
The Role of Histone Deacetylases in Acute Lung Injury—Friend or Foe

Abstract
:1. Introduction
2. 18 HDACs
2.1. Classical HDAC Family
2.1.1. Class I HDACs
2.1.2. Class II HDACs
2.1.3. Class IV HDACs
2.2. Sir2 Family
Class III HDACs
3. Physiological Functions of HDACs
3.1. HDACs Regulate Transcription
3.2. HDACs Regulate Non-Histone Substrates
3.3. HDACs Indirectly Regulate Post-Translational Modifications
4. Advances in the Studies of HDACs in ALI
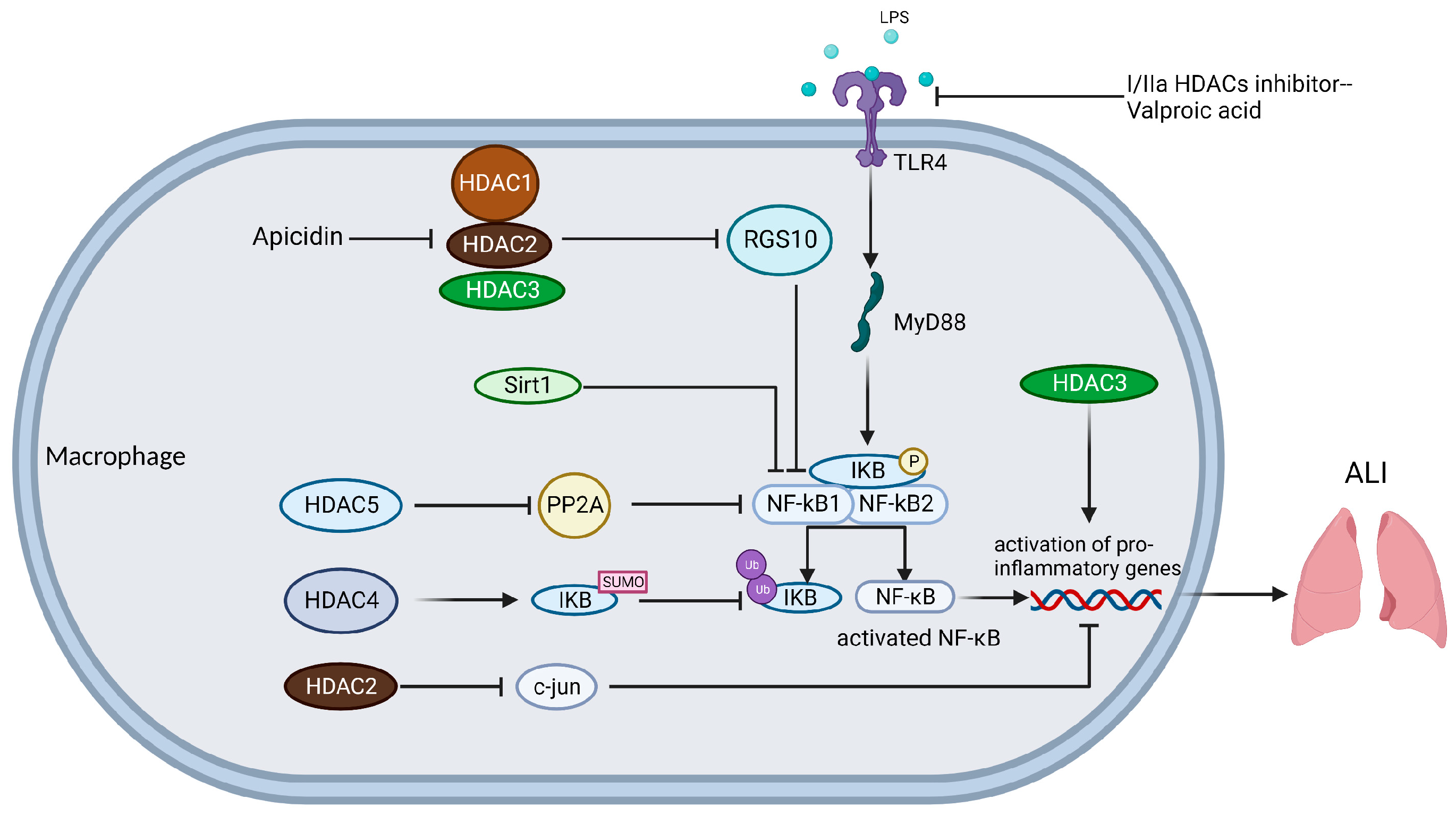
4.1. Macrophages
4.2. Vascular Endothelial Cells (VECs)
4.3. Alveolar Epithelial Cells (AECs)
4.4. Neutrophils
5. Targeting HDACs
5.1. Targeting Classical HDAC Family
5.2. Targeting Sir2 Family
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; LeGall, J.R.; Morris, A.; Spragg, R. Report of the American-European Consensus conference on acute respiratory distress syndrome: Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus Committee. J. Crit. Care 1994, 9, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Long, M.E.; Mallampalli, R.K.; Horowitz, J.C. Pathogenesis of pneumonia and acute lung injury. Clin. Sci. 2022, 136, 747–769. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Mart, M.F.; Ware, L.B. The long-lasting effects of the acute respiratory distress syndrome. Expert Rev. Respir. Med. 2020, 14, 577–586. [Google Scholar] [CrossRef]
- Zhan, Z.; Cai, H.; Cai, H.; Liang, X.; Lai, S.; Luo, Y. Effects of 45° prone position ventilation in the treatment of acute respiratory distress syndrome: A protocol for a randomized controlled trial study. Medicine 2021, 100, e25897. [Google Scholar] [CrossRef]
- Derwall, M.; Martin, L.; Rossaint, R. The acute respiratory distress syndrome: Pathophysiology, current clinical practice, and emerging therapies. Expert Rev. Respir. Med. 2018, 12, 1021–1029. [Google Scholar] [CrossRef]
- Wilson, J.G.; Liu, K.D.; Zhuo, H.; Caballero, L.; McMillan, M.; Fang, X.; Cosgrove, K.; Vojnik, R.; Calfee, C.S.; Lee, J.W.; et al. Mesenchymal stem (stromal) cells for treatment of ARDS: A phase 1 clinical trial. Lancet Respir. Med. 2015, 3, 24–32. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, J.; Lee, J.W. Therapeutic use of mesenchymal stem cell-derived extracellular vesicles in acute lung injury. Transfusion 2019, 59 (Suppl. S1), 876–883. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Li, Y.; Dou, H.; Liang, H.; Hou, Y. SPION-MSCs enhance therapeutic efficacy in sepsis by regulating MSC-expressed TRAF1-dependent macrophage polarization. Stem Cell Res. Ther. 2021, 12, 531. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Thuy, L.T.; Lee, Y.; Piao, C.; Choi, J.S.; Lee, M. Dual-Functional Dendrimer Micelles with Glycyrrhizic Acid for Anti-Inflammatory Therapy of Acute Lung Injury. ACS Appl. Mater. Interfaces 2021, 13, 47313–47326. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ma, Y.; Su, Z.; Zhao, R.; Zhao, X.; Nie, H.G.; Xu, P.; Zhu, L.; Zhang, M.; Li, X.; et al. Meta-Analysis of Preclinical Studies of Fibrinolytic Therapy for Acute Lung Injury. Front. Immunol. 2018, 9, 1898. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, G.; Han, K.; Wang, Y.; Wang, L.; Gao, J.; Zhao, S.; Wang, G.; Chen, S.; Luo, A.; et al. Mitochondrial PKM2 deacetylation by procyanidin B2-induced SIRT3 upregulation alleviates lung ischemia/reperfusion injury. Cell Death Dis. 2022, 13, 594. [Google Scholar] [CrossRef]
- Ahmad, I.; Molyvdas, A.; Jian, M.Y.; Zhou, T.; Traylor, A.M.; Cui, H.; Liu, G.; Song, W.; Agarwal, A.; Jilling, T.; et al. AICAR decreases acute lung injury by phosphorylating AMPK and upregulating heme oxygenase-1. Eur. Respir. J. 2021, 58, 2003694. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Zhou, Y.; Qu, M.; Wang, Y.; Guo, K.; Shen, R.; Sun, Z.; Cata, J.P.; Yang, S.; et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int. J. Biol. Sci. 2022, 18, 3337–3357. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, Z.; Lin, H.; Lei, T.; Zhou, Z.; Huang, W.; Wu, X.; Zuo, L.; Wu, J.; Liu, Y.; et al. TRIM47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice via K63-linked ubiquitination of TRAF2. Signal Transduct. Target. Ther. 2022, 7, 148. [Google Scholar] [CrossRef]
- Nie, Y.; Nirujogi, T.S.; Ranjan, R.; Reader, B.F.; Chung, S.; Ballinger, M.N.; Englert, J.A.; Christman, J.W.; Karpurapu, M. PolyADP-Ribosylation of NFATc3 and NF-κB Transcription Factors Modulate Macrophage Inflammatory Gene Expression in LPS-Induced Acute Lung Injury. J. Innate Immun. 2021, 13, 83–93. [Google Scholar] [CrossRef]
- Colasanti, T.; Fiorito, S.; Alessandri, C.; Serafino, A.; Andreola, F.; Barbati, C.; Morello, F.; Alfè, M.; Di Blasio, G.; Gargiulo, V.; et al. Diesel exhaust particles induce autophagy and citrullination in Normal Human Bronchial Epithelial cells. Cell Death Dis. 2018, 9, 1073. [Google Scholar] [CrossRef]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in epigenetic therapeutics with focus on solid tumors. Clin. Epigenetics 2021, 13, 83. [Google Scholar] [CrossRef]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays News Rev. Mol. Cell. Dev. Biol. 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Aramsangtienchai, P.; Spiegelman, N.A.; He, B.; Miller, S.P.; Dai, L.; Zhao, Y.; Lin, H. HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2016, 11, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Bahl, S.; Seto, E. Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance. Cell. Mol. Life Sci. CMLS 2021, 78, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Yoshida, M.; Kudo, N.; Kosono, S.; Ito, A. Chemical and structural biology of protein lysine deacetylases. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2017, 93, 297–321. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. JASN 2018, 29, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Yao, Y.L.; Sun, J.M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [Google Scholar] [CrossRef] [PubMed]
- Rine, J.; Herskowitz, I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 1987, 116, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef]
- Rundlett, S.E.; Carmen, A.A.; Kobayashi, R.; Bavykin, S.; Turner, B.M.; Grunstein, M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 14503–14508. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef]
- Yang, W.M.; Tsai, S.C.; Wen, Y.D.; Fejer, G.; Seto, E. Functional domains of histone deacetylase-3. J. Biol. Chem. 2002, 277, 9447–9454. [Google Scholar] [CrossRef]
- Li, W.; Kou, J.; Qin, J.; Li, L.; Zhang, Z.; Pan, Y.; Xue, Y.; Du, W. NADPH levels affect cellular epigenetic state by inhibiting HDAC3-Ncor complex. Nat. Metab. 2021, 3, 75–89. [Google Scholar] [CrossRef]
- Szigety, K.M.; Liu, F.; Yuan, C.Y.; Moran, D.J.; Horrell, J.; Gochnauer, H.R.; Cohen, R.N.; Katz, J.P.; Kaestner, K.H.; Seykora, J.T.; et al. HDAC3 ensures stepwise epidermal stratification via NCoR/SMRT-reliant mechanisms independent of its histone deacetylase activity. Genes Dev. 2020, 34, 973–988. [Google Scholar] [CrossRef]
- Van den Wyngaert, I.; de Vries, W.; Kremer, A.; Neefs, J.; Verhasselt, P.; Luyten, W.H.; Kass, S.U. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000, 478, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000, 14, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Richon, V.M.; Rifkind, R.A.; Marks, P.A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 2000, 97, 1056–1061. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7354–7359. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Miska, E.A.; Langley, E.; Reynaud-Deonauth, S.; Kotecha, S.; Towers, N.; Spohr, G.; Kouzarides, T.; Mohun, T.J. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999, 18, 5085–5098. [Google Scholar] [CrossRef]
- Zhou, X.; Marks, P.A.; Rifkind, R.A.; Richon, V.M. Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. USA 2001, 98, 10572–10577. [Google Scholar] [CrossRef]
- Kao, H.Y.; Lee, C.H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 2002, 277, 187–193. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Galleano, I.; Madsen, A.S.; Olsen, C.A. Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol. 2018, 25, 849–856.e8. [Google Scholar] [CrossRef]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar]
- Zheng, W. The Zinc-Dependent HDACs: Non-Histone Substrates and Catalytic Deacylation Beyond Deacetylation. Mini Rev. Med. Chem. 2022, 22, 2478–2485. [Google Scholar] [CrossRef]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Arora, S. Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 2020, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Mittal, A.K.; Kalonia, H.; Madan, S.; Ghosh, S.; Sinha, J.K.; Rajput, S.K. SIRT1 Promotes Neuronal Fortification in Neurodegenerative Diseases through Attenuation of Pathological Hallmarks and Enhancement of Cellular Lifespan. Curr. Neuropharmacol. 2021, 19, 1019–1037. [Google Scholar] [PubMed]
- Roichman, A.; Elhanati, S.; Aon, M.A.; Abramovich, I.; Di Francesco, A.; Shahar, Y.; Avivi, M.Y.; Shurgi, M.; Rubinstein, A.; Wiesner, Y.; et al. Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat. Commun. 2021, 12, 3208. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zang, W.; Wang, J.; Huang, Y.; He, Y.; Yan, L.; Liu, J.; Zheng, W. The chemical biology of sirtuins. Chem. Soc. Rev. 2015, 44, 5246–5264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Yi, F.; Zhang, Y.; Wang, Z.; Wang, Z.; Li, Z.; Zhou, T.; Xu, H.; Liu, J.; Jiang, B.; Li, X.; et al. The deacetylation-phosphorylation regulation of SIRT2-SMC1A axis as a mechanism of antimitotic catastrophe in early tumorigenesis. Sci. Adv. 2021, 7, eabe5518. [Google Scholar] [CrossRef]
- Diao, Z.; Ji, Q.; Wu, Z.; Zhang, W.; Cai, Y.; Wang, Z.; Hu, J.; Liu, Z.; Wang, Q.; Bi, S.; et al. SIRT3 consolidates heterochromatin and counteracts senescence. Nucleic Acids Res. 2021, 49, 4203–4219. [Google Scholar] [CrossRef]
- Guo, Y.; Jia, X.; Cui, Y.; Song, Y.; Wang, S.; Geng, Y.; Li, R.; Gao, W.; Fu, D. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 2021, 41, 101915. [Google Scholar] [CrossRef]
- Ji, Z.; Liu, G.H.; Qu, J. Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genom. Yi Chuan Xue Bao 2022, 49, 287–298. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Leng, H.; Liu, S.; Lei, Y.; Tang, Y.; Gu, S.; Hu, J.; Chen, S.; Feng, J.; Li, Q. FACT interacts with Set3 HDAC and fine-tunes GAL1 transcription in response to environmental stimulation. Nucleic Acids Res. 2021, 49, 5502–5519. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. Targeting the Zinc-Dependent Histone Deacetylases (HDACs) for Drug Discovery. In Chemical Epigenetics; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–27. [Google Scholar]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 6317–6327. [Google Scholar] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314. [Google Scholar] [CrossRef]
- Norton, V.G.; Marvin, K.W.; Yau, P.; Bradbury, E.M. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J. Biol. Chem. 1990, 265, 19848–19852. [Google Scholar] [CrossRef]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef]
- Choi, C.H.; Burton, Z.F.; Usheva, A. Auto-acetylation of transcription factors as a control mechanism in gene expression. Cell Cycle 2004, 3, 114–115. [Google Scholar] [CrossRef]
- Shankaranarayanan, P.; Chaitidis, P.; Kühn, H.; Nigam, S. Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J. Biol. Chem. 2001, 276, 42753–42760. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Qian, M.; Liu, Z.; Tang, X.; Sun, J.; Jiang, Y.; Sun, S.; Cao, X.; Pang, Q.; Liu, B. Deacetylase-independent function of SIRT6 couples GATA4 transcription factor and epigenetic activation against cardiomyocyte apoptosis. Nucleic Acids Res. 2020, 48, 4992–5005. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Izumiya, Y.; Araki, S.; Nakamura, T.; Kimura, Y.; Hanatani, S.; Yamada, T.; Ishida, T.; Yamamoto, M.; Onoue, Y.; et al. Cardiomyocyte Sirt (Sirtuin) 7 Ameliorates Stress-Induced Cardiac Hypertrophy by Interacting With and Deacetylating GATA4. Hypertension 2020, 75, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Gan, H.; Shen, T.; Chupp, D.P.; Taylor, J.R.; Sanchez, H.N.; Li, X.; Xu, Z.; Zan, H.; Casali, P. B cell Sirt1 deacetylates histone and non-histone proteins for epigenetic modulation of AID expression and the antibody response. Sci. Adv. 2020, 6, eaay2793. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jayabal, S.; Ghorbani, M.; Legault, L.M.; McGraw, S.; Watt, A.J.; Yang, X.J. ATAT1 regulates forebrain development and stress-induced tubulin hyperacetylation. Cell. Mol. Life Sci. CMLS 2019, 76, 3621–3640. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Jia, L.F.; Gan, Y.H. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene 2016, 35, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef]
- Fu, J.; Yoon, H.G.; Qin, J.; Wong, J. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol. Cell. Biol. 2007, 27, 4641–4651. [Google Scholar] [CrossRef]
- Liu, Z.; Mai, A.; Sun, J. Lysine acetylation regulates Bruton’s tyrosine kinase in B cell activation. J. Immunol. 2010, 184, 244–254. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Samant, S.A.; Wolfgeher, D.; Trivedi, C.M.; Gupta, M.P. Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes. Mol. Cell. Biol. 2011, 31, 2349–2363. [Google Scholar] [CrossRef]
- Zheng, G.; Yang, Y.C. Sumoylation and acetylation play opposite roles in the transactivation of PLAG1 and PLAGL2. J. Biol. Chem. 2005, 280, 40773–40781. [Google Scholar] [CrossRef]
- Zheng, G.; Yang, Y.C. Acetylation and alternative splicing regulate ZNF76-mediated transcription. Biochem. Biophys. Res. Commun. 2006, 339, 1069–1075. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Jeon, E.J.; Li, Q.L.; Lee, Y.H.; Choi, J.K.; Kim, W.J.; Lee, K.Y.; Bae, S.C. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 2004, 279, 29409–29417. [Google Scholar] [CrossRef] [PubMed]
- Caron, C.; Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. BioEssays News Rev. Mol. Cell. Dev. Biol. 2005, 27, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Luo, Z.Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef]
- Valencia-Sánchez, M.I.; De Ioannes, P.; Wang, M.; Truong, D.M.; Lee, R.; Armache, J.P.; Boeke, J.D.; Armache, K.J. Regulation of the Dot1 histone H3K79 methyltransferase by histone H4K16 acetylation. Science 2021, 371. [Google Scholar] [CrossRef]
- Cutler, J.A.; Perner, F.; Armstrong, S.A. Histone PTM Crosstalk Stimulates Dot1 Methyltransferase Activity. Trends Biochem. Sci. 2021, 46, 522–524. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.B.; Chen, S.Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Halili, M.A.; Andrews, M.R.; Sweet, M.J.; Fairlie, D.P. Histone deacetylase inhibitors in inflammatory disease. Curr. Top. Med. Chem. 2009, 9, 309–319. [Google Scholar] [CrossRef]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Matuschak, G.M.; Lechner, A.J. Acute lung injury and the acute respiratory distress syndrome: Pathophysiology and treatment. Mo. Med. 2010, 107, 252–258. [Google Scholar] [PubMed]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 2015, 16, 36–44. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Climent, M.; Viggiani, G.; Chen, Y.W.; Coulis, G.; Castaldi, A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 4370. [Google Scholar] [CrossRef]
- Yan, J.; Yang, F.; Wang, D.; Lu, Y.; Liu, L.; Wang, Z. MicroRNA-217 modulates inflammation, oxidative stress, and lung injury in septic mice via SIRT1. Free Radic. Res. 2021, 55, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Guan, H.; Zhang, J.L.; Zheng, Z.; Wang, H.T.; Tao, K.; Han, S.C.; Su, L.L.; Hu, D. Acute downregulation of miR-199a attenuates sepsis-induced acute lung injury by targeting SIRT1. Am. J. Physiol. Cell Physiol. 2018, 314, C449–C455. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, T.; Zhang, C.; Ji, H.; Tong, X.; Xia, R.; Wang, W.; Ma, Z.; Shi, X. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit. Care 2021, 25, 356. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L. Depleting microRNA-146a-3p attenuates lipopolysaccharide-induced acute lung injury via up-regulating SIRT1 and mediating NF-κB pathway. J. Drug Target. 2021, 29, 420–429. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef] [PubMed]
- Ferlito, M.; Wang, Q.; Fulton, W.B.; Colombani, P.M.; Marchionni, L.; Fox-Talbot, K.; Paolocci, N.; Steenbergen, C. Hydrogen sulfide [corrected] increases survival during sepsis: Protective effect of CHOP inhibition. J. Immunol. 2014, 192, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, J.; Wang, J.; Chen, M.; Xia, H.; Yao, S.; Zhang, D. SIRT1 ameliorated septic associated-lung injury and macrophages apoptosis via inhibiting endoplasmic reticulum stress. Cell. Signal. 2022, 97, 110398. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Shen, Y.; Xv, G.; Di, Z.; Li, Y.; Li, G. Aloin suppresses lipopolysaccharide-induced acute lung injury by inhibiting NLRP3/NF-κB via activation of SIRT1 in mice. Immunopharmacol. Immunotoxicol. 2020, 42, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.C.; Chao, S.C.; Wu, H.Y.; Chiang, C.L.; Wang, C.C.; Liu, S.H.; Weng, T.I. Salidroside ameliorates sepsis-induced acute lung injury and mortality via downregulating NF-κB and HMGB1 pathways through the upregulation of SIRT1. Sci. Rep. 2017, 7, 12026. [Google Scholar] [CrossRef]
- Liu, Z.; Meng, Y.; Miao, Y.; Yu, L.; Yu, Q. Propofol reduces renal ischemia/reperfusion-induced acute lung injury by stimulating sirtuin 1 and inhibiting pyroptosis. Aging 2020, 13, 865–876. [Google Scholar] [CrossRef]
- Pillai, V.B.; Bindu, S.; Sharp, W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef]
- Kurundkar, D.; Kurundkar, A.R.; Bone, N.B.; Becker, E.J., Jr.; Liu, W.; Chacko, B.; Darley-Usmar, V.; Zmijewski, J.W.; Thannickal, V.J. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 2019, 4, e120722. [Google Scholar] [CrossRef]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef]
- Wang, Q.L.; Yang, L.; Liu, Z.L.; Peng, Y.; Gao, M.; Deng, L.T.; Liu, X.; Xing, W. Sirtuin 6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome via dual mechanisms dependent on and independent of autophagy. Cytotherapy 2022, 24, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, A.; Hu, J.; Kang, J. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol. Cell Biol. 2019, 97, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liu, Q.; Adrianto, I.; Wu, X.; Glassbrook, J.; Khalasawi, N.; Yin, C.; Yi, Q.; Dong, Z.; Geissmann, F.; et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020, 11, 3822. [Google Scholar] [CrossRef]
- Ghiboub, M.; Zhao, J.; Li Yim, A.Y.F.; Schilderink, R.; Verseijden, C.; van Hamersveld, P.H.P.; Duarte, J.M.; Hakvoort, T.B.M.; Admiraal, I.; Harker, N.R.; et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front. Immunol. 2020, 11, 550769. [Google Scholar] [CrossRef]
- Hollinger, S.; Hepler, J.R. Cellular regulation of RGS proteins: Modulators and integrators of G protein signaling. Pharmacol. Rev. 2002, 54, 527–559. [Google Scholar] [CrossRef]
- Almutairi, F.; Tucker, S.L.; Sarr, D.; Rada, B. PI3K/ NF-κB-dependent TNF-α and HDAC activities facilitate LPS-induced RGS10 suppression in pulmonary macrophages. Cell. Signal. 2021, 86, 110099. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Purves, T.; Palmer, S.; Sullenger, B.; Welty-Wolf, K.; Haines, K.; Agarwal, S.; Kasotakis, G. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PLoS ONE 2021, 16, e0245169. [Google Scholar] [CrossRef]
- Traenckner, E.B.; Pahl, H.L.; Henkel, T.; Schmidt, K.N.; Wilk, S.; Baeuerle, P.A. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef]
- Yang, Q.; Tang, J.; Xu, C.; Zhao, H.; Zhou, Y.; Wang, Y.; Yang, M.; Chen, X.; Chen, J. Histone deacetylase 4 inhibits NF-κB activation by facilitating IκBα sumoylation. J. Mol. Cell Biol. 2020, 12, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zheng, R.; Sun, S. Drug Repurposing: Escitalopram attenuates acute lung injury by inhibiting the SIK2/ HDAC4/ NF-κB signaling cascade. Biochem. Biophys. Res. Commun. 2022, 599, 1–8. [Google Scholar] [PubMed]
- Zhao, Y.; Ma, G.; Yang, X. HDAC5 promotes Mycoplasma pneumoniae-induced inflammation in macrophages through NF-κB activation. Life Sci. 2019, 221, 13–19. [Google Scholar] [CrossRef]
- Xu, C.; Tang, J.; Yang, Q.; Zhao, H.; Liu, Y.; Cao, J.; Zhou, Y.; Chen, X.; Chen, J. Histone deacetylase 5 deacetylates the phosphatase PP2A for positively regulating NF-κB signaling. J. Biol. Chem. 2021, 297, 101380. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712–2728.e8. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Rong, S.; Repa, J.J.; St Clair, R.; Parks, J.S.; Mishra, N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Fu, J. HDAC6 Mediates Macrophage iNOS Expression and Excessive Nitric Oxide Production in the Blood During Endotoxemia. Front. Immunol. 2020, 11, 1893. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Youn, G.S.; Lee, K.W.; Choi, S.Y.; Park, J. Overexpression of HDAC6 induces pro-inflammatory responses by regulating ROS-MAPK-NF-κB/AP-1 signaling pathways in macrophages. Free Radic. Biol. Med. 2016, 97, 14–23. [Google Scholar] [CrossRef]
- Zhang, W.B.; Yang, F.; Wang, Y.; Jiao, F.Z.; Zhang, H.Y.; Wang, L.W.; Gong, Z.J. Inhibition of HDAC6 attenuates LPS-induced inflammation in macrophages by regulating oxidative stress and suppressing the TLR4-MAPK/NF-κB pathways. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 117, 109166. [Google Scholar] [CrossRef] [PubMed]
- Youn, G.S.; Park, J.K.; Lee, C.Y.; Jang, J.H.; Yun, S.H.; Kwon, H.Y.; Choi, S.Y.; Park, J. MicroRNA-22 negatively regulates LPS-induced inflammatory responses by targeting HDAC6 in macrophages. BMB Rep. 2020, 53, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Sutton, N.R.; Wang, Z.; Goonewardena, S.N.; Holinstat, M. Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID-19. Eur. J. Pharmacol. 2021, 898, 173988. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, M.; Kretzer, C.; Lindemann, H.; Godmann, M.; Heinze, T.; Werz, O.; Heinzel, T. Biocompatible valproic acid-coupled nanoparticles attenuate lipopolysaccharide-induced inflammation. Int. J. Pharm. 2021, 601, 120567. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Zhou, Z.; Rajasingh, S.; Panda, A.; Sampath, V.; Rajasingh, J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int. J. Biochem. Cell Biol. 2018, 102, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Przanowski, P.; Dabrowski, M.; Ellert-Miklaszewska, A.; Kloss, M.; Mieczkowski, J.; Kaza, B.; Ronowicz, A.; Hu, F.; Piotrowski, A.; Kettenmann, H.; et al. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J. Mol. Med. 2014, 92, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, H.Y.; Kim, M.G.; Jeong, S.; Yun, C.H.; Han, S.H. Short-chain Fatty Acids Inhibit Staphylococcal Lipoprotein-induced Nitric Oxide Production in Murine Macrophages. Immune Netw. 2019, 19, e9. [Google Scholar] [CrossRef]
- Mowery, N.T.; Terzian, W.T.H.; Nelson, A.C. Acute lung injury. Curr. Probl. Surg. 2020, 57, 100777. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, K.; Li, J.; Tang, X.; Lin, J.; Lu, X.; Huang, R.; Yang, B.; Shi, Y.; Ye, D.; et al. Melatonin attenuates choroidal neovascularization by regulating macrophage/microglia polarization via inhibition of RhoA/ROCK signaling pathway. J. Pineal Res. 2020, 69, e12660. [Google Scholar] [CrossRef]
- Kása, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier functi on in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [PubMed]
- Kovacs-Kasa, A.; Gorshkov, B.A.; Kim, K.M.; Kumar, S.; Black, S.M.; Fulton, D.J.; Dimitropoulou, C.; Catravas, J.D.; Verin, A.D. The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Sci. Rep. 2016, 6, 39018. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, L.; Kovacs-Kasa, A.; Verin, A.D.; Fulton, D.; Lucas, R.; Su, Y. Histone deacetylases in vascular permeability and remodeling associate d with acute lung injury. Vessel. Plus 2018, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Geudens, I.; Bruyr, J.; Potente, M.; Bleuart, A.; Lebrun, M.; Simonis, N.; Deroanne, C.; Twizere, J.C.; Soubeyran, P.; et al. PP2A regulatory subunit Bα controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J. 2013, 32, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Kasa, A.; Kovacs, L.; Cherian-Shaw, M.; Patel, V.; Meadows, M.L.; Fulton, D.J.; Su, Y.; Verin, A.D. Inhibition of Class IIa HDACs improves endothelial barrier function in endotoxin-induced acute lung injury. J. Cell. Physiol. 2021, 236, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Hao, S.; Xu, X.; Zhou, J.; Liu, Z.; Lu, H.; Wang, L.; Jin, W.; Li, S. Activation of SIRT1 ameliorates LPS-induced lung injury in mice via decreasing endothelial tight junction permeability. Acta Pharmacol. Sin. 2019, 40, 630–641. [Google Scholar] [CrossRef]
- Tian, Y.G.; Zhang, J. Protective effect of SIRT3 on acute lung injury by increasing manganese superoxide dismutase-mediated antioxidation. Mol. Med. Rep. 2018, 17, 5557–5565. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Qin, Y.; Cao, L.; Hu, L. Sirtuin 6 mitigated LPS-induced human umbilical vein endothelial cells inflammatory responses through modulating nuclear factor erythroid 2-related factor 2. J. Cell. Biochem. 2019, 120, 11305–11317. [Google Scholar] [CrossRef]
- Wyman, A.E.; Nguyen, T.T.T.; Karki, P.; Tulapurkar, M.E.; Zhang, C.O.; Kim, J.; Feng, T.G.; Dabo, A.J.; Todd, N.W.; Luzina, I.G.; et al. SIRT7 deficiency suppresses inflammation, induces EndoMT, and increases vascular permeability in primary pulmonary endothelial cells. Sci. Rep. 2020, 10, 12497. [Google Scholar] [CrossRef]
- Tafrihi, M.; Nakhaei Sistani, R. E-Cadherin/β-Catenin Complex: A Target for Anticancer and Antimetastasis Plants/Plant-derived Compounds. Nutr. Cancer 2017, 69, 702–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Wu, X.Q.; Chen, L.; Hu, H.H.; Wang, Y.N.; Zhao, Y.Y. Poricoic acid A as a modulator of TPH-1 expression inhibits renal fibrosis via modulating protein stability of β-catenin and β-catenin-mediated transcription. Ther. Adv. Chronic Dis. 2020, 11, 2040622320962648. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Shen, M.J.; Wang, H.; Li, Y.; Tang, A.L.; Li, S.; Xiong, M.C.; Guo, Y.; Zhang, G.Q. Sirt3 Maintains Microvascular Endothelial Adherens Junction Integrity to Alleviate Sepsis-Induced Lung Inflammation by Modulating the Interaction of VE-Cadherin and β-Catenin. Oxidative Med. Cell. Longev. 2021, 2021, 8978795. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, J.; Becker, B.F.; Chappell, D. Endothelial Glycocalyx. Crit. Care Clin. 2020, 36, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. JTH 2019, 17, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef]
- Ali, M.M.; Mahmoud, A.M.; Le Master, E.; Levitan, I.; Phillips, S.A. Role of matrix metalloproteinases and histone deacetylase in oxidative stress-induced degradation of the endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H647–H663. [Google Scholar] [CrossRef]
- Ramnath, R.; Foster, R.R.; Qiu, Y.; Cope, G.; Butler, M.J.; Salmon, A.H.; Mathieson, P.W.; Coward, R.J.; Welsh, G.I.; Satchell, S.C. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: A contributor to endothelial cell glycocalyx dysfunction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4686–4699. [Google Scholar] [CrossRef]
- Lipphardt, M.; Song, J.W.; Goligorsky, M.S. Sirtuin 1 and endothelial glycocalyx. Pflug. Arch. Eur. J. Physiol. 2020, 472, 991–1002. [Google Scholar] [CrossRef]
- Wang, X.Y.; Li, X.Y.; Wu, C.H.; Hao, Y.; Fu, P.H.; Mei, H.X.; Chen, F.; Gong, Y.Q.; Jin, S.W.; Li, H. Protectin conjugates in tissue regeneration 1 restores lipopolysaccharide-induced pulmonary endothelial glycocalyx loss via ALX/SIRT1/NF-kappa B axis. Respir. Res. 2021, 22, 193. [Google Scholar] [CrossRef]
- Li, H.; Hao, Y.; Yang, L.L.; Wang, X.Y.; Li, X.Y.; Bhandari, S.; Han, J.; Liu, Y.J.; Gong, Y.Q.; Scott, A.; et al. MCTR1 alleviates lipopolysaccharide-induced acute lung injury by protecting lung endothelial glycocalyx. J. Cell. Physiol. 2020, 235, 7283–7294. [Google Scholar] [CrossRef] [PubMed]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; You, J.; Zhou, Q. Research advance on mechanism and application of HATs and HDACs in epithelial-mesenchymal transition of lung cancer. Zhongguo Fei Ai Za Zhi Chin. J. Lung Cancer 2013, 16, 211–215. [Google Scholar]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Zhu, Y.; Tan, J.; Xie, H.; Wang, J.; Meng, X.; Wang, R. HIF-1α regulates EMT via the Snail and β-catenin pathways in paraquat poisoning-induced early pulmonary fibrosis. J. Cell. Mol. Med. 2016, 20, 688–697. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Carter, C.L.; Lin, C.; Liu, C.Y.; Yang, L.; Liu, Z.R. Phosphorylated p68 RNA helicase activates Snail1 transcription by promoting HDAC1 dissociation from the Snail1 promoter. Oncogene 2010, 29, 5427–5436. [Google Scholar] [CrossRef]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef]
- Sun, J.; Meng, D.; Yu, T.; Li, F.; Zhang, G.; Tian, X.; Zhao, N.; Li, G.; Li, L.; Wang, H.; et al. N-terminal truncated carboxypeptidase E represses E-cadherin expression in lung cancer by stabilizing the Snail-HDAC complex. Am. J. Cancer Res. 2020, 10, 925–938. [Google Scholar]
- Gu, L.Z.; Sun, H.; Chen, J.H. Histone deacetylases 3 deletion restrains PM2.5-induced mice lung injury by regulating NF-κB and TGF-β/Smad2/3 signaling pathways. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 85, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, D.; Wang, L.; Zhao, W.; Han, X. Trichostatin A inhibits radiation-induced epithelial-to-mesenchymal transition in the alveolar epithelial cells. Oncotarget 2017, 8, 101745–101759. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.; Liu, T.; McCall, C.E. Epigenetic coordination of acute systemic inflammation: Potential therapeutic targets. Expert Rev. Clin. Immunol. 2014, 10, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Li, Y.; Xue, Z.; Shao, R.; Li, L.; Zhu, Y.; Zhang, H.; Yang, J. Xuanfei Baidu Decoction reduces acute lung injury by regulating infiltration of neutrophils and macrophages via PD-1/IL17A pathway. Pharmacol. Res. 2022, 176, 106083. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Babes, L.; Rahn, J.J.; Ahn, B.Y.; Goring, K.R.; King, J.C.; Lau, A.; Petri, B.; Hao, X.; Chojnacki, A.K.; et al. Dipeptidase-1 Is an Adhesion Receptor for Neutrophil Recruitment in Lungs and Liver. Cell 2019, 178, 1205–1221.e17. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef]
- Poli, V.; Pui-Yan Ma, V.; Di Gioia, M.; Broggi, A.; Benamar, M.; Chen, Q.; Mazitschek, R.; Haggarty, S.J.; Chatila, T.A.; Karp, J.M.; et al. Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience 2021, 24, 103256. [Google Scholar] [CrossRef]
- Sahakian, E.; Chen, J.; Powers, J.J.; Chen, X.; Maharaj, K.; Deng, S.L.; Achille, A.N.; Lienlaf, M.; Wang, H.W.; Cheng, F.; et al. Essential role for histone deacetylase 11 (HDAC11) in neutrophil biology. J. Leukoc. Biol. 2017, 102, 475–486. [Google Scholar] [CrossRef]
- Hamilton, J.L.; Mohamed, M.F.; Witt, B.R.; Wimmer, M.A.; Shafikhani, S.H. Therapeutic assessment of N-formyl-methionyl-leucyl-phenylalanine (fMLP) in reducing periprosthetic joint infection. Eur. Cells Mater. 2021, 42, 122–138. [Google Scholar] [CrossRef]
- Lu, W.; Yao, X.; Ouyang, P.; Dong, N.; Wu, D.; Jiang, X.; Wu, Z.; Zhang, C.; Xu, Z.; Tang, Y.; et al. Drug Repurposing of Histone Deacetylase Inhibitors That Alleviate Neutrophilic Inflammation in Acute Lung Injury and Idiopathic Pulmonary Fibrosis via Inhibiting Leukotriene A4 Hydrolase and Blocking LTB4 Biosynthesis. J. Med. Chem. 2017, 60, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; He, Y.; Ding, D.; Cui, A.; Bao, H.; Ma, J.; Hou, X.; Li, Y.; Feng, D.; Li, X.; et al. Aging exaggerates acute-on-chronic alcohol-induced liver injury in mice and humans by inhibiting neutrophilic sirtuin 1-C/EBPα-miRNA-223 axis. Hepatology 2022, 75, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Bordbari, S.; Mörchen, B.; Pylaeva, E.; Siakaeva, E.; Spyra, I.; Domnich, M.; Droege, F.; Kanaan, O.; Lang, K.S.; Schadendorf, D.; et al. SIRT1-mediated deacetylation of FOXO3a transcription factor supports pro-angiogenic activity of interferon-deficient tumor-associated neutrophils. Int. J. Cancer 2022, 150, 1198–1211. [Google Scholar] [CrossRef]
- Cao, W.; Zhu, M.Y.; Lee, S.H.; Lee, S.B.; Kim, H.J.; Park, B.O.; Yoon, C.H.; Khadka, D.; Oh, G.S.; Shim, H.; et al. Modulation of Cellular NAD(+) Attenuates Cancer-Associated Hypercoagulability and Thrombosis via the Inhibition of Tissue Factor and Formation of Neutrophil Extracellular Traps. Int. J. Mol. Sci. 2021, 22, 12085. [Google Scholar] [CrossRef]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef]
- Candido, E.P.; Reeves, R.; Davie, J.R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978, 14, 105–113. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [CrossRef]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Bressi, J.C.; de Jong, R.; Wu, Y.; Jennings, A.J.; Brown, J.W.; O’Connell, S.; Tari, L.W.; Skene, R.J.; Vu, P.; Navre, M.; et al. Benzimidazole and imidazole inhibitors of histone deacetylases: Synthesis and biological activity. Bioorganic Med. Chem. Lett. 2010, 20, 3138–3141. [Google Scholar] [CrossRef]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; De Fabiani, E.; Altucci, L.; et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Wald, J.; Lavu, S.; Roberts, J.; Beaumont, C.; Haddad, J.; Elliott, P.; Westphal, C.; Jacobson, E. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br. J. Clin. Pharmacol. 2013, 75, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ma, X.; Yuan, C.; He, Y.; Li, L.; Fang, S.; Xia, W.; He, T.; Qian, S.; Xu, Z.; et al. Discovery of 2-((4,6-dimethylpyrimidin-2-yl)thio)-N-phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. Eur. J. Med. Chem. 2017, 134, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.S.; Scian, M.; Sripathy, S.; Posakony, J.; Lao, U.; Loe, T.K.; Leko, V.; Thalhofer, A.; Schuler, A.D.; Bedalov, A.; et al. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J. Med. Chem. 2014, 57, 3283–3294. [Google Scholar] [CrossRef]
- Disch, J.S.; Evindar, G.; Chiu, C.H.; Blum, C.A.; Dai, H.; Jin, L.; Schuman, E.; Lind, K.E.; Belyanskaya, S.L.; Deng, J.; et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. [Google Scholar] [CrossRef]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure-Activity Relationship, X-ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.; Lam, E.W.; Steegborn, C.; et al. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Khan, M.N.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Family | Class | Homology (S. cerevisiae) | Isoform | Localization | Key Features |
|---|---|---|---|---|---|
| Classical HDAC family (histone deacetylase family) | Class I HDACs | RPD3, HOS | HDAC1 | Nucleus | Exist in NuRD, Sin3A, CoREST, and MiDAC complexes |
| HDAC2 | |||||
| HDAC3 | Nucleus/Cytoplasm | Exist in NCoR/SMRT | |||
| HDAC8 | Long-chain acyl lysine substrates | ||||
| Class IIa HDACs | HDA1 | HDAC4 | Nucleus/ Cytoplasm | HDRP and MEF2-interacting transcriptional repressors | |
| HDAC5 | |||||
| HDAC7 | |||||
| HDAC9 | |||||
| Class IIb HDACs | HDAC6 | Cytoplasm | CD1 and CD2 structural domains | ||
| HDAC10 | |||||
| Class IV HDACs | —— | HDAC11 | Nucleus | Long-chain acyl lysine substrates | |
| Sir2 family (Sirtuin) | Class III HDACs | Sir2 | Sirt1 | Nucleus/ Cytoplasm | NAD+-dependent, Related to ADP/ATP |
| Sirt2 | |||||
| Sirt3 | Nucleus/Mitochondria | ||||
| Sirt4 | Mitochondria | ||||
| Sirt5 | |||||
| SIrt6 | Nucleus | ||||
| Sirt7 |
| Compound Name | Compound Characteristics | Compound Targets | References |
|---|---|---|---|
| HDAC inhibitor | |||
| n-Butyrate | Carboxylic acid | Class I, Class IIa, and Class IV HDACs | [208,209] |
| Trichostatin A (TSA) | Hydroxamic acid, Streptomyces product | Class I, Class II, and Class IV HDACs | [210] |
| Trapoxin | Fungal cyclopeptide, irreversible inhibitor | Class I, Class IIa, and Class IV HDACs | [211] |
| FK228 (romidepsin) | Potent antitumor antibiotic, approved by FDA in 2009 | Class I, Class IIa, and Class IV HDACs | [212] |
| Suberoylanilide hydroxamic acid (SAHA) | Hydroxamic acid, a synthetic compound for induction of erythrogenic differentiation, approved by the FDA in 2006 | Class I, Class II, and Class IV HDACs | [213,214] |
| MS-275 (Entinostat) | Benzamide derivative | HDAC1, HDAC2, HDAC3 | [215,216] |
| Tubacin | —— | HDAC6 | [217] |
| 2-trifluoroacetylthiophene derivatives | Class IIa HDACs specific substrate | Class IIa HDACs | [218] |
| Sirtuin activator | |||
| Piceatannol | Natural product | Sirt1, Sirt3, Sirt5 | [219] |
| Resveratrol | Natural products in grapes and red wines | Sirt1, Sirt3, Sirt5 | [220] |
| 1,4-DHP derivative | Natural products | Sirt1, Sirt2, Sirt3 | [221] |
| SRT1720 | Synthesis, selective | Sirt1 | [222] |
| SRT2104 | Synthesis, selective | Sirt1 | [223] |
| Sirtuin inhibitor | |||
| Cambinol | Natural product | One of the first Sirtuin inhibitors discovered | [224] |
| Ex-527 | Competitive inhibition of nicotinamide binding | Sirt1, Sirt2, Sirt3 | [225] |
| AGK2 | —— | Sirt2 | [226] |
| Compound 28e | Synthetic, selective | Sirt1, Sirt2, Sirt3 | [227] |
| Compound 8 | —— | Sirt1, Sirt2, Sirt3 | [228] |
| ELT-11c | —— | Sirt1, Sirt2, Sirt3 | [229] |
| UBCS0137 (Compound 39) | —— | Sirt1, Sirt2, Sirt3, Sirt5 | [230] |
| Compound 15e | —— | Sirt1, Sirt2, Sirt3 | [231] |
| SirReal2 | —— | Sirt1, Sirt2, Sirt3, Sirt4, Sirt5, Sirt6 | [232] |
| 3′-(3-fluoro-phenethyloxy)-2-anilinobenzamide | Synthetic, selective | Sirt1, Sirt2 | [233] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, G.; Liu, B.; Fu, T.; Liu, Y.; Li, B.; Li, N.; Geng, Q. The Role of Histone Deacetylases in Acute Lung Injury—Friend or Foe. Int. J. Mol. Sci. 2023, 24, 7876. https://doi.org/10.3390/ijms24097876
Luo G, Liu B, Fu T, Liu Y, Li B, Li N, Geng Q. The Role of Histone Deacetylases in Acute Lung Injury—Friend or Foe. International Journal of Molecular Sciences. 2023; 24(9):7876. https://doi.org/10.3390/ijms24097876
Chicago/Turabian StyleLuo, Guoqing, Bohao Liu, Tinglv Fu, Yi Liu, Boyang Li, Ning Li, and Qing Geng. 2023. "The Role of Histone Deacetylases in Acute Lung Injury—Friend or Foe" International Journal of Molecular Sciences 24, no. 9: 7876. https://doi.org/10.3390/ijms24097876