Abstract
Acute kidney injury (AKI) is a global health problem and has recently been recognized as a risk factor for developing chronic kidney disease (CKD). Unfortunately, there are no effective treatments to reduce or prevent AKI, which results in high morbidity and mortality rates. Ischemic preconditioning (IPC) has emerged as a promising strategy to prevent, to the extent possible, renal tissue from AKI. Several studies have used this strategy, which involves short or long cycles of ischemia/reperfusion (IR) prior to a potential fatal ischemic injury. In most of these studies, IPC was effective at reducing renal damage. Since the first study that showed renoprotection due to IPC, several studies have focused on finding the best strategy to activate correctly and efficiently reparative mechanisms, generating different modalities with promising results. In addition, the studies performing remote IPC, by inducing an ischemic process in distant tissues before a renal IR, are also addressed. Here, we review in detail existing studies on IPC strategies for AKI pathophysiology and the proposed triggering mechanisms that have a positive impact on renal function and structure in animal models of AKI and in humans, as well as the prospects and challenges for its clinical application.
1. Generalities of Acute Kidney Injury
Acute kidney injury (AKI) is a very frequent syndrome that occurs in 10–15% of hospitalized patients, and its incidence is considerably higher in critically ill patients, which in turn, increases patient mortality [1,2]. Worldwide, it has been estimated that 13.3 million people per year have at least one episode of AKI, contributing to approximately 1.7 million deaths per year [3]. The most common cause of AKI is related to an ischemic process that induces a decrease in renal oxygenation with the subsequent generation of free radicals [4]. Several types of cells in the nephron are affected; however, the proximal tubular epithelial cells are the most compromised due to their dependence on aerobic oxidative metabolism [5]. Although epithelial cells have the capacity to activate reparative mechanisms, it has been reported that a maladaptive response may occur that leads to tubulointerstitial fibrosis; therefore, clinical and experimental studies have demonstrated that AKI is a risk factor for the development of chronic kidney disease (CKD) [6,7,8,9,10]. CKD is a global health concern since it has been estimated that 1.2 million people die annually due to this pathology [11]. Coupled with this, several studies have reported that 20.1% to 44% of AKI survivors are susceptible to suffering at least another AKI episode that increases both the progression to CKD and mortality, a situation that has already been termed recurrent AKI (rAKI) [12,13,14,15,16,17]. Interestingly and contradictory to clinical observations, experimental studies have shown that repeated and short ischemic insults [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32], as well as two renal ischemia periods of the same magnitude, may induce renal protection, an effect that has been known as ischemic preconditioning (IPC) [33,34,35,36,37,38,39,40,41,42].
2. Pathophysiology of Acute Kidney Injury
AKI is characterized by a transitory loss of kidney function, limited to 7 days of duration, due to a decrease in renal blood flow (RBF). The main causes of AKI are sepsis, a decrease in intravascular volume, renal ischemia, and nephrotoxic drugs, among others. AKI is diagnosed when the serum creatinine is elevated by ≥0.3 mg/dL in 48 h, by an increase of ≥1.5 times the basal values, or by a decrease in the urinary output to <0.5 mL/kg/h during 6 h [43].
During an AKI episode, the hypoxic state damages the endothelial cells, which results in increased production of vasoconstrictor factors such as endothelin-1, prostaglandin H2, angiotensin II, and thromboxane A2. Simultaneously, there is an increase in adhesion molecules (ICAM-1 and/or β-integrin) [44] and in proinflammatory cytokines, such as TNF-α, MCP-1, IL-6, IL-32, IL-1β, IL-18, and TGF-β. These events allow immune cell infiltration and phagocytic cell activation, such as neutrophils and macrophages. Although necessary to clean cell debris and apoptotic cells, their recruitment and activation generate a local release of proinflammatory signals, reactive oxygen species (ROS), and proteases that perpetuate renal dysfunction and tissue injury [45]. As a result of vascular damage, there is a disruption of the extracellular matrix and an alteration of the cytoskeleton that promote modifications in cell–cell interactions, which in turn induce greater vascular permeability. If this inflammatory response is not regulated, it can lead to cell death, drastically reducing the number of vessels and worsening the hypoxic state, ultimately resulting in tubular interstitial fibrosis [46].
The damage to epithelial cells that occurs in AKI appears primarily in the S2 and S3 segments of the proximal tubule. An imbalance in the ATP supply disrupts the cytoskeletal architecture and β-actin and tubulin filaments are disrupted, inducing loss of the brush border, loss of cell polarization, and mislocalization of membrane proteins [47]. The disruption in the localization of transporters and adhesion molecules deregulates the absorption or secretion functions of these segments, as well as alter cell–cell interactions, causing a detachment of both viable and nonviable cells and increasing solute wasting and protein excretion in urine, which, in turn, may combine with the Tamm–Horsfall protein (THP) and fibronectin to form casts that obstruct the tubules, which are a hallmark of AKI [48]. The cells of the tubular epithelium have the ability to proliferate and replace the lost cells, thus, repairing the damaged epithelium [49,50]. If the whole process occurs correctly, the kidney functions are restored in a few days. However, in some cases, maladaptive mechanisms are activated, leading to a failure in kidney repair and favoring progressive injury. Some of these mechanisms are tubular cell arrest in the G2/M cycle [51], chronic inflammation and cell infiltration [52], myofibroblast production, and increased extracellular matrix deposition [53].
3. Long-Term Consequences of AKI
Experimental, epidemiological, and clinical studies have reported that AKI is an independent risk factor for the development of CKD, which has been termed AKI to CKD transition [9]. The course of this transition is determined by the initial insult severity and duration, where the age of the patients is another preponderant factor [54,55]. Many efforts have been made to understand the mechanisms that managed the AKI to CKD transition. The most relevant include chronic hypoxia [56], vascular rarefaction [57] proliferation of epithelial cells with excessive production of TGF-β [7], transdifferentiation of pericytes into myofibroblasts [58], and chronic stress of the endoplasmic reticulum [59]. Many other mechanisms, however, remain to be elucidated.
4. Ischemic Preconditioning (IPC)
In 1986, Murry et al. [60] described that four cycles of 5 min of ischemia in a dog’s left anterior descending (LAD) coronary artery induced protection against an insult of greater magnitude, i.e., 90 min of ischemia, which was evidenced by a reduction in 75% of the myocardial infarction area. Since then, this process has been called ischemic preconditioning (IPC), and it consists of making the tissue tolerant by performing repeated episodes of ischemia, alternated with reperfusion, before a sustained and more severe ischemic damage is produced.
Nevertheless, two years earlier Zager RA, et al. [41] showed in Sprague Dawley rats that a mild or severe bilateral renal ischemia (BRI) of 25 or 40 min, performed 18 or 48 h before a second hit of 40 min of BRI, produced renal protection, which was evidenced by a significant reduction in serum creatinine concentration (SCr) compared to the group with only one IR event. One year later, the same group demonstrated that a 15 min period of IR performed 3.5 to 24 h before a second insult of greater magnitude (25 min) was sufficient to provide renal protection; interestingly, when the interval was shortened to 0.5 h between each episode of IR, kidney protection was not achieved and even further damage resulted [42]. These results strongly suggest that the benefits of IPC require a longer period of time for the protective molecular mechanisms to become established.
Since then, continuous research has been carried out to determine the best model of IPC and its respective protection mechanism with the intention of being used in the clinical setting as a possible therapeutic intervention to reduce or avoid AKI.
5. Impact of One or More Short Cycles of Renal IPC on a Larger Ischemic Insult
It is well known that brief cycles of IR are enough to induce renal preconditioning. Table 1 presents some of the studies that evaluated the effect of one or more brief cycles of IR before a larger ischemic insult is performed. In the 15 studies reported, the effect of 1, 2, 3, or 4 short ischemic cycles, ranging from seconds to 15 min, were evaluated; the most common protocol used was 5 min of ischemia and 5 min of reperfusion. The second ischemic challenge ranged from 30 to 60 min, with half of the studies employing the experimental model of bilateral renal ischemia (BRI) and the other half the unilateral renal ischemia (UIR). As is appreciated in Table 1, there is no consensus on establishing effective renal protection depending on the ischemic period, the number of cycles, the interval between each cycle, or the experimental model used; still, most of these studies observed a clear renoprotection. A meta-analysis carried out by Wever et al. [61] showed that the IPC strategy is strongly influenced by the animal species used, which could explain some differences found in all these studies. For example, Joo et al. [19] and Mahfoudh-Boussaid et al. [24] found an increase in p-Akt, but Li et al. [27] showed a decrease, and in all these three studies, the outcome was the same, renoprotection. Khalid U. et al., compared multiple short cycles of IPC to one of moderate magnitude before a major insult is induced in rats, finding that 4 cycles of 2 min of ischemia separated by 5 min of reperfusion resulted in better renoprotection [28]. To our knowledge, the only study that did not find protection was done on Wistar rats, with IPC consisting of 3 cycles of 5 min of ischemia and 5 min of reperfusion [61]. Notably, most of these studies evaluated the IPC effects only 24 or 48 h after the preconditioning, except for Timsit et al. [23] and Zhang et al. [31], who evaluated the animals after 15 and 42 days, finding major survival of the animals that underwent IPC. Therefore, the impact of short periods of IPC on the long-term consequences of an AKI event remains to be fully defined.

Table 1.
Effect of one or more short cycles of renal ischemic preconditioning on a larger ischemic insult.
6. Effect of Two or Three IR Episodes of Similar Severity
A way for evaluating the impact of the IPC upon a subsequent insult (hit) of similar magnitude has been extensively studied, inducing two BRI episodes of similar magnitude, ranging from 15 to 40 min, in both rats and mice. As shown in Table 2, it is more common to find two insults lasting 30 min and that were spaced from 0.5 h to 15 days. This table highlights the fact that in more than half of these studies, the interval between each IR was 7 days, which makes these studies more comparable. Furthermore, the animals were studied in most cases 24 to 48 h after the last insult. However, little is known about the long-term impact of the IPC. As summarized in Table 2, there was an improvement in renal function in almost all cases when compared to the damage induced by a single BRI episode, showing a window of protection when each ischemia is carried out between 3 to 8 days and even at intervals of 15 days. Only in the study by Dong, Y. et al. [62] was a worsening in renal function reported. The difference between this study and the rest is that the IR only occurred unilaterally, which suggests that different mechanisms are activated between BRI and URI. There are practically no studies that have carried out more than two episodes of renal ischemia and their long-term impact, except one that we recently published. In our study, AKI to CKD transition was evaluated after three mild (20 min) or three severe (45-min) episodes of IR (3IR) and compared with a single moderate or severe IR episode (1IR). The animals were followed for 9 months, and the 1IR group (20 or 45-min) developed CKD as evidenced by progressive proteinuria and renal fibrosis. Interestingly, the long-term consequences of AKI were markedly ameliorated in the 3IR group. Our study shows that renal preconditioning by three cycles of moderate and severe IR remarkably reduced the long-term consequences of AKI [63].
Table 2.
Impact of one or more cycles of IPC of similar magnitude.
7. Remote Ischemic Preconditioning (rIPC)
The renoprotective effect of the IPC does not necessarily have to be generated in the tissue to be protected, but IPC could be performed in distant tissues, developing messengers that travel to different organs activating protective signaling pathways. This kind of precondition is known as remote IPC (rIPC) [64]. This phenomenon has been described first in the heart and brain, and shortly after in the kidney. The great advantage of the rIPC is its potential use in the clinical setting. As shown in Table 3, most of the experimental studies performed short periods of 5 min of ischemia on the hindlimb or femoral artery, and the challenges were mostly BRI of 45 min and evaluated 24 h later. All the studies reported renal function improvement except for the study performed by Kierulf-Lassen, C. et al. [65]. This protection was associated with an increase in the activity of antioxidant enzymes, such as SOD and catalase, preventing oxidative stress and death cell. A downregulation in cytokine levels and infiltration cells is also an important finding. The only two studies that evaluated the chronic effect of rIPC reported opposite results. In one study, there was an inhibition of TGF-β expression, whereas in the other hand, tubulo-interstitial fibrosis was found. This difference may be explained by the different species studied. Additionally, an attempt has been made to elucidate the molecules that could be responsible for exerting remote protection [66,67,68,69]. It has been reported that there is crosstalk between kidneys and various organs, such as intestine, heart, spleen, and brain [65,70,71]. Apparently, the molecules responsible for carrying the message to the kidneys may be the same regardless of which organ the preconditioning stimulus is applied to. Very few of the studies address this question, but the few that do report implied molecules, such as hormones, cytokines, and nitric oxide (NO). It seems that the renoprotective molecules involved must be in the circulation in order to reach distant sites, and one approach that could explain it is through exosomes.
Table 3.
Significance of remote IPC in distant organs on the injury induced by IR.
8. Renoprotective Mechanisms Induced by Short and Long Cycles of IPC
Several mechanisms have been described as responsible for the renoprotection conferred by short or long cycles of IPC shown in Table 1, Table 2 and Table 3. Many of them converge in the different reported studies and are described below.
Reduction of Oxidative Stress: Although there are differences among species, there are conserved mechanisms that are activated by the IPC to protect cells from injury. It is well known that during AKI, there is a failure to produce ATP in proximal tubular cells due to mitochondrial damage. The mitochondria are the main source of reactive oxygen species (ROS) and if their levels are not controlled by antioxidative enzymes, such as catalase or superoxide dismutase (SOD), cells could suffer apoptosis or necrosis. It has been reported, however, that low levels of ROS are required for proper cell function by activation signaling pathways or protein modifications [77,78]. Until a certain point, ROS are also involved in the IPC, since there is an increase in superoxide, hydrogen peroxide, and lipid peroxidation that persisted 8 days later. All these events are accompanied by an increase in 33% of the total antioxidant capacity [25], an elevation in manganese superoxide dismutase (MnSOD) activity, and a decrease in angiotensin II [35,38]. The correct regulation of ROS limits oxidative stress preventing lipid peroxidation, as has been demonstrated by the decrease in malondialdehyde (MDA) levels after IPC [23,27]. The positive involvement of ROS during IPC is partially lost when antioxidant reagents such as MNTMPyP (manganese(III) tetrakis(1-methyl-4-pyridyl) porphyrin) or N-acetylcysteine were administered, while other proteins, such as inducible nitric-oxide synthase (iNOS) or heat shock protein of 25 kDa (HSP25), which are also overexpressed by IPC, were not affected [38,39]. The role of antioxidant enzymes activity seems to be crucial in the rIPC, since SOD and catalase activity increase when the rIPC is performed in the small intestine protecting the kidneys against ischemic injury [70]. It is well known that the decrease in ROS generation prevents apoptosis by inducing antiapoptotic proteins such as Bcl-2 and by reducing BAX and cleaved Caspase 3 levels [66,73]. This reduction of apoptosis could also be explained by the increase in the phosphorylation of Akt and ERK1/2 in the renal tissue of animals with rIPC performed in the heart [79].
Nitric Oxide and pAkt Pathway Involvement: During AKI, the elevation of nitric oxide (NO) seems to be secondary to the enhanced iNOS activity, which is a vasodilator factor. Gene deletion of iNOS or its pharmacological inhibition with L-N6-(1-iminoethyl) lysine (L-NIL) increases kidney susceptibility to a second ischemic insult. Interestingly, the deletion of the eNOS gene has no effect on this susceptibility [39]. Although the protection does not disappear completely, iNOS is important to the late protection because of its sustained expression up to 12 weeks after performing IPC [19]. One of the proofs that demonstrates the importance of NOS enzymes was evidenced in eNOS-deficient mice, where there was no IPC protection [18] or when a nonselective NOS inhibitor, such as N-nitro-L-arginine methyl ester (L-NAME), was administrated, showing a partial loss of IPC protection [18,22,24]. Furthermore, it has been observed that the activity of iNOS progressively increases 24 h after ischemia injury and could explain its crucial role in long-term protection [19]. Medullary congestion and tubular necrosis are prevented by IPC, possibly due to the vasodilatory effects that NO has and the specific localization of eNOS (endothelium) and iNOS (glomerulus and proximal tubules), helping to maintain a better blood flow and reducing the hypoxic risk [22]. Besides inhibited NOS enzymes, a decrease in the stability of HIF-1α has been observed, preventing the expression of its target genes involved in adaptation to low oxygen levels [24], suggesting that NO also helps to stabilize HIF-1α [80].
As was reported before, the role of NO in kidney protection has already been demonstrated by IPC in local tissue but its participation in remote protection is controversial. The main difficulty in proposing NO as a mediator in rIPC is its short half-life, which is around 2 milliseconds or less [81], making it impossible for NO to travel through circulation to distant organs. However, the NO oxidation product, nitrite, which has a half-life of around 60 min [82] and has vasodilator and cytoprotector effects, could be the mediator of NO generated by the rIPC [83]. Regardless of whether NO or nitrite is responsible for the renoprotective effects, Gholampour et al. [67] showed that when rIPC is performed in the left femoral artery, the renoprotection observed after inducing BIR was associated with decreased lipid peroxidation and increased GPX and catalase activity. This antioxidant effect was diminished when L-NAME was administrated, indicating the participation of NO in the renoprotection observed.
Another mechanism involved in the IPC is the participation of Akt and its phosphorylation (p-Akt), which is a serine/threonine kinase that participates in cell survival [24]. The IPC protection has been associated with the activation of the Akt signal pathway, which, in turn, induces antiapoptotic proteins (Bcl-2 and Bcl-x) and prevents DNA fragmentation [36]. Nevertheless, Akt and p-Akt involvement in the IPC seems to depend on the species and the strain studied. While in mice C57BL/6 and Wistar rats, IPC promotes elevation in p-Akt, an effect that is associated with cytoprotection by counteracting apoptosis [19,24], in Sprague Dawley rats, IPC reduces the activation of the Akt pathway, preventing the phosphorylation of NF-κB, a master transcription factor that regulates inflammatory responses; therefore, IPC was associated with a reduction in renal inflammation [27]. These differences may be explained through the particular signal pathway activated for the IPC. On the one hand, it has been found that the protection is through activating the phosphatidylinositol-3 kinase (PI3K)-Akt pathway or through protein kinase C (PKC) in mice [19], and, on the other hand, the overexpression of HO-1, induced by the master transcription factor Nrf2, was observed in rats after the IPC [27].
Heat Shock Proteins Induction: Other mediators involved in the IPC are the heat shock proteins (HSPs) that participate in several cellular functions, such as the correct protein folding, intracellular protein transport, translocation of transcription factors, regulation of cell signaling in inflammation, apoptosis, and proliferation [84]. One of the hallmarks of IR inducing renal injury is the loss of epithelial cell polarization, where HSP25 plays an important role in stabilizing actin microfilaments [85]. In this regard, it has been reported that the induction of HSP25 is dependent on the ischemic intervals; however, there is a greater HSP25 overexpression after the IPC, most likely to counteract the renal damage [38,39,40]. Another HSP involved in the IPC protection is HSP32, also known as heme-oxygenase-1 (HO-1) [27]. Indeed, we demonstrated that 24 h after the third round of ischemic insults with 10-day intervals, there was a significant increment in HO-1 and M2 macrophages, together with an anti-inflammatory response mediated by a decrease in NF-κB-p65 phosphorylation and IL-6. Thus, repeated episodes of IR with 10-day intervals induced long-term renal protection accompanied with HO-1 overexpression and an increase in M2 macrophages [63].
Furthermore, when there is an accumulation of unfolded protein in the endoplasmic reticulum (ER), the unfolded protein response (UPR) is activated. One principal ER chaperone is the 78-kDa glucose-regulated protein (GRP78), also known as binding immunoglobulin protein (BiP), which facilitates the correct folding of proteins and helps to translocate new synthetized peptides into the ER membrane [86]. Interestingly, the IPC has been reported to increase the expression of GRP78/BiP and of proteins, such as tumor necrosis factor receptor-associated factor 2 (TRAF2) and activating transcription factor 4 (ATF4), which are involved in decreasing ER stress-induced apoptosis [24].
Amelioration of Renal Inflammation: It has been reported that the IPC reduced the phosphorylation of mitogen-activated protein kinase 7, 4, and 3/6 (MKK7, MKK4 and MKK3/6, respectively), inhibiting the downstream activation of JNK and p38, which are involved in the induction of adhesion molecules in endothelial cells and proinflammatory cytokines [40]. Several studies have shown that the IPC reduces proinflammatory cytokine expression, including that of TNF-α, IL-6, IL-1β, IL-17, and MCP-1 [25,30,31], as well as downregulation of the TLR4/NF-κB pathway that limits the infiltration of immune cells into the kidney [27,30,32]. Zhang et al. [32] found that the IPC does not seem to prevent immune cell infiltration into the kidney, but inhibits dendritic cell maturation. Splenocytes from animals with IPC had more Tregs and mature CD11c+ macrophages/dendritic cells (DC) mediating immune tolerance by reducing cytokine-secretory responses, including TNF-α, IFN, MCP-1, and IL-6 [34]. Interestingly, this IPC protective effect could be translated to T cell-deficient mice, when Tregs from mice with IPC were transferred [33]. Cho et al. described that the protection conferred by this transfer strategy is mediated by Treg cells [34]. The role of macrophage infiltration in the IPC has also been studied. In BALB/c and C57BL/6 mice, the administration of lipo-clodronate (which depletes macrophages) that reduced the amount of macrophages did not modify the protective effect of IPC [34,37].
Similarly, systemic proinflammatory cytokines, such as IL-1 and IL-6, are reduced after an rIPC, which may decrease immune cell infiltration and pro-apoptotic proteins in remote organs [66,69,71,74,79]. In addition to this, the NF-κB is also inhibited [63,69,71,73], provoking an anti-inflammatory response accompanied by an elevation of IL-10 as an anti-inflammatory cytokine [71]. It has also been reported that the elevation of serum TNF-α promotes renalase expression. Renalase is an amino oxidase that originates in the kidney that can regulate blood pressure and serve as a prosurvival/growth factor [87,88]. Wang et al. demonstrated that renalase is required for kidney protection induced by rIPC, since when a renalase siRNA is administrated, the protection is abolished [66]. TGF-β and its downstream signaling proteins Smad2 and Smad3 are well-known mediators of renal fibrosis in the long consequences of IR [7]. Nevertheless, rIPC avoids the induction of this cytokine with the concomitant decrease in fibronectin, collagen I and III, and α-SMA, and the consequent renal fibrosis [75]. This protection seems to depend on the species and the type of rIPC location, because when rIPC is applied in the limbs of rats, there is no renal fibrosis after three months [75], but when the rIPC is located in the heart of mice, renal fibrosis is worse after four weeks [76].
Involvement of Autophagy in the protection conferred by IPC: Autophagy regulates the renewal and death of senescent or damaged cells. There are three different types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. In general, it has been reported that autophagy-related proteins (ATG) are induced by IPC [89]. Beclin-1, a Bcl-2 interacting protein, is the main upstream regulator of autophagy and served as a scaffold to Vps34, Vps15, and ATG14L, which participate with the vesicle nucleation [90]. In animals undergoing IPC, the induction of Beclin-1 occurs as fast as 6 h post-IR and remains elevated for 24 h [26].
The second step of autophagy is the autophagosome formation ATG12-conjugation system, the increase in microtubule-associated protein 1 light chain 3 (LC3), and phosphatidylethanolamine (LC3-II) expression. These proteins are responsible for elongation and closure of the phagophore membrane, which could be derived from mitochondria, endoplasmic reticulum (ER), Golgi, or plasma membranes. It has been shown that after IPC, the increase in LC3-II and Atg12 promoted autophagy and blocked apoptosis in the kidney [20,27,31]. Moreover, the concentration of LC3-II on the apical side of the proximal tubule cells in animals with IPC has been observed [20]. IPC not only induced LC3-II accumulation but also the maturation of autolysosomes, the final step for the clearance of autophagic cargo. This specific accumulation of autophagosomes on the apical side of proximal tubule cells could be due to the enrichment of ATP-dependent transporters, generating an elevated rate of mitochondria replacement.
The correct and regulated form of damaged mitochondria degradation, known as mitophagy, is another mechanism of the IPC protective effect [20,21,25]. This is mediated by the induction of PINK1 (PTEN induces putative kinase-1) that surrounds damaged mitochondria and, therefore, serves as a sensor, and BNIP3L (BCL2-interacting protein 3-like), and FUNDC1 (FUN14 domain containing 1) that are receptors that initiate mitophagy under hypoxic conditions and interact with UNC-51-like kinase 1 (Ulk1). After IPC, PINK1 was activated, but when its expression was reduced with a shRNA, the mitolysosomes were almost blocked, along with the protection [20]. Interestingly, in renal proximal tubule-conditional Fundc1-deficient mice, the LC3II expression is downregulated together with loss of IPC protection [20,21].
Post-translational modifications involved in the IPC protection. Interestingly, protein acetylation is higher in the renal tissue of aging rats, which results in the abolishment of the IPC protective effect [25]. microRNAs (miRNA) regulate gene expression, not only maintaining cellular functions but also regulating responses to tissue injury. miRNAs bind to mRNAs and inhibit or promote translation. There are some microRNA signatures established during the IPC. Yamamoto. et al. found that some miRNAs, such as, miR-17-3p and miR-19s, inhibit PTEN, whereas miR-34a activates it [29]. Interestingly, microRNAs such as miR-21-5p, miR-22-3p, and miR-222-3p that are involved in fibrosis, angiogenesis, and vascular remodeling are suppressed by IPC [28].
Hormones participation in the IPC. The key molecule responsible for the communication between the brain and the kidney is erythropoietin (EPO) [68]. EPO is a glycoprotein produced in the kidney whose main function is to stimulate the production of red blood cells, but it has also been proposed as a protective hormone against ischemic brain injury [91]. Another protective effect is mediated by the heteromeric complex known as the tissue protective receptor (TPR), a complex made up of the EPO receptor (EPOR) and the common β receptor (βCR), a subunit shared by type 1 cytokines [92]. TPR could activate several signaling pathways, one of which involves PI3K and Akt. Activation of this pathway promotes the phosphorylation of glycogen synthase kinase 3β (GSK3β), inhibiting the mitochondrial permeability transition (MPT) and stabilizing mitochondrial function [68,93]. The heart also has close communication with the kidney, as is the case of rIPC by permanent ligation of the LAD artery, in which chronic hypoxia protects the kidney against UIR. These animals exhibit lower levels of BUN and Kim-1, accompanied by increased EPO, PDK1, glut1, and VEGF mRNA levels. However, these results should be taken with caution, since this protection was not observed 4 weeks after the UIR; instead, worse renal fibrosis was exhibited [76].
Alteration in cellular metabolism. IR injury provokes inflammation, osmotic dysregulation, and energy metabolism perturbation. Some differentially produced metabolites, such as ceramide, acylcarnitine, betaine aldehyde, adenosine, and glucosylceramide, could explain rIPC protection because of their involvement in these pathways [72,93]. Other relevant consequences could be post-translational modifications, as is the increase in protein O-GlcNAcylation, a form of glycan union with proteins due to rIPC. This process has been reported to regulate HSP40 and HSP70 and CHOP expression and attenuate NF-κB activity [73].
Exosomes as a transport mechanism in the rIPC. Exosomes could be a crucial mediator in the renoprotection generated by distant IPC, reaching the damaged tissue from their origin in remote tissue. These microvesicles contain a variety of components, such as protein, RNA, lipids, and enzymes, conferring a role in various cellular processes such as, regulation of gene transcription and translation, inflammation regulation, and cellular communication [94,95]. Pan T et al. [69] demonstrated that in the rIPC, the increased generation of exosomes could reach several organs including the kidneys, which was associated with a reduction in serum creatinine and urinary NGAL. These exosomes contain MiR-21, an antiapoptotic miRNA. Another important finding was that MiR-21 has also been found in serum exosomes from human with rIPC. The exosome content determines the trigger mechanisms and the effects in distant tissues, mainly provoking an anti-inflammatory response and inhibiting NF-κB activity [69].
9. The Remote IPC in the Clinical Setting
AKI is one of the pathologies in which the applicability of rIPC has been proven to be effective by reducing its frequency and severity in patients susceptible to it. Because of rIPC advantages and low invasiveness, several clinical trials have been conducted to evaluate its efficacy and safety. Although some studies report promising results, the clinical utility of rIPC to prevent AKI is still unclear. As it can be appreciated in Table 4, the protocol used in most of the studies using rIPC is practically the same, which involves 3 to 4 cycles of 5 min of arm ischemia, which was achieved through arm compression with a cuff inflated mostly to 200 mmHg. Even though the same protocol was followed in several studies, the results are contrasting, as we will see below.
Table 4.
Results Generated by Clinical trials with remote IPC on AKI incidence.
In 2007, Ali, ZA et al. [96] included 82 patients in a study on repairing abdominal aortic aneurysms (AAA) who were randomized into two groups: one received rIPC through two cycles of intermittent cross-clamping of the common iliac artery for 10 min, followed by a same period of reperfusion, and the other was treated without rIPC. Interestingly, rIPC reduced the incidence of postoperative myocardial injury (27% that can be caused by myocardial infarction), myocardial infarction (22%), and renal impairment (23%). In another study that included 40 AAA patients with rIPC in both lower limbs, the urine albumin/creatinine ratio decreased (46%) in comparison with the control group without rIPC [98]. Most clinical trials seeking to reduce AKI have studied rIPC in patients who have undergone cardiac surgery. Venugopal et al. [102] reported a decrease in AKI incidence by 14.5% when rIPC was applied to 78 patients. Deftereos et al. [97] included 220 patients undergoing cardiac surgery in a study and found that AKI incidence was 29.5% in the control group (n = 109), whereas it was reduced to 12.4% in the rIPC group (n = 111). In a similar study, which included 160 patients under cardiac surgery, the AKI incidence was reduced from 47.5% to 30% in patients with rIPC compared to controls [113]. Zhou, H et al. [114] found that the postoperative AKI incidence of 73.8% in 65 patients decreased to 55.4% in 65 patients with rIPC. Another study involving 28 patients found that in one-half of them with rIPC, there was a 64% decrease in cardiac surgery associated with AKI [108]. Interestingly, Zarbock et al. [105] evaluated the short- and long-term effects of rIPC in 240 patients undergoing cardiac surgery. Half of them received the rIPC strategy, finding not only that AKI cases were significantly reduced in the first 72 h, but also the incidence of long-term adverse renal events was reported after 90 days, such as all-cause of mortality, renal replacement therapy, and persistent renal dysfunction without dialysis [107]. Furthermore, different periods of rIPC were studied in 100 patients subjected to cardiac surgery. Intriguingly, all periods of rIPC studied were associated with an increase in tissue inhibitor of metalloproteinases (TIMP-2) and insulin-like growth factor-binding protein (IGFBP7), which in turn correlated with a reduction in AKI cases [110].
The rIPC has also been evaluated in 178 patients undergoing coronary artery bypass graft (CABG); it was found that AKI incidence decreased by 48% in the rIPC group compared to the control group [101]. In addition, the reduction in renal injury biomarkers, such as cystatin C and NGAL, has been reported after rIPC in 60 patients with CABG [100].
Patients undergoing coronary artery angiography (CAA) are a population at risk of developing contrast-induced AKI (CI-AKI); therefore, several clinical trials have studied whether rIPC could be used as a renal protective intervention. Er et al. [115] randomized 82 patients, half of whom served as controls and the other half received rIPC, finding a decrease in CI-AKI incidence from 40% to 12%, respectively. Savaj, et al. [104] studied 96 diabetic patients who underwent CAA and who were randomly divided equally into the control and rIPC group; in spite of an observed reduction of SCr due to rIPC, the CI-AKI incidence was not different between groups. These results suggest that the heterogeneity of the patients studied prevented seeing a difference in the incidence of AKI. In another study, the benefit of rIPC was evaluated in 60 patients with pre-established CKD and who underwent angiography, half of them without rIPC and the other half with rIPC; a decrease in the incidence of CI-AKI was observed from 26.9% to 7.7%, respectively [111]. Similar results have been reported by Yamanaka et al. [106], Zagidullin et al., [109] Moretti et al. [112], and Elserafy et al., finding a decrease in the incidence of CI-AKI [116]. Additionally, Thielman et al. [103] and Zimmerman et al. [99] studied patients under CABG or valve surgery and observed a SCr decrease due to rIPC of 3 cycles of 5 min of ischemia in the upper arm or lower extremity, respectively.
In addition to these clinical studies that clearly demonstrate that rIPC produces renoprotection, there are a number of studies that report that the rIPC strategy in patients with risk of developing AKI does not exert renal protection or influence the incidence of AKI [117,122,123,124,126,128,129,130,131,132], particularly in CI-AKI [64,125,127,133,135,136] or on renal injury biomarkers [118,119,120,121]. These discrepancies could be due to several factors that reflect the complexity of the clinical trial design and the influence of comorbidities of each studied population. All these variables that impact the response to rIPC were reviewed by McCafferty K et al. [137] in animal models and other organs besides the kidney. In these studies, the included groups were composed of young and healthy animals. Unfortunately, in most of the clinical trials, these conditions do not occur because the patients evaluated exhibit one or more comorbidities, and some of them are elderly. Undoubtedly, more studies are needed to consider these factors and to determinate the influence of each one on the possible renoprotection by rIPC.
Considering the differences in the clinical trials mentioned above, it is crucial to deepen our knowledge of all the possible molecular and cellular mechanisms involved; however, it is necessary to carry out clinical trials designed to assess the real impact of rIPC, pondering the specific influence of comorbidities in the studied population.
Finally, the signaling pathways described so far that participate in the renoprotection conferred by the IPC are summarized in Figure 1. These pathways included actin microfilament stabilization, autophagy induction, and reduction of oxidative stress through the induction of antioxidant enzymes such catalase and SOD. The resulting decrease in the generation of free radicals leads to the inhibition of apoptosis, and, in particular, to the turnover of mitochondria through mitophagy. Coupled with this, the IPC also influences the inflammatory response that is activated during AKI by maintaining an anti-inflammatory profile, which in turn allows controlled removal of damaged cells without activating immune cell infiltration signals and without the production of extracellular matrix proteins. We believe that the knowledge of the signaling pathways involved during the IPC will allow the creation of new proposals for therapies that activate or regulate some of these signaling pathways, without forgetting the great potential that the rIPC has, above all because it is a simple, noninvasive, safe, and low-cost strategy.
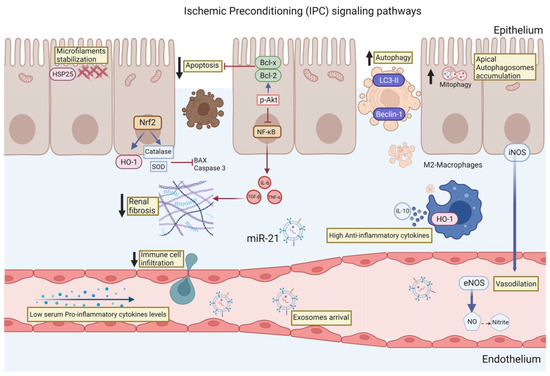
Figure 1.
Main renoprotective signaling pathways induced by the ischemic preconditioning (IPC): (1) The induction of HSP27 allows the stabilization of the microfilament, which blocks epithelial cells from losing their polarization, a classic alteration observed after an IR episode. (2) Increased translocation of the transcription factor Nrf2 to the nucleus, where it induces the transcription of antioxidant enzymes such as catalase, superoxide dismutase (SOD), and heme-oxygenase (HO-1), which prevents the formation of reactive oxygen species (ROS). (3) Inhibition of epithelial cell apoptosis by reducing ROS and increasing the expression of antiapoptotic proteins such as Bcl-x and Bcl-2. (4) Although autophagy, particularly mitophagy, is increased, this allows for better regulation of damaged mitochondria. (5) Decreased activation of the proinflammatory transcription factor NF-κB, which also decreases the release of proinflammatory cytokines such as IL-6 and TNF-α, which, in turn, prevent the generation of renal fibrosis in the long term. (6) Increase of cells with an anti-inflammatory profile such as M2-macropaghes that remove cell debris and secrete anti-inflammatory cytokines such as IL-10. (7) Finally, some molecules such as NO, nitrites, and exosomes seem to be involved in the remote actions of IPC.
Author Contributions
J.A.O.-T. and N.A.B. prepared figures; J.A.O.-T. and N.A.B. drafted manuscript; J.A.O.-T. and N.A.B. edited and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Our study mentioned in the present review were supported by grants from the Mexican Council of Science and Technology (CONACyT) (A1-S-8715 to NAB), from the Universidad Nacional Autónoma de México, UNAM/PAPIIT (IN201619 and IN201022 to NAB), and from Aliados Estratégicos en Salud y Nutrición (to NAB).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This study was performed in partial fulfillment of the requirements for the degree, Juan Antonio Ortega-Trejo is a doctoral student from Programa de Doctorado en Ciencias Bioquímicas, Universidad Nacional Autónoma de México (UNAM) and received a fellowship from CONACYT (572965).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Al-jaghbeer, M.; Dealmeida, D.; Bilderback, A.; Ambrosino, R.; Kellum, J.A. Clinical Decision Support for In-Hospital AKI. J. Am. Soc. Nephrol. 2018, 29, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.J.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epipidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Lewington, A.J.P.; Mehta, R.L.; Cerda, J. Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef]
- See, E.J. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef]
- Rodríguez-Romo, R.; Benítez, K.; Barrera-Chimal, J.; Pérez-Villalva, R.; Gómez, A.; Aguilar-León, D.; Rangel-Santiago, J.F.; Huerta, S.; Gamba, G.; Uribe, N.; et al. AT1 receptor antagonism before ischemia prevents the transition of acute kidney injury to chronic kidney disease. Kidney Int. 2016, 89, 363–373. [Google Scholar] [CrossRef]
- Noble, R.A.; Lucas, B.J.; Selby, N.M. Long-term outcomes in patients with acute kidney injury. Clin. J. Am. Soc. Nephrol. 2020, 15, 423–429. [Google Scholar] [CrossRef]
- García-Ortuño, L.E.; Bobadilla, N.A. Integrative view of the mechanisms that induce acute kidney injury and its transition to chronic kidney disease. Rev. Investig. Clin. 2018, 70, 261–268. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Pérez-Villalva, R.; Ortega, J.A.; Sánchez, A.; Durand, M.; Jaisser, F.; Bobadilla, N.A. Mild ischemic Injury Leads to Long-Term Alterations in the Kidney: Amelioration by Spironolactone Administration. Int. J. Biol. Sci. 2015, 11, 892–900. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Sykes, L.; Asar, O.; Ritchie, J.; Raman, M.; Vasallo, D.; Alderson, H.V.; O’Dnogue, D.J.; Green, D.; Diggle, P.J.; Kalra, P.A. The influence of multiple episodes of acute kidney injury on survival and progression to end stage kidney disease in patients with chronic kidney disease. PLoS ONE 2019, 14, e0219828. [Google Scholar] [CrossRef]
- Thakar, C.V.; Christianson, A.; Himmelfarb, J.; Leonard, A.C. Acute Kidney Injury Episodes and Chronic Kidney Disease Risk in Diabetes Mellitus. Clin. J. Am. Soc. Nephrol. 2011, 6, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Siew, E.D.; Parr, S.K.; Abdel-Kader, K.; Eden, S.K.; Peterson, J.F.; Bansal, N.; Hung, A.M.; Fly, J.; Speroff, T.; Ikizler, T.A.; et al. Predictors of Recurrent AKI. J. Am. Soc. Nephrol. 2016, 27, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.; Marks, A.; Fluck, N.; Levin, A.; Prescott, G.; Black, C. Intermediate and Long-term Outcomes of Survivors of Acute Kidney Injury Episodes: A Large Population-Based Cohort Study. Am. J. Kidney Dis. 2017, 69, 18–28. [Google Scholar] [CrossRef]
- Rodríguez, E.; Arias-Cabrales, C.; Bermejo, S.; Sierra, A.; Burballa, C.; Soler, M.J.; Barrios, C.; Pascual, J. Impact of Recurrent Acute Kidney Injury on Patient Outcomes. Kidney Blood Press. Res. 2018, 43, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.; Geen, J.; Williams, J.D.; Phillips, A.O. Recurrent acute kidney injury: Predictors and impact in a large population-based cohort. Nephrol. Dial. Transplant. 2020, 35, 1361–1369. [Google Scholar] [CrossRef]
- Yamasowa, H.; Shimizu, S.; Inoue, T.; Takaoka, M.; Matsumura, Y. Endothelial Nitric Oxide Contributes to the Renal Protective Effects of Ischemic Preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 153–159. [Google Scholar] [CrossRef]
- Joo, J.D.; Kim, M.; D′Agati, V.D.; Lee, H.T. Ischemic Preconditioning Provides Both Acute and Delayed Protection against Renal Ischemia and Reperfusion Injury in Mice. J. Am. Soc. Nephrol. 2006, 17, 3115–3123. [Google Scholar] [CrossRef]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, P.; Li, R.; Ren, J.; Zhou, H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020, 30, 101415. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xing, B.; Liu, X.; Zhan, B.; Zhou, J.; Zhu, H.; Chen, Z. Similarities Between Ozone Oxidative Preconditioning and Ischemic Preconditioning in Renal Ischemia/Reperfusion Injury. Arch. Med. Res. 2008, 39, 169–178. [Google Scholar] [CrossRef]
- Timsit, M.O.; Gadet, R.; Abdennebi, H.B.; Codas, R.; Petruzzo, P.; Badet, L. Renal Ischemic Preconditioning Improves Recovery of Kidney Function and Decreases α-Smooth Muscle Actin Expression in a Rat Model. J. Urol. 2008, 180, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Mahfoudh-Boussaid, A.; Zaouaali, M.A.; Hadj-Ayed, K.; Miled, A.-H.; Saidane-Mosbahi, D.; Rosello-Carafau, J.; Ben Abdennebi, H. Ischemic preconditioning reduces endoplasmic reticulum stress and upregulates hypoxia inducible factor-1alpha in ischemic kidney: The role of nitric oxide. J. Biomed. Sci. 2012, 19, 1–8. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Pevzner, I.B.; Andrianova, N.V.; Zorova, L.D.; Popkov, V.A.; Solachev, D.N.; Kolosova, N.G.; Plotnikov, E.Y.; Zorov, D.B. The age-associated loss of ischemic preconditioning in the kidney is accompanied by mitochondrial dysfunction, increased protein acetylation and decreased autophagy. Sci. Rep. 2017, 7, 44430. [Google Scholar] [CrossRef]
- Xie, Y.; Jiang, D.; Xiao, J.; Fu, C.; Zhang, Z.; Ye, Z.; Zhang, X. Ischemic preconditioning attenuates ischemia/reperfusion-induced kidney injury by activating autophagy via the SGK1 signaling pathway. Cell Death Dis. 2018, 9, 338. [Google Scholar] [CrossRef]
- Li, J.R.; Ou, Y.C.; Wu, C.C.; Wang, J.D.; Lin, S.Y.; Wang, Y.Y.; Chen, C.J. Ischemic preconditioning improved renal ischemia/reperfusion injury and hyperglycemia. IUBMB Life 2019, 71, 321–329. [Google Scholar] [CrossRef]
- Khalid, U.; Jenkins, R.H.; Andrews, R.; Pino-Chavez, G.; Cossins, B.C.; Chavez, R.; Bowen, T.; Fraser, D.J. Determination of a microRNA signature of protective kidney ischemic preconditioning originating from proximal tubules. Sci. Rep. 2021, 11, 9862. [Google Scholar] [CrossRef]
- Yamamoto, M.; Morita, T.; Ishikawa, M.; Sakamoto, A. Specific microRNAs are involved in the reno-protective effects of sevoflurane preconditioning and ischemic preconditioning against ischemia reperfusion injury in rats. Int. J. Mol. Med. 2020, 45, 1141–1149. [Google Scholar] [CrossRef]
- Choi, H.S.; Hwang, J.K.; Kim, J.G.; Hwang, H.S.; Lee, S.J.; Chang, Y.K.; Kim, J.I.; Moon, I.S. The optimal duration of ischemic preconditioning for renal ischemia-reperfusion injury in mice. Ann. Surg. Treat. Res. 2017, 93, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xia, W.; Duan, J.; Li, X.; Qian, S.; Shen, H. Ischemic Preconditioning Alleviates Mouse Renal Ischemia/Reperfusion Injury by Enhancing Autophagy Activity of Proximal Tubular Cells. Kidney Dis. 2022, 8, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Song, N.; Fang, Y.; Teng, J.; Xu, X.; Hu, J.; Zhang, P.; Chen, R.; Lu, Z.; Yu, X.; et al. Delayed Ischemic Preconditioning Attenuated Renal Ischemia-Reperfusion Injury by Inhibiting Dendritic Cell Maturation. Cell. Physiol. Biochem. 2018, 46, 1807–1820. [Google Scholar] [CrossRef]
- Burne-Taney, M.J.; Liu, M.; Baldwin, W.M.; Racusen, L.; Rabb, H. Decreased Capacity of Immune Cells to Cause Tissue Injury Mediates Kidney Ischemic Preconditioning. J. Immunol. 2006, 176, 7015–7020. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.Y.; Choi, H.M.; Lee, S.Y.; Kim, M.G.; Kim, H.-K.; Jo, S.K. The role of Tregs and CD11c+ macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 2010, 78, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Kim, J.I.; Kim, J.; Park, J.W.; Park, K.M. Angiotensin II Removes Kidney Resistance Conferred by Ischemic Preconditioning. Biomed Res. Int. 2014, 2014, 602149. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, J.; Kim, K.Y.; Kim, J.I.; Cho, M.H.; Park, J.M. Previous ischemia and reperfusion injury results in resistance of the kidney against subsequent ischemia and reperfusion insult in mice; A role for the Akt signal pathway. Nephrol. Dial. Transplant. 2012, 27, 3762–3770. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, J.; Park, Y.K.; Park, K.M. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation 2008, 85, 447–455. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.; Park, K.M. Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. AJP Ren. Physiol. 2010, 298, F158–F166. [Google Scholar] [CrossRef]
- Park, K.M.; Byun, J.Y.; Kramers, C.; Kim, J.I.; Huang, P.L.; Bonventre, J.V. Inducible nitric-oxide synthase is an important contributor to prolonged protective effects of ischemic preconditioning in the mouse kidney. J. Biol. Chem. 2003, 278, 27256–27266. [Google Scholar] [CrossRef]
- Park, K.M.; Chen, A.; Bonventre, J.V. Prevention of Kidney Ischemia/Reperfusion-induced Functional Injury and JNK, p38, and MAPK Kinase Activation by Remote Ischemic Pretreatment. J. Biol. Chem. 2001, 276, 11870–11876. [Google Scholar] [CrossRef]
- Zager, R.A.; Baltes, L.A.; Sharma, H.M.; Jurkowitz, M.S. Responses of the ischemic acute renal failure kidney to additional ischemic events. Kidney Int. 1984, 26, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Jurkowitz, S.; Merola, A.J. Responses of the normal rat kidney to sequential ischemic events. Am. J. Physiol.-Ren. Physiol. 1985, 249, F148–F159. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Lameire, N. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. 2012, 2, 1–138. [Google Scholar]
- Kelly, K.J.; Williams, W.W.; Colvin, R.B.; Bonventre, J.V. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. USA 1994, 91, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Ysebaert, D.K.; De Greef, K.E.; Vercauteren, S.R.; Ghielli, M.; Verpooten, G.A.; Eysken, E.J.; De Broe, M. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 2000, 15, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Hörbelt, M.; Lee, S.Y.; Mang, H.E.; Knipe, N.L.; Sado, Y.; Kribben, A.; Sutton, T.A. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2007, 293, 688–695. [Google Scholar] [CrossRef]
- Mukherjee, K.; Gu, C.; Collins, A.; Mettlen, M.; Samelko, B.; Altintas, M.M.; Sudhini, Y.R.; Wang, X.; Bouley, R.; Brown, D.; et al. Simultaneous stabilization of actin cytoskeleton in multiple nephron-specific cells protects the kidney from diverse injury. Nat. Commun. 2022, 13, 2422. [Google Scholar] [CrossRef]
- Patel, R.; McKenzie, J.; McQueen, E. Tamm-Horsfall Urinary Mucoprotein and Tubular Obstruction by Casts in Acute Renal Failure. Lancet 1964, 283, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef]
- Geng, H.; Lan, R.; Wang, G.; Siddiqi, A.R.; Naski, M.C.; Brooks, A.I.; Barnes, J.L.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Inhibition of Autoregulated TGF-beta Signaling Simultaneously Enhances Proliferation and Differentiation of Kidney Epithelium and Promotes Repair Following Renal Ischemia. Am. J. Pathol. 2009, 174, 1291–1308. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Markó, L.; Vigolo, E.; Hinze, C.; Park, J.K.; Roël, G.; Balogh, A.; Choi, M.; Wübken, A.; Cording, J.; Blasig, I.E.; et al. Tubular epithelial NF-κB activity regulates ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 2658–2669. [Google Scholar] [CrossRef]
- Yamamoto, T.; Noble, N.A.; Miller, D.E.; Border, W.A. Sustained expression of TGF-beta1 underlies development of progressive kidney fibrosis. Kidney Int. 1994, 45, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Amdur, R.L.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011, 79, 1361–1369. [Google Scholar] [CrossRef]
- Hatakeyama, Y.; Horino, T.; Nagata, K.; Matsumoto, T.; Terada, Y.; Okuhara, Y. Transition from acute kidney injury to chronic kidney disease: A single-centre cohort study. Clin. Exp. Nephrol. 2018, 22, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Donohoe, D.L.; Roethe, K.; Mattson, S.L. Chronic renal hypoxia after acute ischemic injury: Effects of L -arginine on hypoxia and secondary damage. Am. J. Physiol.-Ren. Physiol. 2003, 53226, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Tanaka, M.; Humphreys, B.D. Fluorescence Microangiography for Quantitative Assessment of Peritubular Capillary Changes after AKI in Mice. J. Am. Soc. Nephrol. 2014, 25, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chang, F.; Schrimpf, C.; Chen, Y.; Wu, C.; Wu, V.; Chiang, W.; Kuhnert, F.; Kuo, C.J.; Chen, Y.; et al. Targeting Endothelium-Pericyte Cross Talk by Inhibiting VEGF Receptor Signaling Attenuates Kidney Microvascular Rarefaction and Fibrosis. Am. J. Physiol. 2011, 178, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1136–1224. [Google Scholar] [CrossRef]
- Arantes, V.M.; Bueno, R.T.; Módolo, R.P.; Domingues, M.A.C.; de Carvalho, L.R.; do Nascimento Junior, P.; Módolo, N.S.P. Effects of Ischemic Preconditioning and Postconditioning in a Renal Ischemia-Reperfusion Injury Model: A Comparative Experimental Study in Rats. Transplant. Proc. 2018, 50, 3811–3815. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, Q.; Wen, J.; Chen, T.; He, L.; Wang, Y.; Yin, J.; Wu, R.; Xue, R.; Li, S.; et al. Ischemic Duration and Frequency Determines AKI-to-CKD Progression Monitored by Dynamic Changes of Tubular Biomarkers in IRI Mice. Front. Physiol. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Trejo, J.A.; Pérez-Villalva, R.; Sánchez-Navarro, A.; Marquina, B.; Rodríguez-Iturbe, B.; Bobadilla, N.A. Repeated Episodes of Ischemia/Reperfusion Induce Heme-Oxygenase-1 (HO-1) and Anti-Inflammatory Responses and Protects against Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 14573. [Google Scholar] [CrossRef] [PubMed]
- Menting, T.P.; Ergun, M.; Bruintjes, M.H.D.; Wever, K.E.; Lomme, R.M.L.M.; van Goor, H.; Warlé, M.C. Repeated remote ischemic preconditioning and isoflurane anesthesia in an experimental model of renal ischemia-reperfusion injury. BMC Anesthesiol. 2017, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Kierulf-Lassen, C.; Kristensen ML, V.; Birn, H.; Jespersen, B.; Nørregaard, R. No effect of remote ischemic conditioning strategies on recovery from renal ischemia-reperfusion injury and protective molecular mediators. PLoS ONE 2015, 10, e0146109. [Google Scholar] [CrossRef]
- Wang, F.; Yin, J.; Lu, Z.; Zhang, G.; Li, J.; Xing, T.; Zhuang, S.; Wang, N. Limb ischemic preconditioning protects against contrast-induced nephropathy via renalase. EBioMedicine 2016, 9, 356–365. [Google Scholar] [CrossRef]
- Gholampour, F.; Khangah, L.; Vatanparast, J.; Karbalaei-Heidari, H.R.; Owji, S.M.; Bahaoddini, A. The role of nitric oxide in the protective action of remote ischemic per-conditioning against ischemia/reperfusion-induced acute renal failure in rat. Iran. J. Basic Med. Sci. 2018, 21, 600–606. [Google Scholar]
- Silachev, D.N.; Isaev, N.K.; Pevzner, I.B.; Zorova, L.D.; Stelmashook, E.V.; Novikova, S.V.; Plotnikov, E.Y.; Skulachev, V.P.; Zorov, D.B. The Mitochondria-Targeted Antioxidants and Remote Kidney Preconditioning Ameliorate Brain Damage through Kidney-to-Brain Cross-Talk. PLoS ONE 2012, 7, e51553. [Google Scholar] [CrossRef]
- Pan, T.; Jia, P.; Chen, N.; Fang, Y.; Liang, Y.; Guo, M.; Ding, X. Delayed remote ischemic preconditioning confers renoprotection against septic acute kidney injury via exosomal miR-21. Theranostics 2019, 9, 405–423. [Google Scholar] [CrossRef]
- Song, T.; Peng, Y.; Guo, S.; Liu, Y.; Liu, L. Brief Small Intestinal Ischemia Lessens Renal Ischemia-reperfusion Injury in Rats. Comp. Med. 2007, 57, 200–205. [Google Scholar]
- Shen, Y.; Qiu, T.; Liu, X.H.; Zhang, L.; Wang, Z.S.; Zhou, J.Q. Renal ischemia-reperfusion injury attenuated by splenic ischemic preconditioning. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2134–2142. [Google Scholar] [PubMed]
- Cho, K.; Min, S.I.; Ahn, S.; Min, S.K.; Ahn, C.; Yu, K.S.; Jang, I.J.; Cho, J.Y.; Ha, J. Integrative Analysis of Renal Ischemia/Reperfusion Injury and Remote Ischemic Preconditioning in Mice. J. Proteome Res. 2017, 16, 2877–2886. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y.; Zhao, S.; Chen, J.; Jin, S.; Jia, P.; Ding, X. Remote Ischemic Preconditioning Ameliorates Acute Kidney Injury due to Contrast Exposure in Rats through Augmented O-GlcNAcylation. Oxid. Med. Cell. Longev. 2018, 2018, 4895913. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Ghanem, S.; Szabo, B.; Nagy, K.; Pal, N.; Tanczos, B.; Somogyi, V.; Barath, B.; Deak, A.; Matolay, O.; et al. Which remote ischemic preconditioning protocol is favorable in renal ischemia-reperfusion injury in the rat? Clin. Hemorheol. Microcirc. 2020, 76, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liu, J.; Ge, Y.; Zhu, Y.; Zhou, L.; Xu, L.; Xu, Z.; Wu, R.; Jia, R. Remote ischemic preconditioning ameliorates renal fibrosis after ischemia-reperfusion injury via transforming growth factor beta1 (TGF-b1) signalling pathway in rats. Med. Sci. Monit. 2020, 26, 1–9. [Google Scholar]
- Terker, A.S.; Sasaki, K.; Arroyo, J.P.; Niu, A.; Wang, S.; Fan, X.; Zhang, Y.; Nwosisi, S.; Zhang, M.Z.; Harris, R.C. Activation of hypoxia-sensing pathways promotes renal ischemic preconditioning following myocardial infarction. Am. J. Physiol.-Ren. Physiol. 2021, 320, F569–F577. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef]
- Rhee, S.G. H2O2, a Necessary Evil for. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Huang, C.H.; Lai, C.C.; Yang, A.H.; Chiang, S.C. Myocardial preconditioning reduces kidney injury and apoptosis induced by myocardial ischaemia and reperfusion. Eur. J. Cardio-thoracic Surg. 2015, 48, 382–391. [Google Scholar] [CrossRef]
- Zaouali, M.A.; Mosbah, I.B.; Boncompagni, E.; Abdennebi, H.B.; Mitjavila, M.T.; Bartrons, R.; Freitas, I.; Rimola, A.; Roselló-Catafau, J. Hypoxia inducible factor-1 α accumulation in steatotic liver preservation: Role of nitric oxide. World J. Gastroenterol. 2010, 16, 3499–3509. [Google Scholar] [CrossRef]
- Liu, X.; Miller, M.J.S.; Joshi, M.S.; Sadowaska-krowicka, H.; Clark, D.A.; Lancaster, J.R. Diffusion-limited Reaction of Free Nitric Oxide with Erythrocytes. J. Biol. Chem. 1998, 273, 18709–18713. [Google Scholar] [CrossRef]
- Dejam, A.; Hunter, C.J.; Tremonti, C.; Pluta, R.M.; Hon, Y.Y.; Grimes, G.; Partovi, K.; Pelletier, M.M.; Oldfield, E.H.; Iii, R.O.C.; et al. Nitrite Infusion in Humans and Nonhuman Primates Endocrine Effects, Pharmacokinetics, and Tolerance Formation. Circulation 2007, 116, 1821–1831. [Google Scholar] [CrossRef]
- Gonzalez, F.M.; Shiva, S.; Vincent, P.S.; Ringwood, L.A.; Hsu, L.; Hon, Y.Y.; Aletras, A.H.; Iii, R.O.C.; Gladwin, M.T.; Arai, A.E.; et al. Nitrite Anion Provides Potent Cytoprotective and Antiapoptotic Effects as Adjunctive Therapy to Reperfusion for Acute Myocardial Infarction. Circulation 2008, 117, 2986–2994. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P. Roles of Heat-Shock Proteins in Innate and Adaptive Immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Smoyer, W.E.; Ransom, R.; Harris, R.C.; Welsh, M.J.; Lutsch, G.; Benndorf, R. Ischemic acute renal failure induces differential expression of small heat shock proteins. J. Am. Soc. Nephrol. 2000, 11, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Pobre, K.F.R.; Poet, X.G.J.; Hendershot, X.L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, G.; Lu, Z.; Geurts, A.M.; Jacob, H.J.; Cowley, A.W.; Wang, N.; Liang, M. Antithrombin III/SerpinC1 insufficiency exacerbates renal ischemia/reperfusion injury. Kidney Int. 2015, 88, 796–803. [Google Scholar] [CrossRef]
- Du, M.; Huang, K.; Huang, D.; Yang, L.; Gao, L.; Wang, X.; Huang, D.; Li, X.; Wang, C.; Zhang, F.; et al. Renalase is a novel target gene of hypoxia-inducible factor-1 in protection against cardiac ischaemia–reperfusion injury. Cardiovasc. Res. 2015, 105, 182–191. [Google Scholar] [CrossRef]
- Sheng, R.; Qin, Z. The divergent roles of autophagy in ischemia and preconditioning. Acta Pharmacol. Sin. 2015, 3, 411–420. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Ruscher, K.; Freyer, D.; Karsch, M.; Isaev, N.; Megow, D.; Sawitzki, B.; Priller, J.; Dirnagl, U.; Meisel, A. Erythropoietin Is a Paracrine Mediator of Ischemic Tolerance in the Brain: Evidence from an In Vitro Model. J. Neurosci. 2002, 22, 10291–10301. [Google Scholar] [CrossRef] [PubMed]
- Brines, M.; Grasso, G.; Fiordaliso, F.; Sfacteria, A.; Ghezzi, P.; Fratelli, M.; Latini, R.; Xie, Q.; Smart, J.; Pobre, E.; et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta -subunit heteroreceptor. Proc. Natl. Acad. Sci. USA 2004, 101, 14907–14912. [Google Scholar] [CrossRef]
- Moon, C.; Krawczyk, M.; Paik, D.; Coleman, T.; Brines, M.; Juhaszova, M.; Sollot, S.J.; Lakatta, E.G.; Talan, M.I. Erythropoietin, Modified to Not Stimulate Red Blood Cell Production, Retains Its Cardioprotective Properties. J. Pharmacol. Exp. Ther. 2006, 316, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammonds, J.R.; Turbide, C. Vesicle Formation during Reticulocyte Maturation. J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.A.; Callaghan, C.J.; Lim, E.; Ali, A.A.; Nouraei, S.A.R.; Akthar, A.M.; Boyle, J.R.; Varty, K.; Kharbanda, R.K.; Dutka, D.P.; et al. Remote Ischemic Preconditioning Reduces Myocardial and Renal Injury After Elective Abdominal Aortic Aneurysm Repair a Randomized Controlled Trial. Circulation 2007, 116, I-98–I-105. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Tzalamouras, V.; Raisakis, K.; Kossyvakis, C.; Kaoukis, A.; Panagopoulou, V.; Karageorgiou, S.; Avramides, D.; Toutouzas, K.; et al. Renoprotective effect of remote ischemic post-conditioning by intermittent balloon inflations in patients undergoing percutaneous coronary intervention. J. Am. Coll. Cardiol. 2013, 61, 1949–1955. [Google Scholar] [CrossRef]
- Walsh, S.R.; Boyle, J.R.; Tang, T.Y.; Sadat, U.; Cooper, D.G.; Lapsley, M.; Norden, A.G.; Varty, K.; Hayes, P.D.; Gaunt, M.E. Remote Ischemic Preconditioning for Renal and Cardiac Protection During Endovascular Aneurysm Repair: A Randomized Controlled Trial. J. Endovasc. Ther. 2009, 16, 680–689. [Google Scholar] [CrossRef]
- Zimmerman, R.F.; Ezeanuna, P.U.; Kane, J.C.; Cleland, C.D.; Kempananjappa, T.J.; Lucas, F.L.; Kramer, R.S. Ischemic preconditioning at a remote site prevents acute kidney injury in patients following cardiac surgery. Kidney Int. 2011, 80, 861–867. [Google Scholar] [CrossRef]
- Yıldırım, F.; Şenarslan, D.A.; İşkesen, İ.; Ön, U.; Kalp, K.; Böbreği, C. Remote Preconditioning Might Protect the Kidney in Heart Surgery. Göğüs-Kalp-Damar Anestezi 2018, 24, 103–110. [Google Scholar]
- Candilio, L.; Malik, A.; Ariti, C.; Barnand, M.; Di Salvo, C.; Lawrence, D.; Hayward, M.; Yap, J.; Roberts, N.; Sheikh, A.; et al. Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: A randomised controlled clinical trial. Heart 2014, 101, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, V.; Laing, C.M.; Ludman, A.; Yellon, D.M.; Hausenloy, D. Effect of Remote Ischemic Preconditioning on Acute Kidney Injury in Nondiabetic Patients Undergoing Coronary Artery Bypass Graft Surgery: A Secondary Analysis of 2 Small Randomized Trials. Am. J. Kidney Dis. 2010, 56, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, M.; Kottenberg, E.; Boengler, K.; Raffelsieper, C.; Neuhaeuser, M.; Peters, J.; Jakob, H.; Heusch, G. Remote ischemic preconditioning reduces myocardial injury after coronary artery bypass surgery with crystalloid cardioplegic arrest. Basic Res. Cardiol. 2010, 105, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Savaj, S.; Savoj, J.; Jebraili, I. Remote Ischemic Preconditioning for Prevention of Contrast-induced Acute Kidney Injury in Diabetic Patients. Iran. J. Kidney Dis. 2014, 8, 457–460. [Google Scholar]
- Zarbock, A.; Schmidt, C.; Van Aken, H.; Wempe, C.; Martens, S.; Zhan, P.K.; Wolf, B.; Goebel, U.; Schwer, C.I.; Rosenberger, P.; et al. Effect of Remote Ischemic Preconditioning on Kidney Injury Among High-Risk Patients Undergoing Cardiac Surgery. Jama 2015, 313, 2133. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Kawai, Y.; Miyoshi, T.; Mima, T.; Takagaki, K.; Tsukuda, S.; Kazatani, Y.; Nakamura, K.; Ito, H. Remote ischemic preconditioning reduces contrast-induced acute kidney injury in patients with ST-elevation myocardial infarction: A randomized controlled trial. Int. J. Cardiol. 2015, 178, 136–141. [Google Scholar] [CrossRef]
- Zarbock, A.; Kellum, J.A.; Van Aken, H.; Schmidt, C.; Küllmar, M.; Rosenberger, P.; Martens, S.; Görlich, D.; Meersch, M. Long-term Effects of Remote Ischemic Preconditioning on Kidney Function in High-risk Cardiac Surgery Patients. Anesthesiology 2017, 126, 787–798. [Google Scholar] [CrossRef]
- Stokfisz, K.; Ledakowicz-Plak, A.; Zagórski, M.; Jander, S.; Przybylak, K.; Zielinska, M. The clinical utility of remote ischemic preconditioning in protecting against cardiac surgery-associated acute kidney injury: A pilot randomized clinical trial. Adv. Clin. Exp. Med. 2020, 29, 189–196. [Google Scholar] [CrossRef]
- Zagidullin, N.S.; Dunayeva, A.R.; Plechev, V.V.; Gilmanov, A.Z.; Zagidullin, S.Z.; Er, F.; Pavlov, V.N. Nephroprotective effects of remote ischemic preconditioning in coronary angiography. Clin. Hemorheol. Microcirc. 2017, 65, 299–307. [Google Scholar] [CrossRef]
- Meersch, M.; Küllmar, M.; Pavenstädt, H.; Rossaint, J.; Kellum, J.A.; Martens, S.; Klausmeyer, P.; Schmidt, E.A.; Kerschke, L.; Zarbock, A. Effects of Different Doses of Remote Ischemic Preconditioning on Kidney Damage among Patients Undergoing Cardiac Surgery: A Single-Center Mechanistic Randomized Controlled Trial. Crit. Care Med. 2020, 48, E690–E697. [Google Scholar] [CrossRef]
- Igarashi, G.; Iino, K.; Watanabe, H.; Ito, H. Remote ischemic pre-conditioning alleviates contrast-induced acute kidney injury in patients with moderate chronic kidney disease. Circ. J. 2013, 77, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Moretti, C.; Cerrato, E.; Cavallero, E.; Lin, S.; Rossi, M.L.; Picchi, A.; Sanguineti, F.; Urgo, F.; Palazzuoli, A.; Bertaina, M.; et al. The EUROpean and Chinese cardiac and renal Remote Ischemic Preconditioning Study (EURO-CRIPS CardioGroup I): A randomized controlled trial. Int. J. Cardiol. 2018, 257, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Min, J.J.; Cho, Y.J.; Hausenloy, D.J.; Ahn, H.; Kim, K.-H.; Hwang, H.Y.; Hong, D.M.; Jeon, Y. Effects of delayed remote ischemic preconditioning on peri-operative myocardial injury in patients undergoing cardiac surgery—A randomized controlled trial. Int. J. Cardiol. 2017, 227, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, L.; Wang, G.; Zhang, C.; Fang, Z.; Lei, G.; Shi, S.; Li, J. Remote Ischemic Preconditioning Prevents Postoperative Acute Kidney Injury After Open Total Aortic Arch Replacement: A Double-Blind, Randomized, Sham-Controlled Trial. Anesth. Analg. 2019, 129, 287–293. [Google Scholar] [CrossRef]
- Er, F.; Nia, A.M.; Dopp, H.; Hellmich, M.; Dahlem, K.M.; Caglayan, E.; Kubacki, T.; Benzing, T.; Erdmann, E.; Burst, V.; et al. Ischemic preconditioning for prevention of contrast medium-induced nephropathy: Randomized pilot renpro trial (Renal protection trial). Circulation 2012, 126, 296–303. [Google Scholar] [CrossRef]
- Elserafy, A.S.; Okasha, N.; Hegazy, T. Prevention of contrast induced nephropathy by ischemic preconditioning in patients undergoing percutaneous coronary angiography. Egypt. Hear. J. 2018, 70, 107–111. [Google Scholar] [CrossRef]
- Walsh, S.R.; Sadat, U.; Boyle, J.R.; Tang, T.Y.; Lapsley, M.; Norden, A.G.; Gaunt, M.E. Remote Ischemic Preconditioning for Renal Protection During Elective Open Infrarenal Abdominal Aortic Aneurysm Repair: Randomized Controlled Trial. Vasc. Endovasc. Surg. 2010, 44, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Shim, J.K.; Kim, J.C.; Kang, K.S.; Seo, Y.H.; Ahn, K.R.; Kwak, Y.L. Effect of remote ischemic preconditioning on renal dysfunction after complex valvular heart surgery: A randomized controlled trial. J. Thorac. Cardiovasc. Surg. 2011, 142, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, H.; Wang, X.; Zhou, Z.; Luo, A.; Tian, Y. Remote Ischemic Preconfitioning Fails to Improve Early Renal Function of Patients Undergoing Living-Donor Renal Transplantation: A Randomized Controlled Trial. Transplantation 2013, 95, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Luo, W.; Huang, R.; Chen, R.; Gao, Y. Multiorgan protection of remote ischemic perconditioning in valve replacement surgery. J. Surg. Res. 2016, 200, 13–20. [Google Scholar] [CrossRef]
- Pedersen, K.R.; Ravn, B.; Povlsen, V.; Schmidt, M.R.; Erlandsen, J.; Hjortdal, V.E. Failure of remote ischemic preconditioning to reduce the risk of postoperative acute kidney injury in children undergoing operation for complex congenital heart disease: A randomized single-center study Abbreviations and Acronyms. J. Thorac. Cardiovasc. Surg. 2012, 143, 576–583. [Google Scholar] [CrossRef]
- Rahman, I.A.; Mascaro, J.G.; Steeds, R.P.; Frenneaux, M.P.; Nightngale, P.; Gosling, P.; Townsend, P.; Townend, J.N.; Green, D.; Bonser, R.S. Remote Ischemic Preconditioning in Human Coronary Artery Bypass Surgery: From Promise to Disappointment? Circulation 2010, 122, 53–60. [Google Scholar] [CrossRef]
- Young, P.J.; Dalley, P.; Garden, A.; Horrocks, C.; La Flamme, A.; Mahon, B.; Miller, J.; Pilcher, J.; Weatherall, J.; Young, W.; et al. A pilot study investigating the effects of remote ischemic preconditioning in high-risk cardiac surgery using a randomised controlled double-blind protocol. Basic Res. Cardiol. 2012, 107, 256. [Google Scholar] [CrossRef] [PubMed]
- Pinaud, F.; Corbeau, J.-J.; Baufreton, C.; Binuani, J.; De Brux, J.-L.; Fouquet, O.; Angoulvant, D.; Furber, A.; Prunier, F. Remote ischemic preconditioning in aortic valve surgery: Results of a randomized controlled study. J. Cardiol. 2016, 67, 36–41. [Google Scholar] [CrossRef]
- Singh, G.B.; Ann, S.H.; Park, J.; Chung, H.C.; Lee, J.S.; Kim, E.S.; Choi, J.; Lee, J.; Kim, S.J.; Shin, E.S. Remote ischemic preconditioning for the prevention of contrast-induced acute kidney injury in diabetics receiving elective percutaneous coronary intervention. PLoS ONE 2016, 11, e0164256. [Google Scholar]
- Bagheri, S.; Shahbazi, S.; Shafa, M.; Borhani-Haghghi, A.; Kiani, M.; Sagheb, M.M. The Effect of Remote Ischemic Preconditioning on the Incidence of Acute Kidney Injury in Patients Undergoing Coronary Artery Bypass Graft Surgery: A Randomized Controlled Trial. Iran. J. Med. Sci. 2018, 43, 587–595. [Google Scholar]
- Wojciechowska, M.; Zarębiński, M.; Pawluczuk, P.; Gralak-Łachowska, D.; Pawłowski, L.; Loska, W.; Goszczyńska, M.; Flis, K.; Cudnoch-Jędrzejewska, A. Remote ischemic preconditioning in renal protection during elective percutaneous coronary intervention. Adv. Exp. Med. Biol. 2018, 1116, 19–25. [Google Scholar] [PubMed]
- Murphy, N.; Vijayan, A.; Frohlich, J.S.; Farrell, F.O.; Barry, M.; Sheehan, S.; Boylan, J.; Conlon, N. Remote Ischemic Preconditioning Does Not Affect the Incidence of Acute Kidney Injury After Elective Abdominal Aortic Aneurysm Repair. J. Cardiothorac. Vasc. Anesth. 2014, 28, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.M.; Jones, D.A.; Kapur, A.; Wragg, A.; Harwood, S.M.; Mathur, R.; Archbold, R.A.; Uppal, R.; Yaqoob, M.M. Remote ischemic preconditioning has a neutral effect on the incidence of kidney injury after coronary artery bypass graft surgery. Kidney Int. 2015, 87, 473–481. [Google Scholar] [CrossRef]
- Nouraei, S.M.; Baradari, A.G.; Jazayeri, A. Does Remote Ischaemic Preconditioning Protect Kidney and Cardiomyocytes After Coronary Revascularization? A Double Blind Controlled Clinical Trial. Med. Arch. 2016, 70, 373–378. [Google Scholar] [CrossRef]
- Song, J.W.; Lee, W.K.; Lee, S.; Shim, J.K.; Kim, H.J.; Kwak, Y.L. Remote ischaemic conditioning for prevention of acute kidney injury after valvular heart surgery: A randomised controlled trial. Br. J. Anaesth. 2018, 121, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.; Candilo, L.; Evans, R.; Ariti, C.; Jenkins, D.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Stokfisz, K.; Ledakowicz-Polak, A.; Kidawa, M.; Zielinska, M. Remote Ischemic Preconditioning and Contrast-Induced Acute Kidney Injury in Patients Undergoing Elective Percutaneous Coronary Intervention: A Randomized Clinical Trial. Curr. Ther. Res.-Clin. Exp. 2020, 93, 100599. [Google Scholar] [CrossRef] [PubMed]
- Menting, T.P.; terenborg, T.B.; De Waal, Y.; Donders, R.; Wever, K.E.; Lemson, M.S.; Van Der Vliet, J.A.; Wetzels, J.F.; Schultzekool, L.J.; Warlé, M.C. Remote Ischemic Preconditioning to Reduce Contrast-Induced Nephropathy: A Randomized Controlled Trial. Eur. J. Vasc. Endovasc. Surg. 2015, 50, 527–532. [Google Scholar] [CrossRef]
- Valappil, S.P.; Kunjukrishnapillai, S.; Viswanathan, S.; Koshy, A.G.; Gupta, P.N.; Velayudhan, R.V.; Iype, M. Remote ischemic preconditioning for prevention of contrast induced nephropathy—Insights from an Indian study. Indian Heart J. 2018, 70, 857–863. [Google Scholar] [CrossRef]
- Ghaemian, A.; Yazdani, J.; Azizi, S.; Farsavian, A.A.; Nabati, M.; Malekrah, A.; Dabirian, M.; Espahbodi, F.; Mirjani, B.; Mohsenipouya, H.; et al. Remote ischemic preconditioning to reduce contrast-induced acute kidney injury in chronic kidney disease: A randomized controlled trial. BMC Nephrol. 2018, 19, 373. [Google Scholar] [CrossRef]
- Mccafferty, K.; Forbes, S.; Thiemermann, C.; Yaqoob, M.M. The challenge of translating ischemic conditioning from animal models to humans: The role of comorbidities. Dis. Model. Mech. 2014, 7, 1321–1333. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).