
The Values and Perspectives of Organoids in the Field of Metabolic Syndrome



Abstract
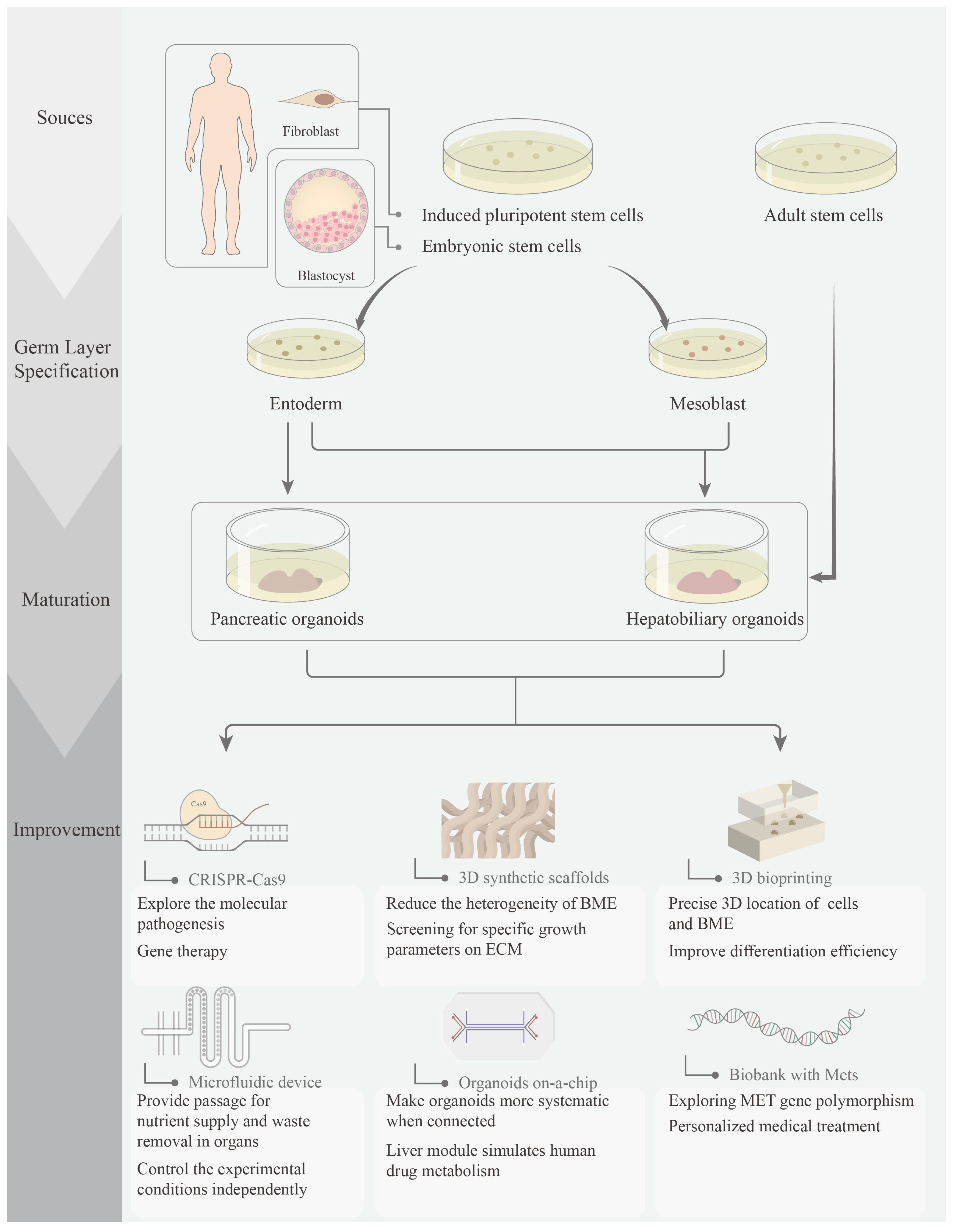
1. Introduction
2. Pancreatic Organoids
2.1. The Secretion of Insulin Requires the Cooperation of Different Cells
2.2. Progress of Pancreatic Organoids
2.3. Pancreatic Organoids and Endogenous Regeneration of Metabolic Syndrome
2.3.1. Islet Regeneration
2.3.2. Oxidative Stress
2.3.3. Circadian Rhythm
3. Hepatobiliary Organoids
3.1. The Formation of Liver Fibrosis Requires the Cooperation of Different Cells
3.2. Progress of Hepatobiliary Organoids
3.2.1. Hepatobiliary Organoids
3.2.2. NAFLD Modelling
3.3. Hepatobiliary Organoids and Endogenous Regeneration of Metabolic Syndrome
3.3.1. Liver Regeneration
3.3.2. Regression of Liver Fibrosis
4. Perspectives of Metabolic Syndrome-Related Organoids
4.1. CRISPR-Based Gene Editing
4.2. 3D Synthetic Scaffolds
4.3. 3D Bioprinting
4.4. Organoids in a Microfluidic Device
4.5. Organoids on a Chip
4.6. Biobanks and MetS
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Aguilar, D.; Deswal, A.; Dunbar, S.B.; Francis, G.S.; Horwich, T.; Jessup, M.; Kosiborod, M.; Pritchett, A.M.; Ramasubbu, K.; et al. Contributory Risk and Management of Comorbidities of Hypertension, Obesity, Diabetes Mellitus, Hyperlipidemia, and Metabolic Syndrome in Chronic Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2016, 134, e535–e578. [Google Scholar] [CrossRef]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2021, 50, 101143. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of Metabolic Syndrome and Dietary Intervention. Int. J. Mol. Sci. 2018, 20, 128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jain, R.; Lammert, E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes. Metab. 2009, 11 (Suppl. S4), 159–167. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef][Green Version]
- Brenot, F.; Herve, P.; Petitpretz, P.; Parent, F.; Duroux, P.; Simonneau, G. Primary pulmonary hypertension and fenfluramine use. Br. Heart J. 1993, 70, 537–541. [Google Scholar] [CrossRef][Green Version]
- Connolly, H.M.; Crary, J.L.; McGoon, M.D.; Hensrud, D.D.; Edwards, B.S.; Edwards, W.D.; Schaff, H.V. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med. 1997, 337, 581–588. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef][Green Version]
- Bhaduri, A.; Andrews, M.G.; Kriegstein, A.R.; Nowakowski, T.J. Are Organoids Ready for Prime Time? Cell Stem Cell 2020, 27, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Castro-Barquero, S.; Ruiz-León, A.M.; Sierra-Pérez, M.; Estruch, R.; Casas, R. Dietary Strategies for Metabolic Syndrome: A Comprehensive Review. Nutrients 2020, 12, 2983. [Google Scholar] [CrossRef]
- Wilkinson, M.J.; Manoogian, E.N.C.; Zadourian, A.; Lo, H.; Fakhouri, S.; Shoghi, A.; Wang, X.; Fleischer, J.G.; Navlakha, S.; Panda, S.; et al. Ten-Hour Time-Restricted Eating Reduces Weight, Blood Pressure, and Atherogenic Lipids in Patients with Metabolic Syndrome. Cell Metab. 2020, 31, 92–104.e105. [Google Scholar] [CrossRef] [PubMed]
- Wewege, M.A.; Thom, J.M.; Rye, K.A.; Parmenter, B.J. Aerobic, resistance or combined training: A systematic review and meta-analysis of exercise to reduce cardiovascular risk in adults with metabolic syndrome. Atherosclerosis 2018, 274, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Aminian, A.; Al-Kurd, A.; Wilson, R.; Bena, J.; Fayazzadeh, H.; Singh, T.; Albaugh, V.L.; Shariff, F.U.; Rodriguez, N.A.; Jin, J.; et al. Association of Bariatric Surgery with Major Adverse Liver and Cardiovascular Outcomes in Patients with Biopsy-Proven Nonalcoholic Steatohepatitis. JAMA 2021, 326, 2031–2042. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef][Green Version]
- Atkinson, M.A.; Campbell-Thompson, M.; Kusmartseva, I.; Kaestner, K.H. Organisation of the human pancreas in health and in diabetes. Diabetologia 2020, 63, 1966–1973. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695–709. [Google Scholar] [CrossRef]
- Ramond, C.; Glaser, N.; Berthault, C.; Ameri, J.; Kirkegaard, J.S.; Hansson, M.; Honoré, C.; Semb, H.; Scharfmann, R. Reconstructing human pancreatic differentiation by mapping specific cell populations during development. eLife 2017, 6, e27564. [Google Scholar] [CrossRef] [PubMed]
- Ramond, C.; Beydag-Tasöz, B.S.; Azad, A.; van de Bunt, M.; Petersen, M.B.K.; Beer, N.L.; Glaser, N.; Berthault, C.; Gloyn, A.L.; Hansson, M.; et al. Understanding human fetal pancreas development using subpopulation sorting, RNA sequencing and single-cell profiling. Development 2018, 145, dev165480. [Google Scholar] [CrossRef][Green Version]
- Nair, G.; Hebrok, M. Islet formation in mice and men: Lessons for the generation of functional insulin-producing β-cells from human pluripotent stem cells. Curr. Opin. Genet. Dev. 2015, 32, 171–180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Capozzi, M.E.; Svendsen, B.; Encisco, S.E.; Lewandowski, S.L.; Martin, M.D.; Lin, H.; Jaffe, J.L.; Coch, R.W.; Haldeman, J.M.; MacDonald, P.E.; et al. β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 2019, 4, e126742. [Google Scholar] [CrossRef]
- Rodriguez-Diaz, R.; Molano, R.D.; Weitz, J.R.; Abdulreda, M.H.; Berman, D.M.; Leibiger, B.; Leibiger, I.B.; Kenyon, N.S.; Ricordi, C.; Pileggi, A.; et al. Paracrine Interactions within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab. 2018, 27, 549–558.e544. [Google Scholar] [CrossRef][Green Version]
- Benninger, R.K.P.; Kravets, V. The physiological role of β-cell heterogeneity in pancreatic islet function. Nat. Rev. Endocrinol. 2022, 18, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef][Green Version]
- Hodson, D.J.; Mitchell, R.K.; Bellomo, E.A.; Sun, G.; Vinet, L.; Meda, P.; Li, D.; Li, W.H.; Bugliani, M.; Marchetti, P.; et al. Lipotoxicity disrupts incretin-regulated human β cell connectivity. J. Clin. Investig. 2013, 123, 4182–4194. [Google Scholar] [CrossRef][Green Version]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef][Green Version]
- Jo, J.; Choi, M.Y.; Koh, D.S. Beneficial effects of intercellular interactions between pancreatic islet cells in blood glucose regulation. J. Theor. Biol. 2009, 257, 312–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.; van de Wetering, M.; Sojoodi, M.; Li, V.S.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef][Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef][Green Version]
- Jennings, R.E.; Berry, A.A.; Strutt, J.P.; Gerrard, D.T.; Hanley, N.A. Human pancreas development. Development 2015, 142, 3126–3137. [Google Scholar] [CrossRef][Green Version]
- Petersen, M.B.K.; Gonçalves, C.A.C.; Kim, Y.H.; Grapin-Botton, A. Recapitulating and Deciphering Human Pancreas Development From Human Pluripotent Stem Cells in a Dish. Curr. Top. Dev. Biol. 2018, 129, 143–190. [Google Scholar] [CrossRef]
- Slack, J.M. Developmental biology of the pancreas. Development 1995, 121, 1569–1580. [Google Scholar] [CrossRef]
- Polak, M.; Bouchareb-Banaei, L.; Scharfmann, R.; Czernichow, P. Early pattern of differentiation in the human pancreas. Diabetes 2000, 49, 225–232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rezania, A.; Bruin, J.E.; Xu, J.; Narayan, K.; Fox, J.K.; O’Neil, J.J.; Kieffer, T.J. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 2013, 31, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D’Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011, 29, 750–756. [Google Scholar] [CrossRef]
- Jennings, R.E.; Berry, A.A.; Gerrard, D.T.; Wearne, S.J.; Strutt, J.; Withey, S.; Chhatriwala, M.; Piper Hanley, K.; Vallier, L.; Bobola, N.; et al. Laser Capture and Deep Sequencing Reveals the Transcriptomic Programmes Regulating the Onset of Pancreas and Liver Differentiation in Human Embryos. Stem Cell Rep. 2017, 9, 1387–1394. [Google Scholar] [CrossRef][Green Version]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef][Green Version]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hrvatin, S.; O’Donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, β cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.-R.; Harb, G.; Poh, Y.-C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Youngblood, R.L.; Sampson, J.P.; Lebioda, K.R.; Shea, L.D. Microporous scaffolds support assembly and differentiation of pancreatic progenitors into β-cell clusters. Acta Biomater. 2019, 96, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Gan, W.J.; Zavortink, M.; Ludick, C.; Templin, R.; Webb, R.; Webb, R.; Ma, W.; Poronnik, P.; Parton, R.G.; Gaisano, H.Y.; et al. Cell polarity defines three distinct domains in pancreatic β-cells. J. Cell Sci. 2017, 130, 143–151. [Google Scholar] [CrossRef][Green Version]
- Almaça, J.; Weitz, J.; Rodriguez-Diaz, R.; Pereira, E.; Caicedo, A. The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metab. 2018, 27, 630–644. [Google Scholar] [CrossRef][Green Version]
- Takahashi, Y.; Sekine, K.; Kin, T.; Takebe, T.; Taniguchi, H. Self-Condensation Culture Enables Vascularization of Tissue Fragments for Efficient Therapeutic Transplantation. Cell Rep. 2018, 23, 1620–1629. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takahashi, Y.; Takebe, T.; Taniguchi, H. Methods for Generating Vascularized Islet-Like Organoids Via Self-Condensation. Curr. Protoc. Stem Cell Biol. 2018, 45, e49. [Google Scholar] [CrossRef] [PubMed]
- Palikuqi, B.; Nguyen, D.-H.T.; Li, G.; Schreiner, R.; Pellegata, A.F.; Liu, Y.; Redmond, D.; Geng, F.; Lin, Y.; Gómez-Salinero, J.M.; et al. Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature 2020, 585, 426–432. [Google Scholar] [CrossRef]
- Malin, S.K.; Finnegan, S.; Fealy, C.E.; Filion, J.; Rocco, M.B.; Kirwan, J.P. β-Cell dysfunction is associated with metabolic syndrome severity in adults. Metab. Syndr. Relat. Disord. 2014, 12, 79–85. [Google Scholar] [CrossRef][Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef][Green Version]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef][Green Version]
- Kulkarni, R.N.; Mizrachi, E.-B.; Ocana, A.G.; Stewart, A.F. Human β-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef][Green Version]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocaña, A. Human β-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef][Green Version]
- Stewart, A.F.; Hussain, M.A.; García-Ocaña, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human β-cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef][Green Version]
- Wei, P.; Li, L.; Qi, H.; Zhou, H.-X.; Deng, C.-Y.; Li, F.-R. Reversible immortalization of Nestin-positive precursor cells from pancreas and differentiation into insulin-secreting cells. Biochem. Biophys. Res. Commun. 2012, 418, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dólleman, S.; Liu, S.; Ackermann, A.M.; Cáceres, E.; Hunter, A.E.; et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017, 25, 911–926. [Google Scholar] [CrossRef][Green Version]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Klein, D.; Lanzoni, G.; García-Santana, C.; Montalvo, A.; Pláceres-Uray, F.; Mazza, E.M.C.; Ricordi, C.; Inverardi, L.A.; et al. P2RY1/ALK3-Expressing Cells within the Adult Human Exocrine Pancreas Are BMP-7 Expandable and Exhibit Progenitor-like Characteristics. Cell Rep. 2018, 22, 2408–2420. [Google Scholar] [CrossRef][Green Version]
- Loomans, C.J.M.; Williams Giuliani, N.; Balak, J.; Ringnalda, F.; van Gurp, L.; Huch, M.; Boj, S.F.; Sato, T.; Kester, L.; de Sousa Lopes, S.M.C.; et al. Expansion of Adult Human Pancreatic Tissue Yields Organoids Harboring Progenitor Cells with Endocrine Differentiation Potential. Stem Cell Rep. 2018, 10, 712–724. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, D.; Wang, J.; Bai, L.; Pan, H.; Feng, H.; Clevers, H.; Zeng, Y.A. Long-Term Expansion of Pancreatic Islet Organoids from Resident Procr+ Progenitors. Cell 2020, 180, 1198–1211. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef][Green Version]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Vernier, M.; Dufour, C.R.; McGuirk, S.; Scholtes, C.; Li, X.; Bourmeau, G.; Kuasne, H.; Park, M.; St-Pierre, J.; Audet-Walsh, E.; et al. Estrogen-related receptors are targetable ROS sensors. Genes Dev. 2020, 34, 544–559. [Google Scholar] [CrossRef]
- Yoshihara, E.; Wei, Z.; Lin, C.S.; Fang, S.; Ahmadian, M.; Kida, Y.; Tseng, T.; Dai, Y.; Yu, R.T.; Liddle, C.; et al. ERRγ Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive β Cells. Cell Metab. 2016, 23, 622–634. [Google Scholar] [CrossRef][Green Version]
- Choi, J.; Oh, T.G.; Jung, H.-W.; Park, K.-Y.; Shin, H.; Jo, T.; Kang, D.-S.; Chanda, D.; Hong, S.; Kim, J.; et al. Estrogen-Related Receptor γ Maintains Pancreatic Acinar Cell Function and Identity by Regulating Cellular Metabolism. Gastroenterology 2022, 163, 239–256. [Google Scholar] [CrossRef]
- Perelis, M.; Marcheva, B.; Ramsey, K.M.; Schipma, M.J.; Hutchison, A.L.; Taguchi, A.; Peek, C.B.; Hong, H.; Huang, W.; Omura, C.; et al. Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 2015, 350, aac4250. [Google Scholar] [CrossRef][Green Version]
- Alvarez-Dominguez, J.R.; Donaghey, J.; Rasouli, N.; Kenty, J.H.R.; Helman, A.; Charlton, J.; Straubhaar, J.R.; Meissner, A.; Melton, D.A. Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell 2020, 26, 108–122. [Google Scholar] [CrossRef][Green Version]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418 Pt 1, 55–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef][Green Version]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef]
- Bedossa, P. Utility and appropriateness of the fatty liver inhibition of progression (FLIP) algorithm and steatosis, activity, and fibrosis (SAF) score in the evaluation of biopsies of nonalcoholic fatty liver disease. Hepatology 2014, 60, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef][Green Version]
- Friedman, S.L. Liver fibrosis -- From bench to bedside. J. Hepatol. 2003, 38 (Suppl. S1), S38–S53. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Cheng, D.; Chai, J.; Wang, H.; Fu, L.; Peng, S.; Ni, X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. [Google Scholar] [CrossRef]
- Wells, R.G. The portal fibroblast: Not just a poor man’s stellate cell. Gastroenterology 2014, 147, 41–47. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.W.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef][Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef][Green Version]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef][Green Version]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.-R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sinton, M.C.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Wernig-Zorc, S.; Thomson, J.P.; Carter, R.N.; Lyall, M.J.; Walker, P.D.; Thakker, A.; Meehan, R.R.; et al. A human pluripotent stem cell model for the analysis of metabolic dysfunction in hepatic steatosis. iScience 2021, 24, 101931. [Google Scholar] [CrossRef]
- McCarron, S.; Bathon, B.; Conlon, D.M.; Abbey, D.; Rader, D.J.; Gawronski, K.; Brown, C.D.; Olthoff, K.M.; Shaked, A.; Raabe, T.D. Functional Characterization of Organoids Derived from Irreversibly Damaged Liver of Patients with NASH. Hepatology 2021, 74, 1825–1844. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Mofrad, P.S.; Contos, M.J.; Sargeant, C.; Luketic, V.A.; Sterling, R.K.; Stravitz, R.T.; Shiffman, M.L.; Clore, J.; Mills, A.S. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1107–1115. [Google Scholar] [CrossRef]
- Wei, J.; Qiu, D.K.; Ma, X. Bile acids and insulin resistance: Implications for treating nonalcoholic fatty liver disease. J. Dig. Dis. 2009, 10, 85–90. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef]
- Meirelles Júnior, R.F.; Salvalaggio, P.; Rezende, M.B.d.; Evangelista, A.S.; Guardia, B.D.; Matielo, C.E.L.; Neves, D.B.; Pandullo, F.L.; Felga, G.E.G.; Alves, J.A.d.S.; et al. Liver transplantation: History, outcomes and perspectives. Einstein 2015, 13, 149–152. [Google Scholar] [CrossRef][Green Version]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43 (Suppl. S1), S45–S53. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Khan, Z. Liver Stem Cells: Experimental Findings and Implications for Human Liver Disease. Gastroenterology 2015, 149, 876–882. [Google Scholar] [CrossRef][Green Version]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef][Green Version]
- Hattoum, A.; Rubin, E.; Orr, A.; Michalopoulos, G.K. Expression of hepatocyte epidermal growth factor receptor, FAS and glypican 3 in EpCAM-positive regenerative clusters of hepatocytes, cholangiocytes, and progenitor cells in human liver failure. Hum. Pathol. 2013, 44, 743–749. [Google Scholar] [CrossRef][Green Version]
- Peng, J.; Li, F.; Wang, J.; Wang, C.; Jiang, Y.; Liu, B.; He, J.; Yuan, K.; Pan, C.; Lin, M.; et al. Identification of a rare Gli1+ progenitor cell population contributing to liver regeneration during chronic injury. Cell Discov. 2022, 8, 118. [Google Scholar] [CrossRef]
- Terai, S.; Tsuchiya, A.; Watanabe, Y.; Takeuchi, S. Transition of clinical and basic studies on liver cirrhosis treatment using cells to seek the best treatment. Inflamm. Regen. 2021, 41, 27. [Google Scholar] [CrossRef]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lämmermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555. [Google Scholar] [CrossRef][Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef][Green Version]
- Takeuchi, S.; Tsuchiya, A.; Iwasawa, T.; Nojiri, S.; Watanabe, T.; Ogawa, M.; Yoshida, T.; Fujiki, K.; Koui, Y.; Kido, T.; et al. Small extracellular vesicles derived from interferon-γ pre-conditioned mesenchymal stromal cells effectively treat liver fibrosis. NPJ Regen. Med. 2021, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Iguchi, T.; Iwasawa, K.; Dunn, A.; Thompson, W.L.; Yoneyama, Y.; Chaturvedi, P.; Zorn, A.M.; Wintzinger, M.; Quattrocelli, M.; et al. En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell 2022, 185, 4216–4232. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; López-Iglesias, C.; Peters, P.J.; Rodríguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- Ranga, A.; Gobaa, S.; Okawa, Y.; Mosiewicz, K.; Negro, A.; Lutolf, M.P. 3D niche microarrays for systems-level analyses of cell fate. Nat. Commun. 2014, 5, 4324. [Google Scholar] [CrossRef][Green Version]
- Raza, A.; Ki, C.S.; Lin, C.-C. The influence of matrix properties on growth and morphogenesis of human pancreatic ductal epithelial cells in 3D. Biomaterials 2013, 34, 5117–5127. [Google Scholar] [CrossRef][Green Version]
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 460–470. [Google Scholar] [CrossRef]
- Correia Carreira, S.; Begum, R.; Perriman, A.W. 3D Bioprinting: The Emergence of Programmable Biodesign. Adv. Healthc. Mater. 2020, 9, e1900554. [Google Scholar] [CrossRef]
- Chakraborty, J.; Chawla, S.; Ghosh, S. Developmental biology-inspired tissue engineering by combining organoids and 3D bioprinting. Curr. Opin. Biotechnol. 2022, 78, 102832. [Google Scholar] [CrossRef]
- Beattie, G.M.; Rubin, J.S.; Mally, M.I.; Otonkoski, T.; Hayek, A. Regulation of proliferation and differentiation of human fetal pancreatic islet cells by extracellular matrix, hepatocyte growth factor, and cell-cell contact. Diabetes 1996, 45, 1223–1228. [Google Scholar] [CrossRef]
- Gage, B.K.; Webber, T.D.; Kieffer, T.J. Initial cell seeding density influences pancreatic endocrine development during in vitro differentiation of human embryonic stem cells. PLoS ONE 2013, 8, e82076. [Google Scholar] [CrossRef][Green Version]
- Memon, B.; Karam, M.; Al-Khawaga, S.; Abdelalim, E.M. Enhanced differentiation of human pluripotent stem cells into pancreatic progenitors co-expressing PDX1 and NKX6.1. Stem Cell Res. Ther. 2018, 9, 15. [Google Scholar] [CrossRef][Green Version]
- Bernal, P.N.; Delrot, P.; Loterie, D.; Li, Y.; Malda, J.; Moser, C.; Levato, R. Volumetric Bioprinting of Complex Living-Tissue Constructs within Seconds. Adv. Mater. 2019, 31, e1904209. [Google Scholar] [CrossRef][Green Version]
- Daly, A.C.; Davidson, M.D.; Burdick, J.A. 3D bioprinting of high cell-density heterogeneous tissue models through spheroid fusion within self-healing hydrogels. Nat. Commun. 2021, 12, 753. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Wang, H.; Zhao, M.; Tao, T.; Zhang, X.; Qin, J. A Droplet Microfluidic System to Fabricate Hybrid Capsules Enabling Stem Cell Organoid Engineering. Adv. Sci. 2020, 7, 1903739. [Google Scholar] [CrossRef][Green Version]
- Koike, H.; Iwasawa, K.; Ouchi, R.; Maezawa, M.; Giesbrecht, K.; Saiki, N.; Ferguson, A.; Kimura, M.; Thompson, W.L.; Wells, J.M.; et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature 2019, 574, 112–116. [Google Scholar] [CrossRef]
- Vunjak-Novakovic, G.; Ronaldson-Bouchard, K.; Radisic, M. Organs-on-a-chip models for biological research. Cell 2021, 184, 4597–4611. [Google Scholar] [CrossRef]
- Tao, T.; Deng, P.; Wang, Y.; Zhang, X.; Guo, Y.; Chen, W.; Qin, J. Microengineered Multi-Organoid System from hiPSCs to Recapitulate Human Liver-Islet Axis in Normal and Type 2 Diabetes. Adv. Sci. 2022, 9, e2103495. [Google Scholar] [CrossRef]
- Abou Ziki, M.D.; Mani, A. Metabolic syndrome: Genetic insights into disease pathogenesis. Curr. Opin. Lipidol. 2016, 27, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Mattheisen, M.; Dahm, S.; Nürnberg, P.; Roe, C.; Johnson, J.; Cox, N.J.; Wichmann, H.E.; Wienker, T.F.; Schulze, J.; et al. A German genome-wide linkage scan for type 2 diabetes supports the existence of a metabolic syndrome locus on chromosome 1p36.13 and a type 2 diabetes locus on chromosome 16p12.2. Diabetologia 2007, 50, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef][Green Version]


{kind=link}
| Initial Cells | Advantages | Disadvantages | Year |
|---|---|---|---|
| hPSCs | The first strategy for producing functional cells from hPSCs developed a 6-step process by including predetermined components. | Insufficient maturity; GSIS is not high enough. Not suitable for long-term cultivation. | 2014 [42] |
| hPSCs | It was demonstrated that endocrine cell clustering is a crucial stage in the development of hPSC-derived cells in culture. | Insufficient maturity; GSIS is not high enough. Not suitable for long-term cultivation. | 2019 [43] |
| hPSCs | Improved control over islet organoid size and cell–cell interactions was demonstrated in islet organoids cultivated in a microporous scaffold. | Insufficient maturity; GSIS is not high enough. Not suitable for long-term cultivation. | 2019 [47] |
| iPSCs | In vitro, WNT4 enhanced GSIS and markedly boosted mitochondrial content and oxidative metabolism. Ex vivo interferon stimulation led to reduced T cell activation and graft rejection as well as endogenous PD-L1 expression. | Insufficient maturity. | 2020 [48] |
| iPSC, HUVECs, hMSCs | Compared to non-vascularized islets, the gene expression patterns of vascularized islet organoids more closely resemble those of native islets. | Insufficient maturity, GSIS is not high enough. | 2018 [51,52] |
| Initial Cells | Advantages | Disadvantages | Year |
|---|---|---|---|
| Adult human EpCAM+ ductal cells | Demonstrate how primary human bile duct cells can be easily differentiated into 3D organoids in vitro using bipotent stem cells and how long-term cultured cells can maintain their genetic integrity. | Simple function and simple cell composition. | 2015 [86] |
| Ductal cells derived from NASH patients’ liver | In contrast to organoids made from healthy sources, it accurately captures the pathological traits of NASH. | Only for the NASH study. | 2015 [96] |
| hiPSCs | Improve the repeatability and scalability of organoids and provide a fully hiPSC-based platform for the generation of organ buds. | Simple function and simple cell composition. | 2017 [88] |
| hiPSCs | IPSCs should be encouraged to co-differentiate into the hepatic, biliary, and mesodermal lineages. | Lack of HSCs and Kupffer cells. Insufficient maturity. | 2019 [89] |
| hiPSCs | Create a repeatable procedure to produce multicellular human liver organoids with hepatocyte, stellate, and Kupffer-like cell types. Organoids that had received FFA therapy replicated important aspects of steatohepatitis, including steatosis, inflammation, and fibrosis phenotypes. | Its functional activity remained undetermined. The inter- or intra-batch organoid variability affects the sHLO phenotype such as fibrosis. | 2019 [89] |
| hiPSCs | The use of patient-specific iPSC, the foregut stage’s storage capability, assay throughput, and multiplexed readouts for examining how other parameters, such as mitochondrial stress, interact. | Insufficient maturity. Lack of adaptive immune components. | 2020 [92] |
| hiPSCs, HUVECs, hMSCs | The initial study demonstrated that PSC-derived human organoids could be vascularized and functional. | Simple function and simple cell composition. | 2013 [87] |
| hPSCs | The first to demonstrate that hepatic and biliary cells may develop from hepatoblast cells and the first to produce organoids at a high rate that are uniform in terms of size, shape, and composition. | Lack of HSCs and Kupffer cells. Insufficient maturity. | 2020 [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.; Ding, M.; Zheng, Y.-W. The Values and Perspectives of Organoids in the Field of Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 8125. https://doi.org/10.3390/ijms24098125
Tan C, Ding M, Zheng Y-W. The Values and Perspectives of Organoids in the Field of Metabolic Syndrome. International Journal of Molecular Sciences. 2023; 24(9):8125. https://doi.org/10.3390/ijms24098125
Chicago/Turabian StyleTan, Chen, Min Ding, and Yun-Wen Zheng. 2023. "The Values and Perspectives of Organoids in the Field of Metabolic Syndrome" International Journal of Molecular Sciences 24, no. 9: 8125. https://doi.org/10.3390/ijms24098125
APA StyleTan, C., Ding, M., & Zheng, Y.-W. (2023). The Values and Perspectives of Organoids in the Field of Metabolic Syndrome. International Journal of Molecular Sciences, 24(9), 8125. https://doi.org/10.3390/ijms24098125