new class of mechanically activated ion channels. Med. Sci. M/S 2012, 28, 1056–1057. [Google Scholar]
- Wu, J.; Lewis, A.H.; Grandl, J. Touch, tension, and transduction-the function and regulation of piezo ion channels. Trends Biochem. Sci. 2017, 42, 57–71. [Google Scholar] [CrossRef]
- Servin-Vences, M.R.; Moroni, M.; Lewin, G.R.; Poole, K. Direct measurement of trpv4 and piezo1 activity reveals multiple mechanotransduction pathways in chondrocytes. eLife 2017, 6, e21074. [Google Scholar] [CrossRef]
- Gracey, E.; Burssens, A.; Cambre, I.; Schett, G.; Lories, R.; McInnes, I.B.; Asahara, H.; Elewaut, D. Tendon and ligament mechanical loading in the pathogenesis of inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.M.; Hart, D.A. The interface of mechanical loading and biological variables as they pertain to the development of tendinosis. J. Musculoskelet. Neuronal Interact. 2011, 11, 94–105. [Google Scholar] [PubMed]
- Maeda, T.; Sakabe, T.; Sunaga, A.; Sakai, K.; Rivera, A.L.; Keene, D.R.; Sasaki, T.; Stavnezer, E.; Iannotti, J.; Schweitzer, R.; et al. Conversion of mechanical force into tgf-beta-mediated biochemical signals. Curr. Biol. CB 2011, 21, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, D.; Itoigawa, Y.; Nojiri, H.; Sano, H.; Itoi, E.; Saijo, Y.; Kaneko, K.; Shimizu, T. Contribution of oxidative stress to the degeneration of rotator cuff entheses. J. Shoulder Elb. Surg. 2014, 23, 628–635. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, N.; Wang, X.; Chen, W.; Zhang, Q. Trpv4 and piezo channels mediate the mechanosensing of chondrocytes to the biomechanical microenvironment. Membranes 2022, 12, 237. [Google Scholar] [CrossRef]
- Wang, B.; Ke, W.; Wang, K.; Li, G.; Ma, L.; Lu, S.; Xiang, Q.; Liao, Z.; Luo, R.; Song, Y.; et al. Mechanosensitive ion channel piezo1 activated by matrix stiffness regulates oxidative stress-induced senescence and apoptosis in human intervertebral disc degeneration. Oxidative Med. Cell. Longev. 2021, 2021, 8884922. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, P.; Li, C.; Lu, Y.; Kou, Y.; Liu, H.; Guo, J.; Li, M. Periostin regulates lps-induced apoptosis via nrf2/ho-1 pathway in periodontal ligament fibroblasts. Oral Dis. 2022. [Google Scholar] [CrossRef]
- Chung, J.H.; Kim, Y.S.; Noh, K.; Lee, Y.M.; Chang, S.W.; Kim, E.C. Deferoxamine promotes osteoblastic differentiation in human periodontal ligament cells via the nuclear factor erythroid 2-related factor-mediated antioxidant signaling pathway. J. Periodontal Res. 2014, 49, 563–573. [Google Scholar] [CrossRef]
- Lu, K.; Zhou, M.; Wang, L.; Wang, Y.; Tang, H.; He, G.; Wang, H.; Tang, C.; He, J.; Wang, W.; et al. N-acetyl-l-cysteine facilitates tendon repair and promotes the tenogenic differentiation of tendon stem/progenitor cells by enhancing the integrin alpha5/beta1/pi3k/akt signaling. BMC Mol. Cell Biol. 2023, 24, 1. [Google Scholar] [CrossRef]
- Beckmann, R.; Houben, A.; Tohidnezhad, M.; Kweider, N.; Fragoulis, A.; Wruck, C.J.; Brandenburg, L.O.; Hermanns-Sachweh, B.; Goldring, M.B.; Pufe, T.; et al. Mechanical forces induce changes in vegf and vegfr-1/sflt-1 expression in human chondrocytes. Int. J. Mol. Sci. 2014, 15, 15456–15474. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Hoffmann, B.; Goltz, K.; Schnakenberg, U.; Jahr, H.; Merkel, R.; Schulze-Tanzil, G.; Pufe, T.; Tohidnezhad, M. Different frequency of cyclic tensile strain relates to anabolic/catabolic conditions consistent with immunohistochemical staining intensity in tenocytes. Int. J. Mol. Sci. 2020, 21, 1082. [Google Scholar] [CrossRef]
- Popov, C.; Burggraf, M.; Kreja, L.; Ignatius, A.; Schieker, M.; Docheva, D. Mechanical stimulation of human tendon stem/progenitor cells results in upregulation of matrix proteins, integrins and mmps, and activation of p38 and erk1/2 kinases. BMC Mol. Biol. 2015, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Kweider, N.; Rath, W.; Huppertz, B.; Wruck, C.J.; Jumakuliev, G.; Beckmann, R.; Pufe, T.; Kadyrov, M. Pp015. Differential expression of nrf2 and vegf in human placental beds from normal and pregnancies complicated with preeclampsia and iugr. Pregnancy Hypertens. 2012, 2, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Tohidnezhad, M.; Varoga, D.; Wruck, C.J.; Brandenburg, L.O.; Seekamp, A.; Shakibaei, M.; Sonmez, T.T.; Pufe, T.; Lippross, S. Platelet-released growth factors can accelerate tenocyte proliferation and activate the anti-oxidant response element. Histochem. Cell Biol. 2011, 135, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Tohidnezhad, M.; Wruck, C.J.; Slowik, A.; Kweider, N.; Beckmann, R.; Bayer, A.; Houben, A.; Brandenburg, L.O.; Varoga, D.; Sonmez, T.T.; et al. Role of platelet-released growth factors in detoxification of reactive oxygen species in osteoblasts. Bone 2014, 65, 9–17. [Google Scholar] [CrossRef]
- Gilbert, S.J.; Blain, E.J. Chapter 4-cartilage mechanobiology: How chondrocytes respond to mechanical load. In Mechanobiology in Health and Disease; Verbruggen, S.W., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 99–126. [Google Scholar]
- Hodgkinson, T.; Amado, I.N.; O’Brien, F.J.; Kennedy, O.D. The role of mechanobiology in bone and cartilage model systems in characterizing initiation and progression of osteoarthritis. APL Bioeng. 2022, 6, 011501. [Google Scholar] [CrossRef]
- Deng, R.; Hua, X.; Li, J.; Chi, W.; Zhang, Z.; Lu, F.; Zhang, L.; Pflugfelder, S.C.; Li, D.Q. Oxidative stress markers induced by hyperosmolarity in primary human corneal epithelial cells. PLoS ONE 2015, 10, e0126561. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Yang, K.; Guo, S.; Wang, J.; Feng, C.; Chen, H. Hyper-osmolarity environment-induced oxidative stress injury promotes nucleus pulposus cell senescence in vitro. Biosci. Rep. 2019, 39, BSR20191711. [Google Scholar] [CrossRef] [PubMed]
- Jahr, H.; Matta, C.; Mobasheri, A. Physicochemical and biomechanical stimuli in cell-based articular cartilage repair. Curr. Rheumatol. Rep. 2015, 17, 22. [Google Scholar] [CrossRef]
- Caron, M.M.; van der Windt, A.E.; Emans, P.J.; van Rhijn, L.W.; Jahr, H.; Welting, T.J. Osmolarity determines the in vitro chondrogenic differentiation capacity of progenitor cells via nuclear factor of activated t-cells 5. Bone 2013, 53, 94–102. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, A.E.; Haak, E.; Das, R.H.; Kops, N.; Welting, T.J.; Caron, M.M.; van Til, N.P.; Verhaar, J.A.; Weinans, H.; Jahr, H. Physiological tonicity improves human chondrogenic marker expression through nuclear factor of activated t-cells 5 in vitro. Arthritis Res. Ther. 2010, 12, R100. [Google Scholar] [CrossRef]
- Jahr, H.; van der Windt, A.E.; Timur, U.T.; Baart, E.B.; Lian, W.S.; Rolauffs, B.; Wang, F.S.; Pufe, T. Physosmotic induction of chondrogenic maturation is tgf-beta dependent and enhanced by calcineurin inhibitor fk506. Int. J. Mol. Sci. 2022, 23, 5110. [Google Scholar] [CrossRef]
- Tan Timur, U.; Caron, M.; van den Akker, G.; van der Windt, A.; Visser, J.; van Rhijn, L.; Weinans, H.; Welting, T.; Emans, P.; Jahr, H. Increased tgf-beta and bmp levels and improved chondrocyte-specific marker expression in vitro under cartilage-specific physiological osmolarity. Int. J. Mol. Sci. 2019, 20, 795. [Google Scholar] [CrossRef]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Investig. A J. Tech. Methods Pathol. 2016, 96, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits wnt-beta-catenin signalling through dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Korswagen, H.C. Regulation of the wnt/beta-catenin pathway by redox signaling. Dev. Cell 2006, 10, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Peng, X.; Chen, Y.; Tang, X.; Qin, Y.; He, M.; Chen, W.; Chen, H. Piezo1 alleviates acetaminophen-induced acute liver injury by activating nrf2 and reducing mitochondrial reactive oxygen species. Biochem. Biophys. Res. Commun. 2023, 652, 88–94. [Google Scholar] [CrossRef]
- Zhu, S.; Makosa, D.; Miller, B.; Griffin, T.M. Glutathione as a mediator of cartilage oxidative stress resistance and resilience during aging and osteoarthritis. Connect. Tissue Res. 2020, 61, 34–47. [Google Scholar] [CrossRef]
- Wruck, C.J.; Fragoulis, A.; Gurzynski, A.; Brandenburg, L.O.; Kan, Y.W.; Chan, K.; Hassenpflug, J.; Freitag-Wolf, S.; Varoga, D.; Lippross, S.; et al. Role of oxidative stress in rheumatoid arthritis: Insights from the nrf2-knockout mice. Ann. Rheum. Dis. 2011, 70, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Rellmann, Y.; Eidhof, E.; Dreier, R. Review: Er stress-induced cell death in osteoarthritic cartilage. Cell. Signal. 2021, 78, 109880. [Google Scholar] [CrossRef]
- Forman, H.J.; Augusto, O.; Brigelius-Flohe, R.; Dennery, P.A.; Kalyanaraman, B.; Ischiropoulos, H.; Mann, G.E.; Radi, R.; Roberts, L.J., 2nd; Vina, J.; et al. Even free radicals should follow some rules: A guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015, 78, 233–235. [Google Scholar] [CrossRef]
- Kadiiska, M.B.; Gladen, B.C.; Baird, D.D.; Dikalova, A.E.; Sohal, R.S.; Hatch, G.E.; Jones, D.P.; Mason, R.P.; Barrett, J.C. Biomarkers of oxidative stress study: Are plasma antioxidants markers of ccl(4) poisoning? Free Radic. Biol. Med. 2000, 28, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Kadiiska, M.B.; Gladen, B.C.; Baird, D.D.; Germolec, D.; Graham, L.B.; Parker, C.E.; Nyska, A.; Wachsman, J.T.; Ames, B.N.; Basu, S.; et al. ; et al. Biomarkers of oxidative stress study ii: Are oxidation products of lipids, proteins, and DNA markers of ccl4 poisoning? Free Radic. Biol. Med. 2005, 38, 698–710. [Google Scholar] [CrossRef]
- Kadiiska, M.B.; Gladen, B.C.; Baird, D.D.; Graham, L.B.; Parker, C.E.; Ames, B.N.; Basu, S.; Fitzgerald, G.A.; Lawson, J.A.; Marnett, L.J.; et al. Biomarkers of oxidative stress study iii. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in ccl4 poisoning. Free Radic. Biol. Med. 2005, 38, 711–718. [Google Scholar] [CrossRef]
- Kadiiska, M.B.; Hatch, G.E.; Nyska, A.; Jones, D.P.; Hensley, K.; Stocker, R.; George, M.M.; Van Thiel, D.H.; Stadler, K.; Barrett, J.C.; et al. Biomarkers of oxidative stress study iv: Ozone exposure of rats and its effect on antioxidants in plasma and bronchoalveolar lavage fluid. Free Radic. Biol. Med. 2011, 51, 1636–1642. [Google Scholar] [CrossRef]
- Kadiiska, M.B.; Basu, S.; Brot, N.; Cooper, C.; Saari Csallany, A.; Davies, M.J.; George, M.M.; Murray, D.M.; Jackson Roberts, L., 2nd; Shigenaga, M.K.; et al. Biomarkers of oxidative stress study v: Ozone exposure of rats and its effect on lipids, proteins, and DNA in plasma and urine. Free Radic. Biol. Med. 2013, 61, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Kadiiska, M.B.; Peddada, S.; Herbert, R.A.; Basu, S.; Hensley, K.; Jones, D.P.; Hatch, G.E.; Mason, R.P. Biomarkers of oxidative stress study vi. Endogenous plasma antioxidants fail as useful biomarkers of endotoxin-induced oxidative stress. Free Radic. Biol. Med. 2015, 81, 100–106. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fragoulis, A.; Tohidnezhad, M.; Kubo, Y.; Wruck, C.J.; Craveiro, R.B.; Bock, A.; Wolf, M.; Pufe, T.; Jahr, H.; Suhr, F. The Contribution of the Nrf2/ARE System to Mechanotransduction in Musculoskeletal and Periodontal Tissues. Int. J. Mol. Sci. 2023, 24, 7722. https://doi.org/10.3390/ijms24097722
Fragoulis A, Tohidnezhad M, Kubo Y, Wruck CJ, Craveiro RB, Bock A, Wolf M, Pufe T, Jahr H, Suhr F. The Contribution of the Nrf2/ARE System to Mechanotransduction in Musculoskeletal and Periodontal Tissues. International Journal of Molecular Sciences. 2023; 24(9):7722. https://doi.org/10.3390/ijms24097722
Chicago/Turabian StyleFragoulis, Athanassios, Mersedeh Tohidnezhad, Yusuke Kubo, Christoph Jan Wruck, Rogerio Bastos Craveiro, Anna Bock, Michael Wolf, Thomas Pufe, Holger Jahr, and Frank Suhr. 2023. "The Contribution of the Nrf2/ARE System to Mechanotransduction in Musculoskeletal and Periodontal Tissues" International Journal of Molecular Sciences 24, no. 9: 7722. https://doi.org/10.3390/ijms24097722
APA StyleFragoulis, A., Tohidnezhad, M., Kubo, Y., Wruck, C. J., Craveiro, R. B., Bock, A., Wolf, M., Pufe, T., Jahr, H., & Suhr, F. (2023). The Contribution of the Nrf2/ARE System to Mechanotransduction in Musculoskeletal and Periodontal Tissues. International Journal of Molecular Sciences, 24(9), 7722. https://doi.org/10.3390/ijms24097722