

Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation

 , , , , , ,
, , , , , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Metabolic Risk Factors and Atherosclerosis Parameters Are Improved by Atorvastatin
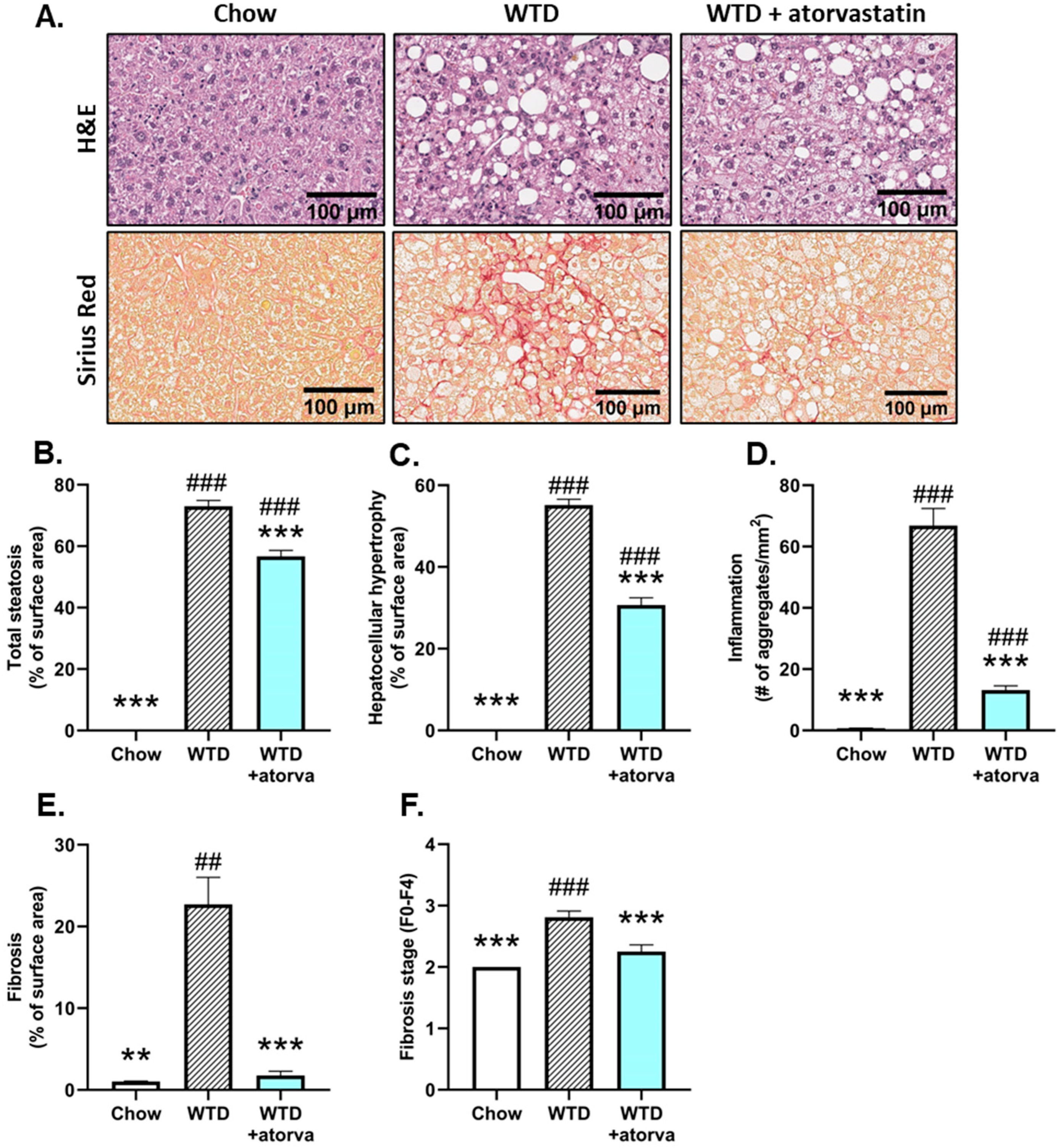
2.2. Atorvastatin Mitigates Hepatic Steatosis and Strongly Reduces Hepatic Inflammation and Fibrosis
2.3. Atorvastatin Decreases Hepatic Cholesterol Content, Strongly Reduces the Formation of Hepatic Cholesterol Crystals and Improves the Lipidomic Profile
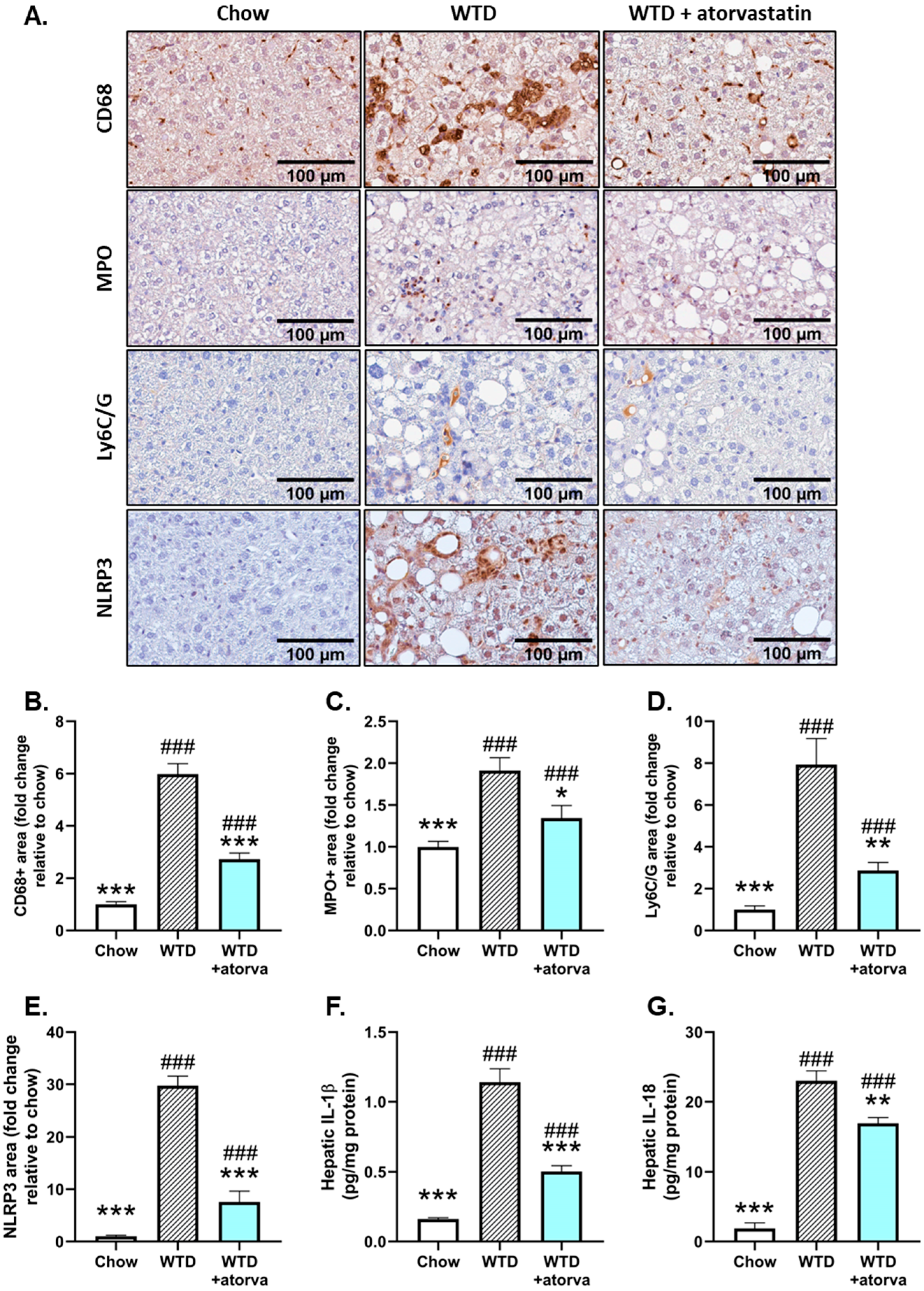
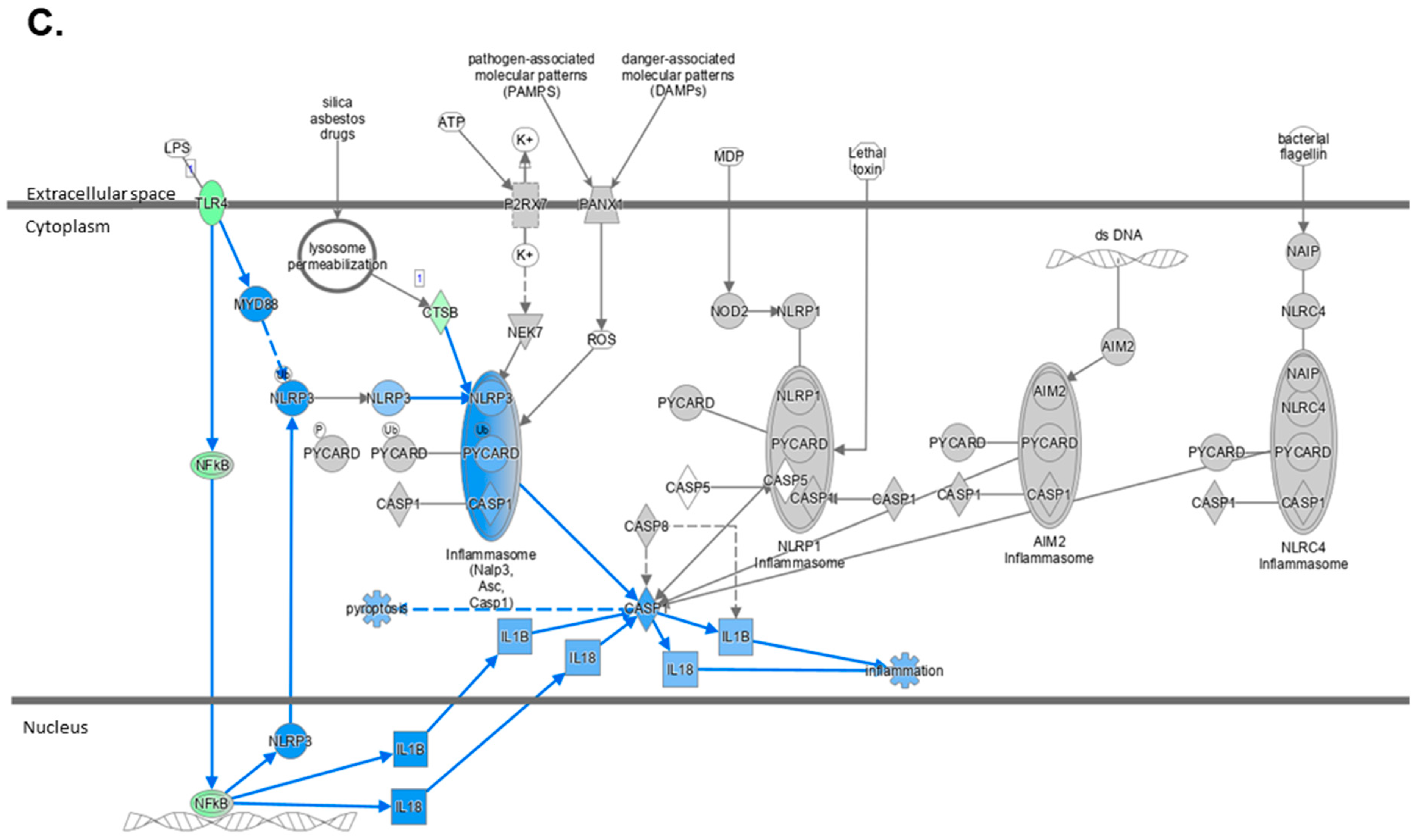
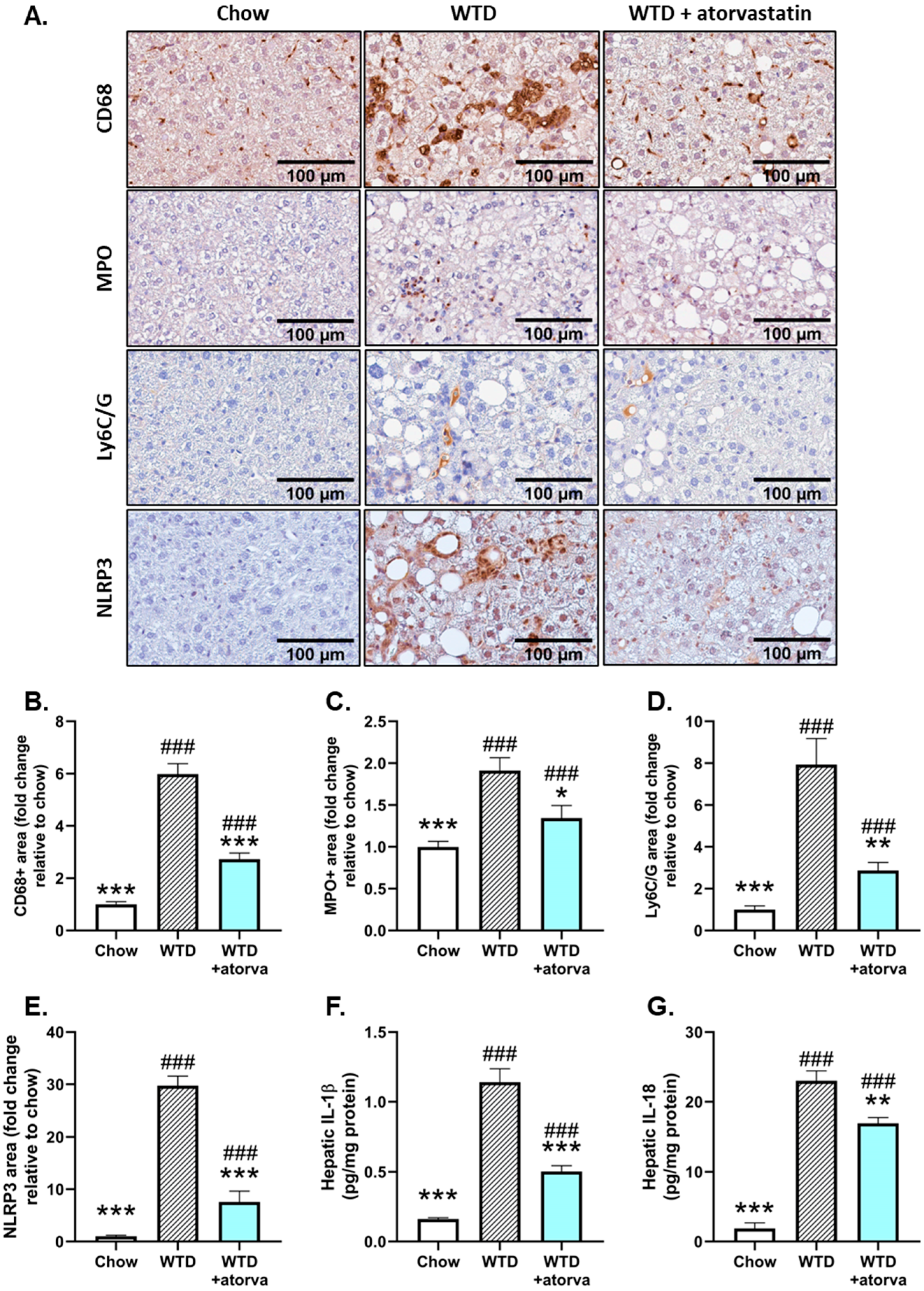
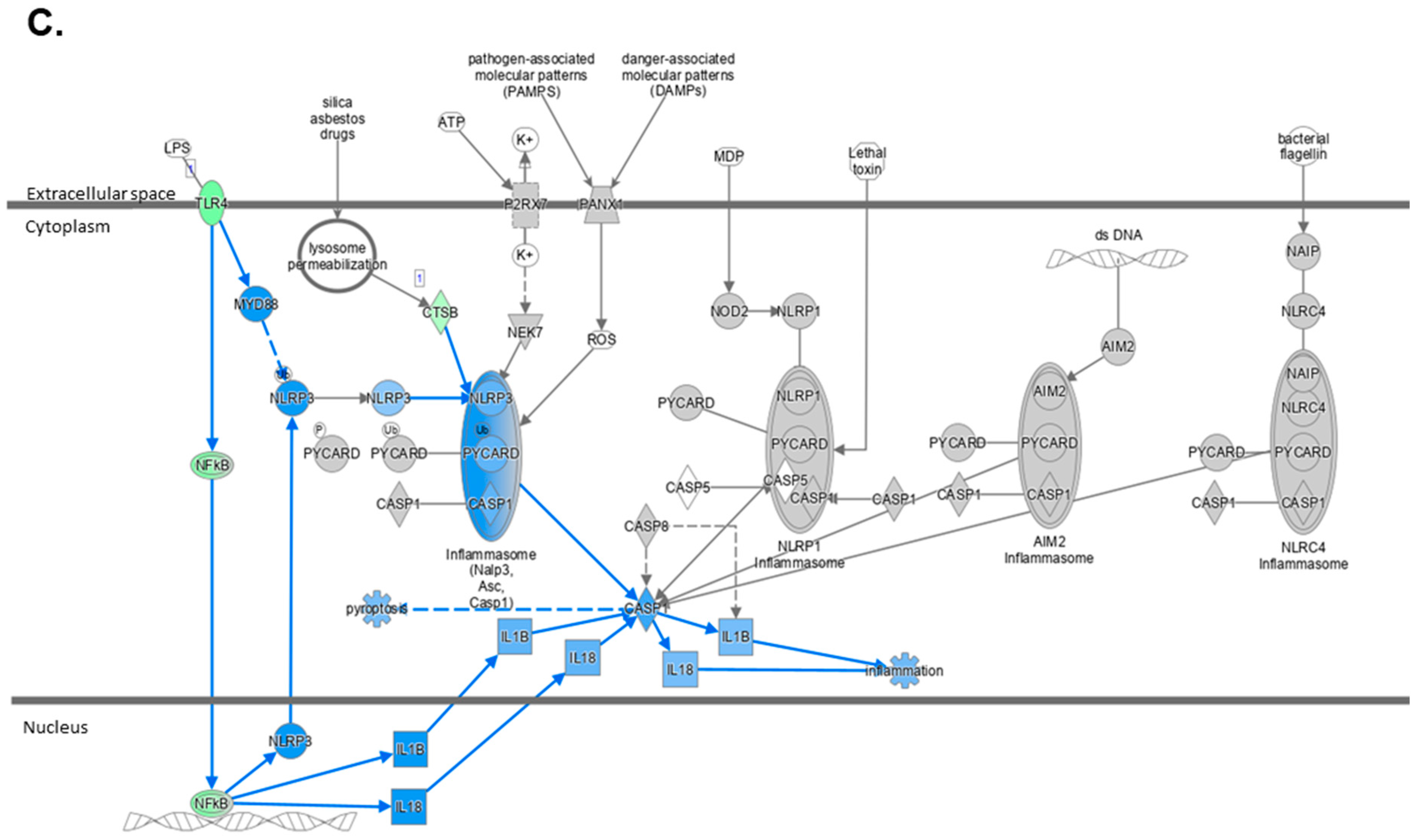
2.4. Atorvastatin Reduces Immune Cell Infiltration and NLRP3 Inflammasome Pathway Activity
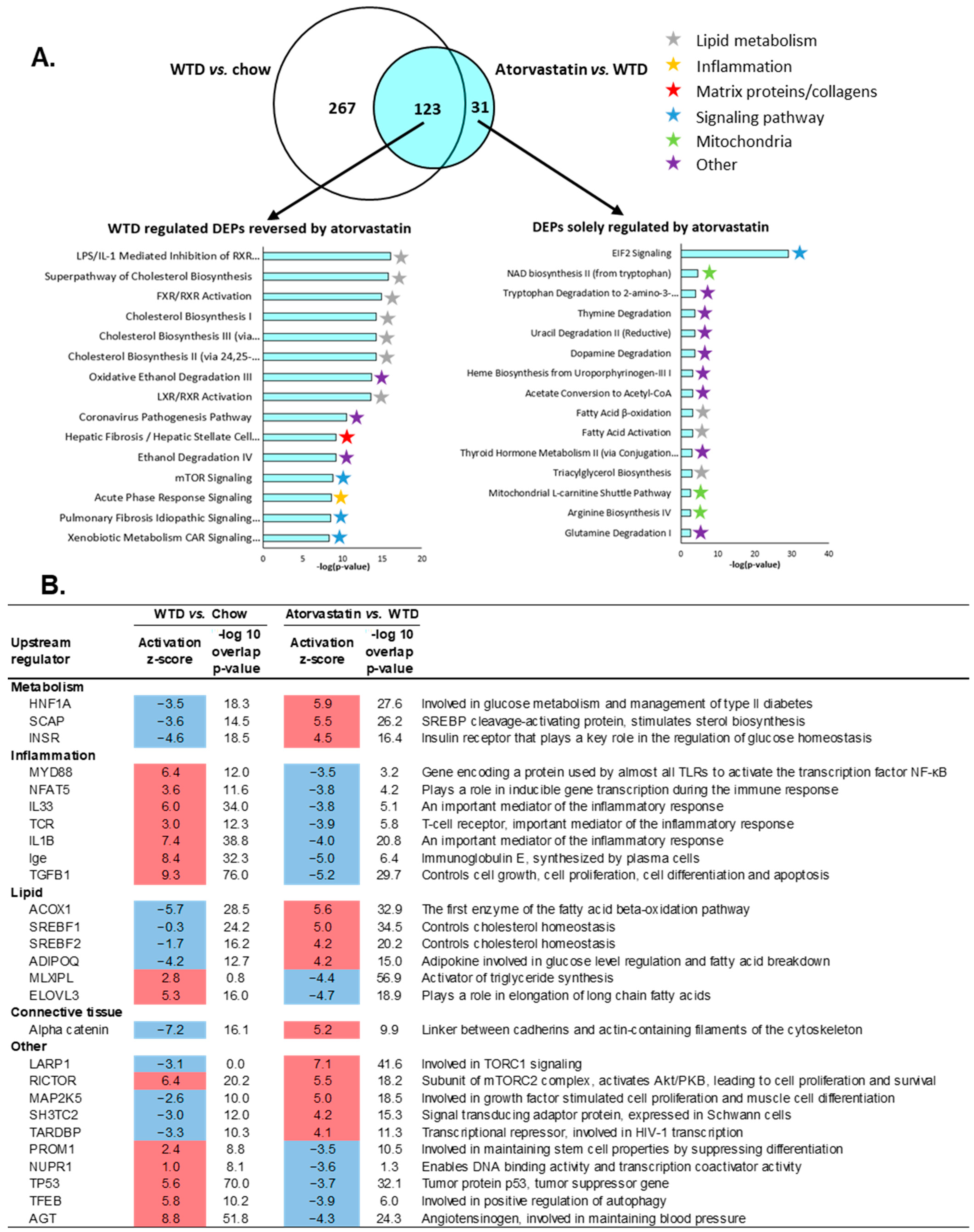
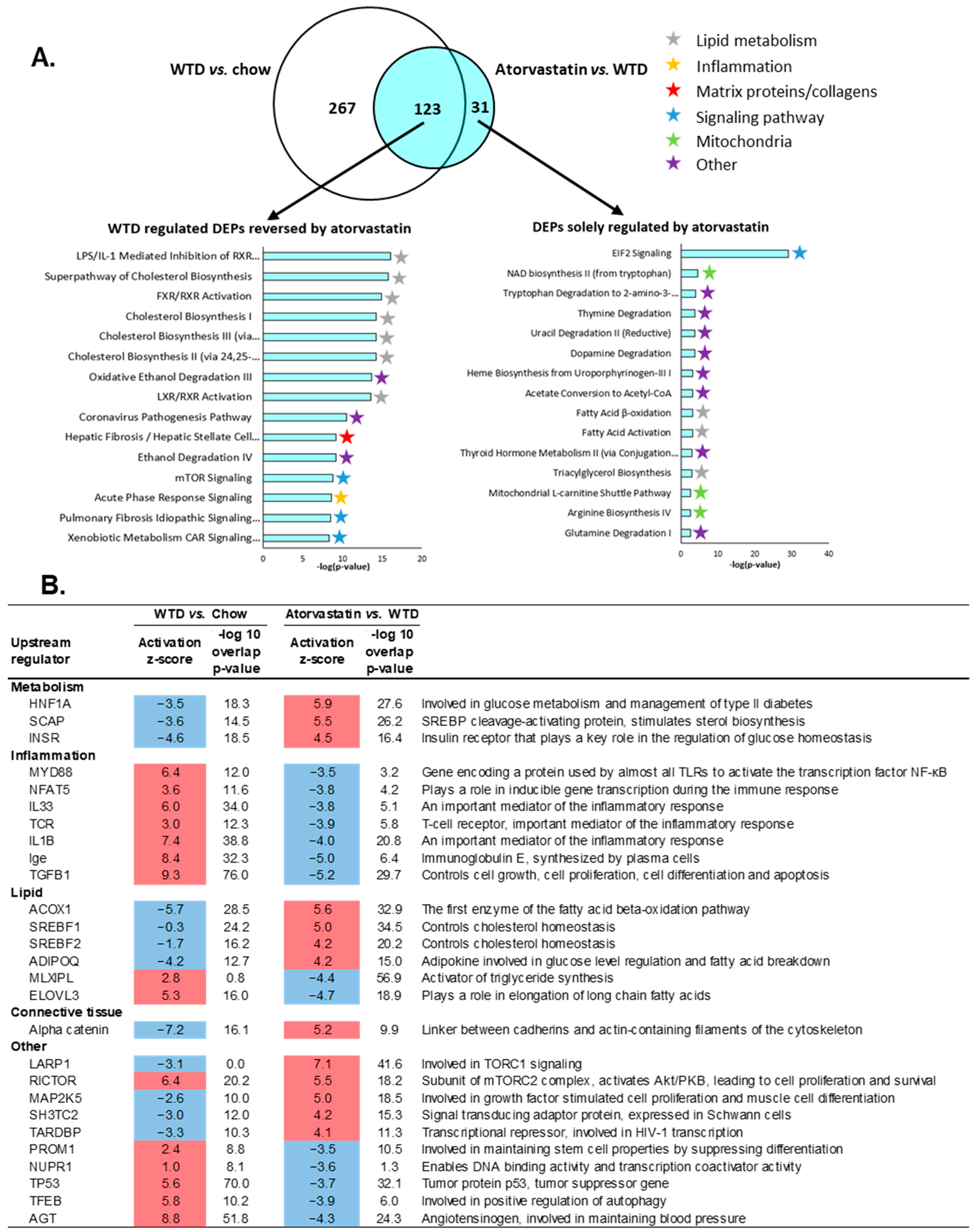
2.5. The Transcriptome Profile of WTD-Fed Mice Is Ameliorated by Atorvastatin
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Biochemical Analyses in Plasma and Liver Tissue
4.3. Histological Assessment of NASH
4.4. Histological Assessment of Atherosclerosis
4.5. Immunohistochemistry
4.6. Hepatic Lipidomic Analysis
4.7. Gene Expression and Pathway Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.M.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111, 154170. [Google Scholar] [CrossRef] [PubMed]
- Van de Ree, M.A.; Huisman, M.V.; Princen, H.M.G.; Meinders, A.E.; Kluft, C.; Berk-Planken, I.; Hoogerbrugge, N.; Jansen, H.; Stolk, R.P.; Van Venrooij, F.V.; et al. Strong decrease of high sensitivity C-reactive protein with high-dose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis 2003, 166, 129–135. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Havekes, L.; Kleemann, R.; Emeis, J.; Fontijn, R.; Horrevoets, A.; Kooistra, T.; Princen, H. Rosuvastatin reduces atherosclerosis development beyond and independent of its plasma cholesterol-lowering effect in APOE*3-Leiden transgenic mice: Evidence for antiinflammatory effects of rosuvastatin. Circulation 2003, 108, 1368–1374. [Google Scholar]
- Kleemann, R.; Verschuren, L.; De Rooij, B.J.; Lindeman, J.; De Maat, M.P.M.; Szalai, A.J.; Princen, H.M.G.; Kooistra, T. Evidence for anti-inflammatory activity of statins and PPARalpha activators in human C-reactive protein transgenic mice in vivo and in cultured human hepatocytes in vitro. Blood 2004, 103, 4188–4194. [Google Scholar] [CrossRef]
- Tuñón, J.; Badimon, L.; Bochaton-Piallat, M.-L.; Cariou, B.; Daemen, M.J.; Egido, J.; Evans, P.; Hoefer, I.E.; Ketelhuth, D.; Lutgens, E.; et al. Identifying the anti-inflammatory response to lipid lowering therapy: A position paper from the working group on atherosclerosis and vascular biology of the European Society of Cardiology. Cardiovasc. Res. 2018, 115, 10–19. [Google Scholar] [CrossRef]
- Nelson, A.; Torres, D.M.; Morgan, A.E.; Fincke, C.; Harrison, S.A. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenter. 2009, 43, 990–994. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Zadelaar, S.; Kleemann, R.; Verschuren, L.; Weij, J.D.V.-V.D.; van der Hoorn, J.; Princen, H.M.; Kooistra, T. Mouse Models for Atherosclerosis and Pharmaceutical Modifiers. Arter. Thromb. Vasc. Biol. 2007, 27, 1706–1721. [Google Scholar] [CrossRef]
- Kühnast, S.; Fiocco, M.; van der Hoorn, J.W.A.; Princen, H.M.G.; Jukema, J.W. Innovative pharmaceutical interventions in cardiovascular disease: Focusing on the contribution of non-HDL-C/LDL-C-lowering versus HDL-C-raisingA systematic review and meta-analysis of relevant preclinical studies and clinical trials. Eur. J. Pharmacol. 2015, 763, 48–63. [Google Scholar] [CrossRef]
- Pouwer, M.G.; Heinonen, S.E.; Behrendt, M.; Andréasson, A.C.; Van Koppen, A.; Menke, A.L.; Pieterman, E.J.; van den Hoek, A.M.; Jukema, J.W.; Leighton, B.; et al. The APOE*3-Leiden Heterozygous Glucokinase Knockout Mouse as Novel Translational Disease Model for Type 2 Diabetes, Dyslipidemia, and Diabetic Atherosclerosis. J. Diabetes Res. 2019, 2019, 9727952. [Google Scholar] [CrossRef]
- Ason, B.; van der Hoorn, J.W.A.; Chan, J.; Lee, E.; Pieterman, E.J.; Nguyen, K.K.; Di, M.; Shetterly, S.; Tang, J.; Yeh, W.-C.; et al. PCSK9 inhibition fails to alter hepatic LDLR, circulating cholesterol, and atherosclerosis in the absence of ApoE. J. Lipid Res. 2014, 55, 2370–2379. [Google Scholar] [CrossRef]
- Van de Poll, S.W.E.; Romer, T.J.; Volger, O.L.; Delsing, D.J.; Bakker Schut, T.C.; Princen, H.M.; Havekes, L.M.; Jukema, J.W.; van Der Laarse, A.; Puppels, G.J. Raman Spectroscopic Evaluation of the Effects of Diet and Lipid-Lowering Therapy on Atherosclerotic Plaque Development in Mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1630–1635. [Google Scholar] [CrossRef]
- Morrison, M.C.; Liang, W.; Mulder, P.; Verschuren, L.; Pieterman, E.J.; Toet, K.; Heeringa, P.; Wielinga, P.Y.; Kooistra, T.; Kleemann, R. Mirtoselect, an anthocyanin-rich bilberry extract, attenuates non-alcoholic steatohepatitis and associated fibrosis in ApoE(∗)3Leiden mice. J. Hepatol. 2015, 62, 1180–1186. [Google Scholar] [CrossRef]
- Zimmer, M.; Bista, P.; Benson, E.L.; Lee, D.Y.; Liu, F.; Picarella, D.; Vega, R.B.; Vu, C.B.; Yeager, M.; Ding, M.; et al. CAT-2003: A novel sterol regulatory element-binding protein inhibitor that reduces steatohepatitis, plasma lipids, and atherosclerosis in apolipoprotein E*3-Leiden mice. Hepatol. Commun. 2017, 1, 311–325. [Google Scholar] [CrossRef]
- Kooistra, T.; Verschuren, L.; De Vries-van der Weij, J.; Koenig, W.; Toet, K.; Princen, H.M.G.; Kleemann, R. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: Evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2322–2330. [Google Scholar] [CrossRef]
- Kühnast, S.; van der Hoorn, J.W.A.; Pieterman, E.J.; Hoek, A.M.V.D.; Sasiela, W.J.; Gusarova, V.; Peyman, A.; Schäfer, H.-L.; Schwahn, U.; Jukema, J.W.; et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J. Lipid Res. 2014, 55, 2103–2112. [Google Scholar] [CrossRef]
- Pouwer, M.G.; Pieterman, E.J.; Worms, N.; Keijzer, N.; Jukema, J.W.; Gromada, J.; Gusarova, V.; Princen, H.M.G. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J. Lipid Res. 2020, 61, 365–375. [Google Scholar] [CrossRef]
- Edwards, J.E.; Moore, R.A. Statins in hypercholesterolaemia: A dose-specific meta-analysis of lipid changes in randomised, double blind trials. BMC Fam. Pract. 2003, 4, 18. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Tzefos, M.; Olin, J.L. 3-hydroxyl-3-methylglutaryl coenzyme A reductase inhibitor use in chronic liver disease: A therapeutic controversy. J. Clin. Lipidol. 2011, 5, 450–459. [Google Scholar] [CrossRef]
- Fatima, K.; Moeed, A.; Waqar, E.; Atif, A.R.; Kamran, A.; Rizvi, H.; Suri, N.F.; Haider, H.; Shuja, S.H.; Khalid, M.; et al. Efficacy of statins in treatment and development of non-alcoholic fatty liver disease and steatohepatitis: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2021, 46, 101816. [Google Scholar] [CrossRef]
- Pastori, D.; Pani, A.; Di Rocco, A.; Menichelli, D.; Gazzaniga, G.; Farcomeni, A.; D’Erasmo, L.; Angelico, F.; Del Ben, M.; Baratta, F. Statin liver safety in non-alcoholic fatty liver disease: A systematic review and metanalysis. Br. J. Clin. Pharmacol. 2022, 88, 441–451. [Google Scholar] [CrossRef]
- Salami, J.A.; Warraich, H.; Valero-Elizondo, J.; Spatz, E.S.; Desai, N.R.; Rana, J.S.; Virani, S.S.; Blankstein, R.; Khera, A.; Blaha, M.J.; et al. National Trends in Statin Use and Expenditures in the US Adult Population From 2002 to 2013: Insights From the Medical Expenditure Panel Survey. JAMA Cardiol. 2017, 2, 56–65. [Google Scholar] [CrossRef]
- Boutari, C.; Pappas, P.D.; Anastasilakis, D.; Mantzoros, C.S. Statins’ efficacy in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 2195–2206. [Google Scholar] [CrossRef]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.-I.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef]
- Eslami, L.; Merat, S.; Malekzadeh, R.; Nasseri-Moghaddam, S.; Aramin, H. Statins for non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Cochrane Database Syst. Rev. 2013, 12, CD008623. [Google Scholar] [CrossRef]
- Li, Q.; Dhyani, M.; Grajo, J.R.; Sirlin, C.; Samir, A.E. Current status of imaging in nonalcoholic fatty liver disease. World J. Hepatol. 2018, 10, 530. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Bril, F.; Sanchez, P.P.; Lomonaco, R.; Orsak, R.B.; Hecht, R.J.; Tio, F.; Cusi, K. Liver Safety of Statins in Prediabetes or T2DM and Nonalcoholic Steatohepatitis: Post Hoc Analysis of a Randomized Trial. J. Clin. Endocrinol. Metab. 2017, 102, 2950–2961. [Google Scholar] [CrossRef]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef]
- Verschuren, L.; Kleemann, R.; Offerman, E.H.; Szalai, A.J.; Emeis, S.J.; Princen, H.M.G.; Kooistra, T. Effect of Low Dose Atorvastatin Versus Diet-Induced Cholesterol Lowering on Atherosclerotic Lesion Progression and Inflammation in Apolipoprotein E*3–Leiden Transgenic Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 161–167. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Stavro, P.M.; Suárez, M.; Arola-Arnal, A.; Van Duyvenvoorde, W.; Kooistra, T.; Wielinga, P.Y.; Kleemann, R. Replacement of Dietary Saturated Fat by PUFA-Rich Pumpkin Seed Oil Attenuates Non-Alcoholic Fatty Liver Disease and Atherosclerosis Development, with Additional Health Effects of Virgin over Refined Oil. PLoS ONE 2015, 10, e0139196. [Google Scholar] [CrossRef]
- Delsing, D.J.; Offerman, E.H.; Van Duyvenvoorde, W.V.; Van Der Boom, H.; De Wit, E.C.; Gijbels, M.J.; van Der Laarse, A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M. Acyl-CoA:Cholesterol Acyltransferase Inhibitor Avasimibe Reduces Atherosclerosis in Addition to Its Cholesterol-Lowering Effect in ApoE*3-Leiden Mice. Circulation 2001, 103, 1778–1786. [Google Scholar] [CrossRef]
- Delsing, D.J.M.; Jukema, J.W.; Van de Wiel, M.A.; Emeis, J.J.; Van der Laarse, A.; Havekes, L.M.; Princen, H.M.G. Differential effects of amlodipine and atorvastatin treatment and their combination on atherosclerosis in ApoE*3-Leiden transgenic mice. J. Cardiovasc. Pharmacol. 2003, 42, 63–70. [Google Scholar] [CrossRef]
- Park, H.-S.; Jang, J.E.; Ko, M.S.; Woo, S.H.; Kim, B.J.; Kim, H.S.; Park, H.S.; Park, I.-S.; Koh, E.H.; Lee, K.-U. Statins Increase Mitochondrial and Peroxisomal Fatty Acid Oxidation in the Liver and Prevent Non-Alcoholic Steatohepatitis in Mice. Diabetes Metab. J. 2016, 40, 376–385. [Google Scholar] [CrossRef]
- Lastuvkova, H.; Faradonbeh, F.A.; Schreiberova, J.; Hroch, M.; Mokry, J.; Faistova, H.; Nova, Z.; Hyspler, R.; Sa, I.C.I.; Nachtigal, P.; et al. Atorvastatin Modulates Bile Acid Homeostasis in Mice with Diet-Induced Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2021, 22, 6468. [Google Scholar] [CrossRef]
- Yarahmadi, S.; Farahmandian, N.; Fadaei, R.; Koushki, M.; Bahreini, E.; Karima, S.; Tond, S.B.; Rezaei, A.; Nourbakhsh, M.; Fallah, S. Therapeutic Potential of Resveratrol and Atorvastatin Following High-Fat Diet Uptake-Induced Nonalcoholic Fatty Liver Disease by Targeting Genes Involved in Cholesterol Metabolism and miR33. DNA Cell Biol. 2023, 42, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamaguchi, K.; Nakajima, T.; Nishikawa, T.; Jo, M.; Mitsumoto, Y.; Kimura, H.; Nishimura, T.; Tochiki, N.; Yasui, K.; et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Pereira, E.N.G.; De Araujo, B.P.; Rodrigues, K.L.; Silvares, R.R.; Martins, C.S.M.; Flores, E.E.I.; Fernandes-Santos, C.; Daliry, A. Simvastatin Improves Microcirculatory Function in Nonalcoholic Fatty Liver Disease and Downregulates Oxidative and ALE-RAGE Stress. Nutrients 2022, 14, 716. [Google Scholar] [CrossRef]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef]
- Boland, A.J.; Gangadharan, N.; Kavanagh, P.; Hemeryck, L.; Kieran, J.; Barry, M.; Walsh, P.T.; Lucitt, M. Simvastatin Suppresses Interleukin Iβ Release in Human Peripheral Blood Mononuclear Cells Stimulated With Cholesterol Crystals. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 509–517. [Google Scholar] [CrossRef]
- Massonnet, B.; Normand, S.; Moschitz, R.; Delwail, A.; Favot, L.; Garcia, M.; Bourmeyster, N.; Cuisset, L.; Grateau, G.; Morel, F.; et al. Pharmacological inhibitors of the mevalonate pathway activate pro-IL-1 processing and IL-1 release by human monocytes. Eur. Cytokine Netw. 2009, 20, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. Active Metabolite of Atorvastatin Inhibits Membrane Cholesterol Domain Formation by an Antioxidant Mechanism. J. Biol. Chem. 2006, 281, 9337–9345. [Google Scholar] [CrossRef]
- Satoh, M.; Tabuchi, T.; Itoh, T.; Nakamura, M. NLRP3 inflammasome activation in coronary artery disease: Results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin. Sci. 2014, 126, 233–241. [Google Scholar] [CrossRef]
- Mitchell, P.; Marette, A. Statin-Induced Insulin Resistance through Inflammasome Activation: Sailing between Scylla and Charybdis. Diabetes 2014, 63, 3569–3571. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef]
- Ooi, G.J.; Meikle, P.J.; Huynh, K.; Earnest, A.; Roberts, S.K.; Kemp, W.; Parker, B.L.; Brown, W.; Burton, P.; Watt, M.J. Hepatic lipidomic remodeling in severe obesity manifests with steatosis and does not evolve with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 524–535. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Post, S.M.; de Crom, R.; van Haperen, R.; van Tol, A.; Princen, H.M. Increased Fecal Bile Acid Excretion in Transgenic Mice with Elevated Expression of Human Phospholipid Transfer Protein. Arter. Thromb. Vasc. Biol. 2003, 23, 892–897. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a General NAFLD Scoring System for Rodent Models and Comparison to Human Liver Pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic Fatty Liver Disease: Pathology and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Kühnast, S.; Van Der Tuin, S.J.L.; Van Der Hoorn, J.W.; Van Klinken, J.B.; Simic, B.; Pieterman, E.; Havekes, L.M.; Landmesser, U.; Lüscher, T.F.; Van Dijk, K.W.; et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J. 2015, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kühnast, S.; van der Hoorn, J.W.; Hoek, A.M.V.D.; Havekes, L.M.; Liau, G.; Jukema, J.W.; Princen, H.M. Aliskiren inhibits atherosclerosis development and improves plaque stability in APOE*3Leiden.CETP transgenic mice with or without treatment with atorvastatin. J. Hypertens. 2012, 30, 107–116. [Google Scholar] [CrossRef]
- Su, B.; Bettcher, L.F.; Hsieh, W.-Y.; Hornburg, D.; Pearson, M.J.; Blomberg, N.; Giera, M.; Snyder, M.P.; Raftery, D.; Bensinger, S.J.; et al. A DMS Shotgun Lipidomics Workflow Application to Facilitate High-Throughput, Comprehensive Lipidomics. J. Am. Soc. Mass Spectrom. 2021, 32, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Ghorasaini, M.; Mohammed, Y.; Adamski, J.; Bettcher, L.; Bowden, J.A.; Cabruja, M.; Contrepois, K.; Ellenberger, M.; Gajera, B.; Haid, M.; et al. Cross-Laboratory Standardization of Preclinical Lipidomics Using Differential Mobility Spectrometry and Multiple Reaction Monitoring. Anal. Chem. 2021, 93, 16369–16378. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gol, L.; Yu, W.-M.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-.Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chow | WTD | WTD + Atorvastatin | |
|---|---|---|---|
| Body weight (g) | 25.9 ± 0.8 | 25.9 ± 0.3 | 25.5 ± 0.5 |
| Food intake (kCal/mouse/day) | 12.7 ± 0.2 *** | 11.3 ± 0.2 | 11.4 ± 0.1 |
| Plasma cholesterol (mM) | 2.4 ± 0.1 *** | 27.8 ± 1.6 | 15.8 ± 0.7 *** |
| Plasma triglycerides (mM) | 2.6 ± 0.2 ** | 5.0 ± 0.4 | 2.8 ± 0.2 *** |
| Liver weight (g) | 1.3 ± 0.1 *** | 2.5 ± 0.1 | 1.8 ± 0.1 *** |
| Plasma alanine transaminase (U/L) | 50.8 ± 6.0 *** | 283.2 ± 21.1 | 143.8 ± 16.5 *** |
| Plasma aspartate transaminase (U/L) | 110.2 ± 14.9 *** | 782.8 ± 48.2 | 521.2 ± 59.0 ** |
| Plasma serum amyloid A (µg/mL) | 11.6 ± 0.4 *** | 23.6 ± 1.6 | 13.7 ± 0.9 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inia, J.A.; Stokman, G.; Pieterman, E.J.; Morrison, M.C.; Menke, A.L.; Verschuren, L.; Caspers, M.P.M.; Giera, M.; Jukema, J.W.; van den Hoek, A.M.; et al. Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation. Int. J. Mol. Sci. 2023, 24, 7818. https://doi.org/10.3390/ijms24097818
Inia JA, Stokman G, Pieterman EJ, Morrison MC, Menke AL, Verschuren L, Caspers MPM, Giera M, Jukema JW, van den Hoek AM, et al. Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation. International Journal of Molecular Sciences. 2023; 24(9):7818. https://doi.org/10.3390/ijms24097818
Chicago/Turabian StyleInia, José A., Geurt Stokman, Elsbet J. Pieterman, Martine C. Morrison, Aswin L. Menke, Lars Verschuren, Martien P. M. Caspers, Martin Giera, J. Wouter Jukema, Anita M. van den Hoek, and et al. 2023. "Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation" International Journal of Molecular Sciences 24, no. 9: 7818. https://doi.org/10.3390/ijms24097818
APA StyleInia, J. A., Stokman, G., Pieterman, E. J., Morrison, M. C., Menke, A. L., Verschuren, L., Caspers, M. P. M., Giera, M., Jukema, J. W., van den Hoek, A. M., & Princen, H. M. G. (2023). Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation. International Journal of Molecular Sciences, 24(9), 7818. https://doi.org/10.3390/ijms24097818