

Rational Design of Daunorubicin C-14 Hydroxylase Based on the Understanding of Its Substrate-Binding Mechanism
 , , ,
, , , 
Abstract
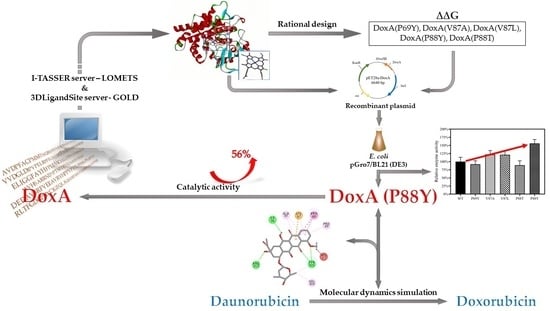
1. Introduction
2. Results and Discussion
2.1. The Heterogenous Expression of DoxA and Analysis of Its Catalytic Activity
2.2. Protein Sequence Analysis and Three-Dimensional Model Construction of DoxA Based on I-TASSER
2.3. Identification of the Mutant Sites and Establishment of Screening Libraries
2.4. Understanding the Relationship between the Enzyme Structural Property and Its Substrate-Binding Efficiency
3. Materials and Methods
3.1. Strains and Cultivation
3.2. Plasmid Construction
3.3. Enzyme Purification
3.4. Enzyme Activity Analysis
3.5. Molecular Docking Analysis
3.6. The Calculation of Conformational Free Energy
3.7. Molecular Dynamics Simulation
3.8. MM-PBSA Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sritharan, S.; Sivalingam, N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef] [PubMed]
- Arcamone, F.; Animati, F.; Capranico, G.; Lombardi, P.; Pratesi, G.; Manzini, S.; Supino, R.; Zunino, F. New developments in antitumor anthracyclines. Pharmacol. Ther. 1997, 76, 117–124. [Google Scholar] [CrossRef]
- Niraula, N.P.; Kim, S.-H.; Sohng, J.K.; Kim, E.-S. Biotechnological doxorubicin production: Pathway and regulation engineering of strains for enhanced production. Appl. Microbiol. Biotechnol. 2010, 87, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tian, X.; Wu, Y.; Shen, X.; Yang, S.; Chen, S. Enhanced doxorubicin production by Streptomyces peucetius using a combination of classical strain mutation and medium optimization. Prep. Biochem. Biotechnol. 2018, 48, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Grein, A. Antitumor Anthracyclines Produced by Streptomyces peucetius. In Advances in Applied Microbiology; Laskin, A.I., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; Volume 32, pp. 203–214. [Google Scholar]
- Hutchinson, C.R.; Colombo, A.L. Genetic engineering of doxorubicin production in Streptomyces peucetius: A review. J. Ind. Microbiol. Biotechnol. 1999, 23, 647–652. [Google Scholar] [CrossRef]
- Shrestha, B.; Pokhrel, A.R.; Darsandhari, S.; Parajuli, P.; Sohng, J.K.; Pandey, R.P. Engineering Streptomyces peucetius for Doxorubicin and Daunorubicin Biosynthesis. In Pharmaceuticals from Microbes: The Bioengineering Perspective; Arora, D., Sharma, C., Jaglan, S., Lichtfouse, E., Eds.; Springer International Publishing: Cham, Switzerland, 2019; Volume 26, pp. 191–209. [Google Scholar]
- Parajuli, N.; Viet, H.T.; Ishida, K.; Tong, H.T.; Lee, H.C.; Liou, K.; Sohng, J.K. Identification and characterization of the afsR homologue regulatory gene from Streptomyces peucetius ATCC 27952. Res. Microbiol. 2005, 156, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Lee, C.-B.; Sohng, J.K. Precursor for biosynthesis of sugar moiety of doxorubicin depends on rhamnose biosynthetic pathway in Streptomyces peucetius ATCC 27952. Appl. Microbiol. Biotechnol. 2010, 85, 1565–1574. [Google Scholar] [CrossRef]
- Han Ah, R.; Park Je, W.; Lee Mi, K.; Ban Yeon, H.; Yoo Young, J.; Kim Eun, J.; Kim, E.; Kim, B.-G.; Sohng Jae, K.; Yoon Yeo, J. Development of a Streptomyces venezuelae-Based Combinatorial Biosynthetic System for the Production of Glycosylated Derivatives of Doxorubicin and Its Biosynthetic Intermediates. Appl. Environ. Microbiol. 2011, 77, 4912–4923. [Google Scholar] [CrossRef]
- Malla, S.; Niraula, N.P.; Liou, K.; Sohng, J.K. Improvement in doxorubicin productivity by overexpression of regulatory genes in Streptomyces peucetius. Res. Microbiol. 2010, 161, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Craney, A.; Ozimok, C.; Pimentel-Elardo, S.M.; Capretta, A.; Nodwell, J.R. Chemical Perturbation of Secondary Metabolism Demonstrates Important Links to Primary Metabolism. Chem. Biol. 2012, 19, 1020–1027. [Google Scholar] [CrossRef]
- Rimal, H.; Lee, S.-W.; Lee, J.-H.; Oh, T.-J. Understanding of real alternative redox partner of Streptomyces peucetius DoxA: Prediction and validation using in silico and in vitro analyses. Arch. Biochem. Biophys. 2015, 585, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, D.; Wang, Q.; Li, J.; Li, H.-L.; Pan, L. Functional expression and purification of DoxA, a key cytochrome P450 from Streptomyces peucetius ATCC 27952. PeerJ 2022, 10, e14373. [Google Scholar] [CrossRef]
- Lomovskaya, N.; Otten Sharee, L.; Doi-Katayama, Y.; Fonstein, L.; Liu, X.-C.; Takatsu, T.; Inventi-Solari, A.; Filippini, S.; Torti, F.; Colombo Anna, L.; et al. Doxorubicin Overproduction in Streptomyces peucetius: Cloning and Characterization of the dnrU Ketoreductase and dnrV Genes and the doxA Cytochrome P-450 Hydroxylase Gene. J. Bacteriol. 1999, 181, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, L.; Shao, J.; Zhu, Z.; Zhang, L. Structure-driven protein engineering for production of valuable natural products. Trends Plant Sci. 2023, 28, 460–470. [Google Scholar] [CrossRef]
- Pramanik, S.; Contreras, F.; Davari, M.D.; Schwaneberg, U. Protein Engineering by Efficient Sequence Space Exploration Through Combination of Directed Evolution and Computational Design Methodologies. In Protein Engineering; Wiley-VCH GmbH: Weinheim, Germany, 2021; pp. 153–176. [Google Scholar]
- Biz, A.; Proulx, S.; Xu, Z.; Siddartha, K.; Mulet Indrayanti, A.; Mahadevan, R. Systems biology based metabolic engineering for non-natural chemicals. Biotechnol. Adv. 2019, 37, 107379. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Maranas, C.D. From directed evolution to computational enzyme engineering—A review. AIChE J. 2020, 66, e16847. [Google Scholar] [CrossRef]
- Xiong, W.; Liu, B.; Shen, Y.; Jing, K.; Savage, T.R. Protein engineering design from directed evolution to de novo synthesis. Biochem. Eng. J. 2021, 174, 108096. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, X.; Li, Q.; Zhu, Y. Computational design of variants for cephalosporin C acylase from Pseudomonas strain N176 with improved stability and activity. Appl. Microbiol. Biotechnol. 2017, 101, 621–632. [Google Scholar] [CrossRef]
- Widmann, M.; Pleiss, J.; Samland, A.K. Computational tools for rational protein engineering of aldolases. Comput. Struct. Biotechnol. J. 2012, 2, e201209016. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, X.; Zhu, Y. Computational design of enzyme–ligand binding using a combined energy function and deterministic sequence optimization algorithm. J. Mol. Model 2015, 21, 191. [Google Scholar] [CrossRef]
- Fraser, M.E.; Joyce, M.A.; Ryan, D.G.; Wolodko, W.T. Two Glutamate Residues, Glu 208α and Glu 197β, Are Crucial for Phosphorylation and Dephosphorylation of the Active-Site Histidine Residue in Succinyl-CoA Synthetase. Biochemistry 2002, 41, 537–546. [Google Scholar] [CrossRef]
- Weiße, R.H.J.; Faust, A.; Schmidt, M.; Schönheit, P.; Scheidig, A.J. Structure of NDP-forming Acetyl-CoA synthetase ACD1 reveals a large rearrangement for phosphoryl transfer. Proc. Natl. Acad. Sci. USA 2016, 113, E519–E528. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.-L.; Westlake, A.C.G.; Nickerson, D.P. Protein engineering of cytochrome P450cam. In Metal Sites in Proteins and Models: Iron Centres; Hill, H.A.O., Sadler, P.J., Thomson, A.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1997; Volume 88, pp. 175–207. [Google Scholar]
- Stevenson, J.-A.; Westlake, A.C.G.; Whittock, C.; Wong, L.-L. The Catalytic Oxidation of Linear and Branched Alkanes by Cytochrome P450cam. J. Am. Chem. Soc. 1996, 118, 12846–12847. [Google Scholar] [CrossRef]
- Ost, T.W.B.; Miles, C.S.; Murdoch, J.; Cheung, Y.-F.; Reid, G.A.; Chapman, S.K.; Munro, A.W. Rational re-design of the substrate binding site of flavocytochrome P450 BM3. FEBS Lett. 2000, 486, 173–177. [Google Scholar] [CrossRef]
- Ba, L.; Li, P.; Zhang, H.; Duan, Y.; Lin, Z. Semi-rational engineering of cytochrome P450sca-2 in a hybrid system for enhanced catalytic activity: Insights into the important role of electron transfer. Biotechnol. Bioeng. 2013, 110, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Offei, S.D.; Aondo Jia, U.T.; Estevez, J.; Perez, Y.; Arman, H.D.; Yoshimoto, F.K. A synthesis of a rationally designed inhibitor of cytochrome P450 8B1, a therapeutic target to treat obesity. Steroids 2022, 178, 108952. [Google Scholar] [CrossRef]
- He, J.; Huang, X.; Xue, J.; Zhu, Y. Computational redesign of penicillin acylase for cephradine synthesis with high kinetic selectivity. Green Chem. 2018, 20, 5484–5490. [Google Scholar] [CrossRef]
- Huang, X.; Han, K.; Zhu, Y. Systematic optimization model and algorithm for binding sequence selection in computational enzyme design. Protein Sci. 2013, 22, 929–941. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; Ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in silico ADMET platform: A journey of machine learning over the past two decades. Drug Discov. Today 2020, 25, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Anjum, F.; Ali, F.; Mohammad, T.; Shafie, A.; Akhtar, O.; Abdullaev, B.; Hassan, I. Discovery of Natural Compounds as Potential Inhibitors of Human Carbonic Anhydrase II: An Integrated Virtual Screening, Docking, and Molecular Dynamics Simulation Study. OMICS J. Integr. Biol. 2021, 25, 513–524. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume 443, pp. 365–382. [Google Scholar]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Wang, W.; Gan, N.; Sun, Q.; Wu, D.; Gan, R.; Zhang, M.; Tang, P.; Li, H. Study on the interaction of ertugliflozin with human serum albumin in vitro by multispectroscopic methods, molecular docking, and molecular dynamics simulation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 219, 83–90. [Google Scholar] [CrossRef]
- Biggs, B.W.; Lim, C.G.; Sagliani, K.; Shankar, S.; Stephanopoulos, G.; De Mey, M.; Ajikumar, P.K. Overcoming heterologous protein interdependency to optimize P450-mediated Taxol precursor synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Q.; Wang, W.; Tan, X.-L.; Wang, X.-Q.; Dong, H. Comparative Analysis of Recombinant Cytochrome P450 CYP9A61 from Cydia pomonella Expressed in Escherichia coli and Pichia pastoris. J. Agric. Food Chem. 2017, 65, 2337–2344. [Google Scholar] [CrossRef]
- Peng, S.; Chu, Z.; Lu, J.; Li, D.; Wang, Y.; Yamg, S.; Zhang, Y. Overexpression of chaperones GroEL/ES from Escherichia coli enhances the indigo biotransformation production of cytochrome P450 BM3 mutant. Biotechnol. Lett. 2022. [Google Scholar] [CrossRef]
- Hu, B.; Zhao, X.; Wang, E.; Zhou, J.; Li, J.; Chen, J.; Du, G. Efficient heterologous expression of cytochrome P450 enzymes in microorganisms for the biosynthesis of natural products. Crit. Rev. Biotechnol. 2023, 43, 227–241. [Google Scholar] [CrossRef]
- Shimizu, T.; Lengalova, A.; Martínek, V.; Martínková, M. Heme: Emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 2019, 48, 5624–5657. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Choi, K.-Y. α, ω-Oxyfunctionalization of C12 alkanes via whole-cell biocatalysis of CYP153A from Marinobacter aquaeolei and a new CYP from Nocardia farcinica IFM10152. Biochem. Eng. J. 2020, 156, 107524. [Google Scholar] [CrossRef]
- Honda, Y.; Nanasawa, K.; Fujii, H. Coexpression of 5-Aminolevulinic Acid Synthase Gene Facilitates Heterologous Production of Thermostable Cytochrome P450, CYP119, in Holo Form in Escherichia coli. Chembiochem 2018, 19, 2156–2159. [Google Scholar] [CrossRef]
- Wu, D.; Zhu, C.; Zhu, B. Expression of daunorubicin C-14 hydroxylase gene in Streptomyces lividans TK24. Chin. J. Pharm. 2004, 35, 13–16. [Google Scholar] [CrossRef]
- Zaj¹c, M.; Dobrowolski, L.; Zió³kowska, G.; Zalewski, P.; Piekarski, M.; Krause, A.; Uszak, J. Development and Validation of RP HPLC Method for Determination of Novel Derivatives of Daunorubicin. Chem. Anal. 2009, 54, 907–917. [Google Scholar]
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Odia, T.; Adebiyi, M. Predicting the structure of Anopheles gambiae cytochrome P450 protein using computational methods. In Proceedings of the ISCB Africa ASBCB Conference on Bioinformatics, Dar es Salaam, Tanzania, 9–11 March 2015. [Google Scholar]
- Lepesheva, G.I.; Waterman, M.R. Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Johnson, E.F. Structures of Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Springer: Boston, MA, USA, 2005; pp. 87–114. [Google Scholar]
- Watanabe, Y.; Fukuyoshi, S.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Takahashi, O.; Oda, A. Prediction of three-dimensional structures and structural flexibilities of wild-type and mutant cytochrome P450 1A2 using molecular dynamics simulations. J. Mol. Graph. Model. 2016, 68, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. A Protocol for Computer-Based Protein Structure and Function Prediction. J. Vis. Exp. 2011, 57, e3259. [Google Scholar] [CrossRef]
- Rajakumara, E.; Saniya, D.; Bajaj, P.; Rajeshwari, R.; Giri, J.; Davari, M.D. Hijacking Chemical Reactions of P450 Enzymes for Altered Chemical Reactions and Asymmetric Synthesis. Int. J. Mol. Sci. 2023, 24, 214. [Google Scholar] [CrossRef] [PubMed]
- McGreig, J.E.; Uri, H.; Antczak, M.; Sternberg, M.J.E.; Michaelis, M.; Wass, M.N. 3DLigandSite: Structure-based prediction of protein–ligand binding sites. Nucleic Acids Res. 2022, 50, W13–W20. [Google Scholar] [CrossRef]
- Sun, Y.; Zeng, W.; Benabbas, A.; Ye, X.; Denisov, I.; Sligar, S.G.; Du, J.; Dawson, J.H.; Champion, P.M. Investigations of Heme Ligation and Ligand Switching in Cytochromes P450 and P420. Biochemistry 2013, 52, 5941–5951. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Martin, M.V.; Guengerich, F.P. Random Mutagenesis of Human Cytochrome P450 2A6 and Screening with Indole Oxidation Products1. Arch. Biochem. Biophys. 2001, 395, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic Structure of Compound I in Human Isoforms of Cytochrome P450 from QM/MM Modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Sciortino, G.; Rodríguez-Guerra Pedregal, J.; Lledós, A.; Garribba, E.; Maréchal, J.-D. Prediction of the interaction of metallic moieties with proteins: An update for protein-ligand docking techniques. J. Comput. Chem. 2018, 39, 42–51. [Google Scholar] [CrossRef]
- Komeda, H.; Yamasaki-Yashiki, S.; Hoshino, K.; Asano, Y. Identification and characterization of d-xylose reductase involved in pentose catabolism of the zygomycetous fungus Rhizomucor pusillus. J. Biosci. Bioeng. 2015, 119, 57–64. [Google Scholar] [CrossRef]
- Bonjoch, N.P.; Tamayo, P.R. Protein Content Quantification by Bradford Method. In Handbook of Plant Ecophysiology Techniques; Reigosa Roger, M.J., Ed.; Springer: Dordrecht, The Netherlands, 2001; pp. 283–295. [Google Scholar]
- Mohammad, T.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification and evaluation of bioactive natural products as potential inhibitors of human microtubule affinity-regulating kinase 4 (MARK4). J. Biomol. Struct. Dyn. 2019, 37, 1813–1829. [Google Scholar] [CrossRef]
- Amera, G.M.; Khan, R.J.; Pathak, A.; Jha, R.K.; Jain, M.; Muthukumaran, J.; Singh, A.K. Structure based drug designing and discovery of promising lead molecules against UDP-N-acetylenolpyruvoylglucosamine reductase (MurB): A potential drug target in multi-drug resistant Acinetobacter baumannii. J. Mol. Graph. Model. 2020, 100, 107675. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | ΔΔG | Mutants | ΔΔG |
|---|---|---|---|
| DoxA(I96L) | −5.12 | DoxA(P88Y) | −12.24 |
| DoxA(I96V) | 0.92 | DoxA(P88T) | −8.06 |
| DoxA(I96F) | −1.94 | DoxA(K67A) | 4.11 |
| DoxA(Y305V) | 2.76 | DoxA(K67R) | 0.72 |
| DoxA(Y305F) | 0.68 | DoxA(K67H) | 4.70 |
| DoxA(Y305W) | −1.01 | DoxA(A97L) | 3.52 |
| DoxA(P69V) | 0.02 | DoxA(A97I) | 2.36 |
| DoxA(P69Y) | −6.10 | DoxA(A97T) | 1.39 |
| DoxA(P69T) | −0.49 | DoxA(L67I) | 4.68 |
| DoxA(V87A) | −8.13 | DoxA(L67A) | 4.58 |
| DoxA(V87I) | −2.86 | DoxA(L67S) | 3.61 |
| DoxA(V87L) | −9.04 |
| Binding Free Energy | DoxA | DoxA (P69Y) | DoxA (V87A) | DoxA (V87L) | DoxA (P88T) | DoxA (P88Y) |
|---|---|---|---|---|---|---|
| Gvan | −54.10 | −58.04 | −57.62 | −58.48 | −57.54 | −52.90 |
| Gele | −257.77 | −277.60 | −304.01 | −294.50 | −227.14 | −277.32 |
| Gpol | 248.42 | 272.77 | 289.08 | 285.83 | 224.04 | 262.99 |
| Gnp | −7.58 | −8.03 | −7.65 | −7.27 | −7.88 | −7.44 |
| Ggas | −311.87 | −335.64 | −361.63 | −352.98 | −284.68 | −330.22 |
| Gsolv | 240.84 | 264.74 | 281.43 | 278.56 | 216.16 | 255.55 |
| ΔGbinding | −71.03 | −70.90 | −80.20 | −74.42 | −68.52 | −74.67 |
| Residues | DoxA | DoxA (P69Y) | DoxA (V87A) | DoxA (V87L) | DoxA (P88T) | DoxA (P88Y) |
|---|---|---|---|---|---|---|
| Pro 69 | −0.83 | −0.51 | −0.48 | −0.53 | −0.37 | −0.82 |
| Val 87 | −0.29 | −0.30 | −0.29 | −0.80 | −1.21 | −0.95 |
| Pro 88 | −0.02 | −0.23 | −0.89 | −0.05 | −0.02 | −0.10 |
| The total | −1.14 | −1.04 | −1.66 | −1.38 | −1.60 | −1.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Gao, L.-X.; Chen, W.; Zhong, J.-J.; Qian, C.; Zhou, W.-W. Rational Design of Daunorubicin C-14 Hydroxylase Based on the Understanding of Its Substrate-Binding Mechanism. Int. J. Mol. Sci. 2023, 24, 8337. https://doi.org/10.3390/ijms24098337
Zhang J, Gao L-X, Chen W, Zhong J-J, Qian C, Zhou W-W. Rational Design of Daunorubicin C-14 Hydroxylase Based on the Understanding of Its Substrate-Binding Mechanism. International Journal of Molecular Sciences. 2023; 24(9):8337. https://doi.org/10.3390/ijms24098337
Chicago/Turabian StyleZhang, Jing, Ling-Xiao Gao, Wei Chen, Jian-Jiang Zhong, Chao Qian, and Wen-Wen Zhou. 2023. "Rational Design of Daunorubicin C-14 Hydroxylase Based on the Understanding of Its Substrate-Binding Mechanism" International Journal of Molecular Sciences 24, no. 9: 8337. https://doi.org/10.3390/ijms24098337
APA StyleZhang, J., Gao, L.-X., Chen, W., Zhong, J.-J., Qian, C., & Zhou, W.-W. (2023). Rational Design of Daunorubicin C-14 Hydroxylase Based on the Understanding of Its Substrate-Binding Mechanism. International Journal of Molecular Sciences, 24(9), 8337. https://doi.org/10.3390/ijms24098337