
Human Tissues Exhibit Diverse Composition of Translation Machinery

 , and
, and 
Abstract
1. Introduction
2. Results
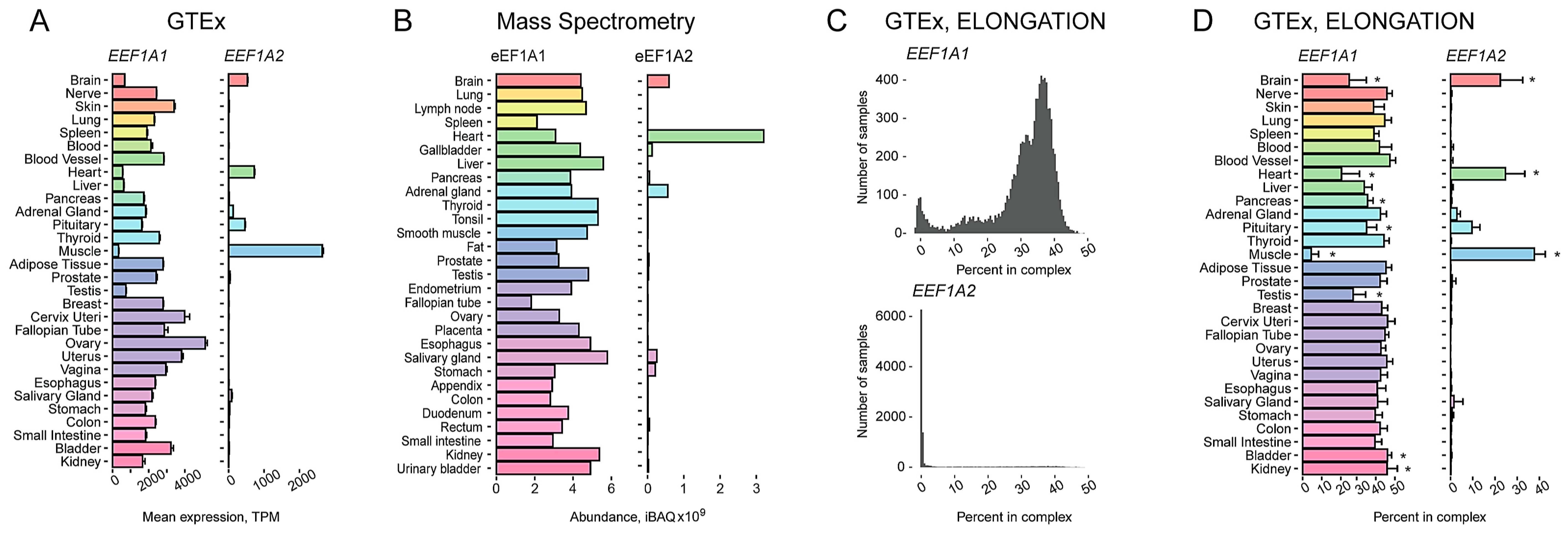
2.1. Characterization of a Tissue-Specific Expression Pattern of Two Human eEF1A Paralogs Using an Integrated Transcriptomic and Proteomic Data Analysis
2.2. Tissue Specificity of Translation-Associated Proteins Encoded in Sex Chromosomes
2.3. A Number of Translation Factors Have a Pronounced Tissue-Specific Representation within Corresponding Functional Groups
2.4. New Striking Examples of Tissue-Specific Expression of Genes Encoding Translation Factors
2.5. Genes Encoding Some ARSases Also Have a Pronounced Tissue-Specific Expression
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Leushkin, E.; Liechti, A.; Ovchinnikova, S.; Mossinger, K.; Bruning, T.; Rummel, C.; Grutzner, F.; Cardoso-Moreira, M.; Janich, P.; et al. Transcriptome and translatome co-evolution in mammals. Nature 2020, 588, 642–647. [Google Scholar] [CrossRef]
- Genuth, N.R.; Barna, M. Heterogeneity and specialized functions of translation machinery: From genes to organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef]
- Shi, Z.; Barna, M. Translating the genome in time and space: Specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu. Rev. Cell Dev. Biol. 2015, 31, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Ingolia, N.T. The Growing Toolbox for Protein Synthesis Studies. Trends Biochem. Sci. 2017, 42, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.P.; Dougherty, J.D.; Heiman, M.; Schmidt, E.F.; Stevens, T.R.; Ma, G.; Bupp, S.; Shrestha, P.; Shah, R.D.; Doughty, M.L.; et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008, 135, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Heiman, M.; Schaefer, A.; Gong, S.; Peterson, J.D.; Day, M.; Ramsey, K.E.; Suarez-Farinas, M.; Schwarz, C.; Stephan, D.A.; Surmeier, D.J.; et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 2008, 135, 738–748. [Google Scholar] [CrossRef]
- Sanz, E.; Yang, L.; Su, T.; Morris, D.R.; McKnight, G.S.; Amieux, P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13939–13944. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Hussmann, J.A.; Weissman, J.S. Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol. 2019, 11, a032698. [Google Scholar] [CrossRef]
- Gonzalez, C.; Sims, J.S.; Hornstein, N.; Mela, A.; Garcia, F.; Lei, L.; Gass, D.A.; Amendolara, B.; Bruce, J.N.; Canoll, P.; et al. Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 10924–10936. [Google Scholar] [CrossRef]
- Gao, X.; Wan, J.; Liu, B.; Ma, M.; Shen, B.; Qian, S.B. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2015, 12, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, A.S.; Meerson, M.B.; Gerashchenko, M.V.; Kulakovskiy, I.V.; Dmitriev, S.E.; Gladyshev, V.N. Multifaceted deregulation of gene expression and protein synthesis with age. Proc. Natl. Acad. Sci. USA 2020, 117, 15581–15590. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, M.V.; Peterfi, Z.; Yim, S.H.; Gladyshev, V.N. Translation elongation rate varies among organs and decreases with age. Nucleic Acids Res. 2021, 49, e9. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Toyama, B.H.; Harris, M.S.; Bock, T.; Iskar, M.; Bork, P.; Ingolia, N.T.; Hetzer, M.W.; Beck, M. Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats. Cell Syst. 2015, 1, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Szekely, V.; Arpat, A.B.; Janich, P.; Gatfield, D. Translational contributions to tissue specificity in rhythmic and constitutive gene expression. Genome Biol. 2017, 18, 116. [Google Scholar] [CrossRef]
- Hernandez-Alias, X.; Benisty, H.; Schaefer, M.H.; Serrano, L. Translational efficiency across healthy and tumor tissues is proliferation-related. Mol. Syst. Biol. 2020, 16, e9275. [Google Scholar] [CrossRef] [PubMed]
- Kames, J.; Alexaki, A.; Holcomb, D.D.; Santana-Quintero, L.V.; Athey, J.C.; Hamasaki-Katagiri, N.; Katneni, U.; Golikov, A.; Ibla, J.C.; Bar, H.; et al. TissueCoCoPUTs: Novel Human Tissue-Specific Codon and Codon-Pair Usage Tables Based on Differential Tissue Gene Expression. J. Mol. Biol. 2020, 432, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Carney, T.D.; Maracci, C.; Yatsenko, A.S.; Shcherbata, H.R.; Rodnina, M.V. Tissue-Specific Regulation of Translational Readthrough Tunes Functions of the Traffic Jam Transcription Factor. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gupta, V.; Warner, J.R. Ribosome-omics of the human ribosome. Rna 2014, 20, 1004–1013. [Google Scholar] [CrossRef]
- Guimaraes, J.C.; Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016, 17, 236. [Google Scholar] [CrossRef]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Waldman, Y.Y.; Tuller, T.; Shlomi, T.; Sharan, R.; Ruppin, E. Translation efficiency in humans: Tissue specificity, global optimization and differences between developmental stages. Nucleic Acids Res. 2010, 38, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Francoeur, A.M.; Liu, S.; Wang, E. Tissue-specific expression in mammalian brain, heart, and muscle of S1, a member of the elongation factor-1 alpha gene family. J. Biol. Chem. 1992, 267, 24064–24068. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Bourbeau, D.; Chen, E.; Petroulakis, E.; Pan, J.; Xu, S.; Wang, E. Characterization of elongation factor-1A (eEF1A-1) and eEF1A-2/S1 protein expression in normal and wasted mice. J. Biol. Chem. 2001, 276, 22915–22922. [Google Scholar] [CrossRef] [PubMed]
- Feral, C.; Guellaen, G.; Pawlak, A. Human testis expresses a specific poly(A)-binding protein. Nucleic Acids Res. 2001, 29, 1872–1883. [Google Scholar] [CrossRef]
- Seli, E.; Lalioti, M.D.; Flaherty, S.M.; Sakkas, D.; Terzi, N.; Steitz, J.A. An embryonic poly(A)-binding protein (ePAB) is expressed in mouse oocytes and early preimplantation embryos. Proc. Natl. Acad. Sci. USA 2005, 102, 367–372. [Google Scholar] [CrossRef]
- Guzeloglu-Kayisli, O.; Lalioti, M.D.; Babayev, E.; Torrealday, S.; Karakaya, C.; Seli, E. Human embryonic poly(A)-binding protein (EPAB) alternative splicing is differentially regulated in human oocytes and embryos. Mol. Hum. Reprod. 2014, 20, 59–65. [Google Scholar] [CrossRef]
- Jenkins, Z.A.; Haag, P.G.; Johansson, H.E. Human eIF5A2 on chromosome 3q25-q27 is a phylogenetically conserved vertebrate variant of eukaryotic translation initiation factor 5A with tissue-specific expression. Genomics 2001, 71, 101–109. [Google Scholar] [CrossRef]
- Evsikov, A.V.; Marin de Evsikova, C. Evolutionary origin and phylogenetic analysis of the novel oocyte-specific eukaryotic translation initiation factor 4E in Tetrapoda. Dev. Genes Evol. 2009, 219, 111–118. [Google Scholar] [CrossRef]
- Xu, J.; Watkins, R.; Arnold, A.P. Sexually dimorphic expression of the X-linked gene Eif2s3x mRNA but not protein in mouse brain. Gene Expr. Patterns 2006, 6, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, C.; Toutain, B.; Le Gall, J.Y.; Le Treut, A.; Guenet, L. Promoter analysis of the human translation termination factor 1 gene. Gene 2003, 316, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Urbero, B.; Eurwilaichitr, L.; Stansfield, I.; Tassan, J.P.; Le Goff, X.; Kress, M.; Tuite, M.F. Expression of the release factor eRF1 (Sup45p) gene of higher eukaryotes in yeast and mammalian tissues. Biochimie 1997, 79, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Si, K.; Das, K.; Maitra, U. Characterization of multiple mRNAs that encode mammalian translation initiation factor 5 (eIF-5). J. Biol. Chem. 1996, 271, 16934–16938. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, S.; Imai, M.; Mizutani, M.; Kikuchi, Y.; Hanaoka, F.; Ui, M.; Katada, T. Molecular cloning of a novel member of the eukaryotic polypeptide chain-releasing factors (eRF). Its identification as eRF3 interacting with eRF1. J. Biol. Chem. 1998, 273, 22254–22259. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Salhi, S.; Le Goff, C.; Viranaicken, W.; Diop, D.; Jean-Jean, O. Involvement of human release factors eRF3a and eRF3b in translation termination and regulation of the termination complex formation. Mol. Cell. Biol. 2005, 25, 5801–5811. [Google Scholar] [CrossRef]
- Cao, Y.; Portela, M.; Janikiewicz, J.; Doig, J.; Abbott, C.M. Characterisation of translation elongation factor eEF1B subunit expression in mammalian cells and tissues and co-localisation with eEF1A2. PLoS ONE 2014, 9, e114117. [Google Scholar] [CrossRef] [PubMed]
- Kaitsuka, T.; Tomizawa, K.; Matsushita, M. Transformation of eEF1Bdelta into heat-shock response transcription factor by alternative splicing. EMBO Rep. 2011, 12, 673–681. [Google Scholar] [CrossRef]
- Wells, J.; Henkler, F.; Leversha, M.; Koshy, R. A mitochondrial elongation factor-like protein is over-expressed in tumours and differentially expressed in normal tissues. FEBS Lett. 1995, 358, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Q.; Liu, X.L.; Zhao, J.; Ma, L. Differential and Tissue-specific Expression of Mitochondrial Elongation Factor Tu and Ts during Development. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao 1999, 31, 37–40. [Google Scholar]
- Jang, C.; Hui, S.; Zeng, X.; Cowan, A.J.; Wang, L.; Chen, L.; Morscher, R.J.; Reyes, J.; Frezza, C.; Hwang, H.Y.; et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 2019, 30, 594–606.e3. [Google Scholar] [CrossRef] [PubMed]
- Neinast, M.D.; Jang, C.; Hui, S.; Murashige, D.S.; Chu, Q.; Morscher, R.J.; Li, X.; Zhan, L.; White, E.; Anthony, T.G.; et al. Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids. Cell Metab. 2019, 29, 417–429.e4. [Google Scholar] [CrossRef]
- Li, X.; Hui, S.; Mirek, E.T.; Jonsson, W.O.; Anthony, T.G.; Lee, W.D.; Zeng, X.; Jang, C.; Rabinowitz, J.D. Circulating metabolite homeostasis achieved through mass action. Nat. Metab. 2022, 4, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Velic, A.; Macek, B.; Lundberg, E.; Kampf, C.; Nagaraj, N.; Uhlen, M.; Cox, J.; Mann, M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteom. 2013, 12, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014, 83, 779–812. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Kumar, A.; Herbein, G. The eEF1A Proteins: At the Crossroads of Oncogenesis, Apoptosis, and Viral Infections. Front. Oncol. 2015, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Negrutskii, B.S.; Shalak, V.F.; Novosylna, O.V.; Porubleva, L.V.; Lozhko, D.M.; El’skaya, A.V. The eEF1 family of mammalian translation elongation factors. BBA Adv. 2023, 3, 100067. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wolfraim, L.A.; Wang, E. Differential expression of S1 and elongation factor-1 alpha during rat development. J. Biol. Chem. 1993, 268, 24453–24459. [Google Scholar] [CrossRef] [PubMed]
- “Chromosome Y: Chromosome Summary—Homo Sapiens”. Ensembl Release 88. 2017-03-29. Available online: https://grch37.ensembl.org/Homo_sapiens/Location/Chromosome?r=Y (accessed on 5 March 2023).
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Berletch, J.B.; Ma, W.; Yang, F.; Shendure, J.; Noble, W.S.; Disteche, C.M.; Deng, X. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015, 11, e1005079. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Jankowsky, E. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Rosner, A.; Rinkevich, B. The DDX3 subfamily of the DEAD box helicases: Divergent roles as unveiled by studying different organisms and in vitro assays. Curr. Med. Chem. 2007, 14, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, T.; Iida, H.; Fukumura, J.; Nishimoto, T. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Exp. Cell Res. 2004, 300, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Venkataramanan, S.; Calviello, L.; Wilkins, K.; Floor, S.N. DDX3X and DDX3Y are redundant in protein synthesis. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Kotov, A.A.; Olenkina, O.M.; Godneeva, B.K.; Adashev, V.E.; Olenina, L.V. Progress in understanding the molecular functions of DDX3Y (DBY) in male germ cell development and maintenance. Biosci. Trends 2017, 11, 46–53. [Google Scholar] [CrossRef]
- Ramathal, C.; Angulo, B.; Sukhwani, M.; Cui, J.; Durruthy-Durruthy, J.; Fang, F.; Schanes, P.; Turek, P.J.; Orwig, K.E.; Reijo Pera, R. DDX3Y gene rescue of a Y chromosome AZFa deletion restores germ cell formation and transcriptional programs. Sci. Rep. 2015, 5, 15041. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Endo, T.; Isotani, A.; Ogawa, M.; Ikawa, M. An azoospermic factor gene, Ddx3y and its paralog, Ddx3x are dispensable in germ cells for male fertility. J. Reprod. Dev. 2019, 65, 121–128. [Google Scholar] [CrossRef]
- Lennox, A.L.; Hoye, M.L.; Jiang, R.; Johnson-Kerner, B.L.; Suit, L.A.; Venkataramanan, S.; Sheehan, C.J.; Alsina, F.C.; Fregeau, B.; Aldinger, K.A.; et al. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron 2020, 106, 404–420.e8. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, I.E.; Ellis, P.S.; Mazeyrat, S.; Duthie, S.; Brockdorff, N.; Mattei, M.G.; Gavin, M.A.; Affara, N.A.; Brown, G.M.; Simpson, E.; et al. Characterization of genes encoding translation initiation factor eIF-2gamma in mouse and human: Sex chromosome localization, escape from X-inactivation and evolution. Hum. Mol. Genet. 1998, 7, 1725–1737. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Riel, J.M.; Stoytcheva, Z.; Ward, M.A. Two Y genes can replace the entire Y chromosome for assisted reproduction in the mouse. Science 2014, 343, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Skopkova, M.; Hennig, F.; Shin, B.S.; Turner, C.E.; Stanikova, D.; Brennerova, K.; Stanik, J.; Fischer, U.; Henden, L.; Muller, U.; et al. EIF2S3 Mutations Associated with Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum. Mutat. 2017, 38, 409–425. [Google Scholar] [CrossRef]
- Maddirevula, S.; Awartani, K.; Coskun, S.; AlNaim, L.F.; Ibrahim, N.; Abdulwahab, F.; Hashem, M.; Alhassan, S.; Alkuraya, F.S. A genomics approach to females with infertility and recurrent pregnancy loss. Hum. Genet. 2020, 139, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Guzeloglu-Kayisli, O.; Lalioti, M.D.; Aydiner, F.; Sasson, I.; Ilbay, O.; Sakkas, D.; Lowther, K.M.; Mehlmann, L.M.; Seli, E. Embryonic poly(A)-binding protein (EPAB) is required for oocyte maturation and female fertility in mice. Biochem. J. 2012, 446, 47–58. [Google Scholar] [CrossRef]
- Ozturk, S.; Sozen, B.; Uysal, F.; Bassorgun, I.C.; Usta, M.F.; Akkoyunlu, G.; Demir, N. The poly(A)-binding protein genes, EPAB, PABPC1, and PABPC3 are differentially expressed in infertile men with non-obstructive azoospermia. J. Assist. Reprod. Genet. 2016, 33, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wei, X.; Chen, Y. Loss of PABPC1 is compensated by elevated PABPC4 and correlates with transcriptome changes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Blanco, P.; Sargent, C.A.; Boucher, C.A.; Howell, G.; Ross, M.; Affara, N.A. A novel poly(A)-binding protein gene (PABPC5) maps to an X-specific subinterval in the Xq21.3/Yp11.2 homology block of the human sex chromosomes. Genomics 2001, 74, 1–11. [Google Scholar] [CrossRef]
- Elia, M. Organ and tissue contribution to metabolic rate. In Energy Metabolism: Tissue Determinants and Cellular Corollaries; Kinney, J.M., Tucker, H.N., Eds.; Raven Press: New York, NY, USA, 1992; pp. 61–80. [Google Scholar]
- Zinoviev, A.; Goyal, A.; Jindal, S.; LaCava, J.; Komar, A.A.; Rodnina, M.V.; Hellen, C.U.T.; Pestova, T.V. Functions of unconventional mammalian translational GTPases GTPBP1 and GTPBP2. Genes Dev. 2018, 32, 1226–1241. [Google Scholar] [CrossRef]
- Ishimura, R.; Nagy, G.; Dotu, I.; Zhou, H.; Yang, X.L.; Schimmel, P.; Senju, S.; Nishimura, Y.; Chuang, J.H.; Ackerman, S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 2014, 345, 455–459. [Google Scholar] [CrossRef]
- Senju, S.; Nishimura, Y. Identification of human and mouse GP-1, a putative member of a novel G-protein family. Biochem. Biophys. Res. Commun. 1997, 231, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Yoshida, K.; Hida, M.; Kato, H.; Uchida, K.; Yamaguchi, R.; Tateyama, S.; Sugano, S. Cloning, expression analysis, and chromosomal mapping of GTPBP2, a novel member of the G protein family. Gene 2000, 256, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Coldwell, M.J.; Sack, U.; Cowan, J.L.; Barrett, R.M.; Vlasak, M.; Sivakumaran, K.; Morley, S.J. Multiple isoforms of the translation initiation factor eIF4GII are generated via use of alternative promoters, splice sites and a non-canonical initiation codon. Biochem. J. 2012, 448, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gradi, A.; Imataka, H.; Svitkin, Y.V.; Rom, E.; Raught, B.; Morino, S.; Sonenberg, N. A novel functional human eukaryotic translation initiation factor 4G. Mol. Cell. Biol. 1998, 18, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Vazquez-Pianzola, P. Functional diversity of the eukaryotic translation initiation factors belonging to eIF4 families. Mech. Dev. 2005, 122, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Palmer, K.; Handel, M.A. Mutation of Eif4g3, encoding a eukaryotic translation initiation factor, causes male infertility and meiotic arrest of mouse spermatocytes. Development 2010, 137, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Shatsky, I.N.; Dmitriev, S.E.; Andreev, D.E.; Terenin, I.M. Transcriptome-wide studies uncover the diversity of modes of mRNA recruitment to eukaryotic ribosomes. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 164–177. [Google Scholar] [CrossRef]
- Wortham, N.C.; Proud, C.G. eIF2B: Recent structural and functional insights into a key regulator of translation. Biochem. Soc. Trans. 2015, 43, 1234–1240. [Google Scholar] [CrossRef]
- Wortham, N.C.; Stewart, J.D.; Harris, S.; Coldwell, M.J.; Proud, C.G. Stoichiometry of the eIF2B complex is maintained by mutual stabilization of subunits. Biochem. J. 2016, 473, 571–580. [Google Scholar] [CrossRef]
- Wolf, D.A.; Lin, Y.; Duan, H.; Cheng, Y. eIF-Three to Tango: Emerging functions of translation initiation factor eIF3 in protein synthesis and disease. J. Mol. Cell Biol. 2020, 12, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Cate, J.H. Human eIF3: From ‘blobology’ to biological insight. Philos. Trans. R. Soc. B 2017, 372, 20160176. [Google Scholar] [CrossRef] [PubMed]
- Valasek, L.S.; Zeman, J.; Wagner, S.; Beznoskova, P.; Pavlikova, Z.; Mohammad, M.P.; Hronova, V.; Herrmannova, A.; Hashem, Y.; Gunisova, S. Embraced by eIF3: Structural and functional insights into the roles of eIF3 across the translation cycle. Nucleic Acids Res. 2017, 45, 10948–10968. [Google Scholar] [CrossRef] [PubMed]
- Kubacka, D.; Miguel, R.N.; Minshall, N.; Darzynkiewicz, E.; Standart, N.; Zuberek, J. Distinct features of cap binding by eIF4E1b proteins. J. Mol. Biol. 2015, 427, 387–405. [Google Scholar] [CrossRef] [PubMed]
- Minshall, N.; Reiter, M.H.; Weil, D.; Standart, N. CPEB interacts with an ovary-specific eIF4E and 4E-T in early Xenopus oocytes. J. Biol. Chem. 2007, 282, 37389–37401. [Google Scholar] [CrossRef]
- Yang, G.; Xin, Q.; Feng, I.; Dean, J. Germ-cell specific eIF4E1B regulates maternal RNA translation to ensure zygotic genome activation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kozlov, E.; Shidlovskii, Y.V.; Gilmutdinov, R.; Schedl, P.; Zhukova, M. The role of CPEB family proteins in the nervous system function in the norm and pathology. Cell Biosci. 2021, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Kamenska, A.; Lu, W.T.; Kubacka, D.; Broomhead, H.; Minshall, N.; Bushell, M.; Standart, N. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Res. 2014, 42, 3298–3313. [Google Scholar] [CrossRef] [PubMed]
- Dostie, J.; Ferraiuolo, M.; Pause, A.; Adam, S.A.; Sonenberg, N. A novel shuttling protein, 4E-T, mediates the nuclear import of the mRNA 5′ cap-binding protein, eIF4E. EMBO J. 2000, 19, 3142–3156. [Google Scholar] [CrossRef]
- Clement, P.M.; Johansson, H.E.; Wolff, E.C.; Park, M.H. Differential expression of eIF5A-1 and eIF5A-2 in human cancer cells. FEBS J. 2006, 273, 1102–1114. [Google Scholar] [CrossRef]
- Loughran, G.; Firth, A.E.; Atkins, J.F.; Ivanov, I.P. Translational autoregulation of BZW1 and BZW2 expression by modulating the stringency of start codon selection. PLoS ONE 2018, 13, e0192648. [Google Scholar] [CrossRef]
- Fijalkowska, D.; Verbruggen, S.; Ndah, E.; Jonckheere, V.; Menschaert, G.; Van Damme, P. eIF1 modulates the recognition of suboptimal translation initiation sites and steers gene expression via uORFs. Nucleic Acids Res. 2017, 45, 7997–8013. [Google Scholar] [CrossRef]
- Andreev, D.E.; Dmitriev, S.E.; Loughran, G.; Terenin, I.M.; Baranov, P.V.; Shatsky, I.N. Translation control of mRNAs encoding mammalian translation initiation factors. Gene 2018, 651, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.P.; Loughran, G.; Sachs, M.S.; Atkins, J.F. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc. Natl. Acad. Sci. USA 2010, 107, 18056–18060. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.P.; Green, R. Roadblocks and resolutions in eukaryotic translation. Nat. Reviews. Mol. Cell Biol. 2018, 19, 526–541. [Google Scholar] [CrossRef]
- Mills, E.W.; Wangen, J.; Green, R.; Ingolia, N.T. Dynamic Regulation of a Ribosome Rescue Pathway in Erythroid Cells and Platelets. Cell Rep. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- O’Connell, A.E.; Gerashchenko, M.V.; O’Donohue, M.F.; Rosen, S.M.; Huntzinger, E.; Gleeson, D.; Galli, A.; Ryder, E.; Cao, S.; Murphy, Q.; et al. Mammalian Hbs1L deficiency causes congenital anomalies and developmental delay associated with Pelota depletion and 80S monosome accumulation. PLoS Genet. 2019, 15, e1007917. [Google Scholar] [CrossRef]
- Kaminska, M.; Havrylenko, S.; Decottignies, P.; Gillet, S.; Le Marechal, P.; Negrutskii, B.; Mirande, M. Dissection of the structural organization of the aminoacyl-tRNA synthetase complex. J. Biol. Chem. 2009, 284, 6053–6060. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, T.P.; Raben, N.; Miller, F.W. A novel gene oriented in a head-to-head configuration with the human histidyl-tRNA synthetase (HRS) gene encodes an mRNA that predicts a polypeptide homologous to HRS. Biochem. Biophys. Res. Commun. 1995, 210, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ruan, Z.R.; Wang, Y.; Huang, Q.; Xue, M.Q.; Zhou, X.L.; Wang, E.D. A threonyl-tRNA synthetase-like protein has tRNA aminoacylation and editing activities. Nucleic Acids Res. 2018, 46, 3643–3656. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef]
- Bortoluzzi, S.; d’Alessi, F.; Romualdi, C.; Danieli, G.A. Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics 2001, 17, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.M.; Kurylo, C.M.; Dass, R.A.; Bojmar, L.; Lyden, D.; Vincent, C.T.; Blanchard, S.C. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018, 4, eaao0665. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Jovanovic, M.; Slavov, N. Ribosome Stoichiometry: From Form to Function. Trends Biochem. Sci. 2019, 44, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Slavov, N.; Semrau, S.; Airoldi, E.; Budnik, B.; van Oudenaarden, A. Differential Stoichiometry among Core Ribosomal Proteins. Cell Rep. 2015, 13, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Reviews. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ye, Y.; Gong, J.; Ruan, H.; Liu, C.J.; Xiang, Y.; Cai, C.; Guo, A.Y.; Ling, J.; Diao, L.; et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun. Biol. 2018, 1, 234. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Chan, L.L.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Farache, D.; Antine, S.P.; Lee, A.S.Y. Moonlighting translation factors: Multifunctionality drives diverse gene regulation. Trends Cell Biol. 2022, 32, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 2014, 37, 17–38. [Google Scholar] [CrossRef]
- Lashkevich, K.A.; Dmitriev, S.E. mRNA Targeting, Transport and Local Translation in Eukaryotic Cells: From the Classical View to a Diversity of New Concepts. Mol. Biol. 2021, 55, 507–537. [Google Scholar] [CrossRef]
- Allen, S.R.; Stewart, R.K.; Rogers, M.; Ruiz, I.J.; Cohen, E.; Laederach, A.; Counter, C.M.; Sawyer, J.K.; Fox, D.T. Distinct responses to rare codons in select Drosophila tissues. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alias, X.; Benisty, H.; Serrano, L.; Schaefer, M.H. Using protein-per-mRNA differences among human tissues in codon optimization. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex or Group | Protein Names |
|---|---|
| eIF2 | EIF2S1, EIF2S2, EIF2S3 |
| eIF2B | EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5 |
| eIF3 | EIF3A, EIF3B, EIF3C, EIF3D, EIF3E, EIF3F, EIF3G, EIF3H, EIF3I, EIF3J, EIF3J, EIF3K, EIF3L, EIF3M |
| Multifactor complex (MFC) | EIF1, EIF1AX, EIF1AY, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, EIF3A, EIF3B, EIF3C, EIF3D, EIF3E, EIF3F, EIF3G, EIF3H, EIF3I, EIF3J, EIF3J, EIF3K, EIF3L, EIF3M, EIF5 |
| eIF4s | EIF4A1, EIF4A2, EIF4B, EIF4H, EIF4E, EIF4E1B, EIF4E3, EIF4G1, EIF4G3 |
| eIF4, 4EBP | EIF4EBP1, EIF4EBP2, EIF4EBP3, EIF4A1, EIF4A2, EIF4B, EIF4H, EIF4E, EIF4E1B, EIF4E3, EIF4G1, EIF4G3 |
| INITIATION | EIF1, EIF1AX, EIF1AY, EIF1B, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, EIF3A, EIF3B, EIF3C, EIF3D, EIF3E, EIF3F, EIF3G, EIF3H, EIF3I, EIF3J, EIF3J, EIF3K, EIF3L, EIF3M, EIF4A1, EIF4A2, EIF4B, EIF4H, EIF4E, EIF4E1B, EIF4E3, EIF4G1, EIF4G3, DDX3X, DDX3Y, DHX29, EIF4EBP1, EIF4EBP2, EIF4EBP3, EIF5, EIF5B, PABPC1 |
| INITIATION+ | EIF1, EIF1AX, EIF1AY, EIF1B, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, EIF3A, EIF3B, EIF3C, EIF3D, EIF3E, EIF3F, EIF3G, EIF3H, EIF3I, EIF3J, EIF3J, EIF3K, EIF3L, EIF3M, EIF4A1, EIF4A2, EIF4B, EIF4H, EIF4E, EIF4E1B, EIF4E3, EIF4G1, EIF4G3, DDX3X, DDX3Y, DHX29, EIF4EBP1, EIF4EBP2, EIF4EBP3, EIF5, EIF5B, EIF6, PABPC1, ABCE1, EIF2D, MCTS1, DENR, MCTS2P, BZW1, BZW2, EIF4E2, EIF4G2, CTIF, PAIP1, PAIP2, EIF2A, ABCF1, NCBP1, NCBP2 |
| eEF1B | EEF1B2, EEF1D, EEF1G |
| eEF1H | EEF1A1, EEF1A2, EEF1B2, EEF1D, EEF1G, VARS1 |
| ELONGATION | EEF1A1, EEF1A2, EEF1B2, EEF1B2P2, EEF1D, EEF1G, EEF2, EEFSEC, EIF5A, EIF5A2 |
| ELONGATION+ | EEF1A1, EEF1A2, EEF1B2, EEF1B2P2, EEF1D, EEF1G, EEF2, DPH1, DPH2, DPH3, DPH5, DPH6, DPH7, EEFSEC, EIF5A, EIF5A2, EIF5AL1, DHPS, DOHH |
| ELONGATION+ GTPBPs | GTPBP1, GTPBP2, EEF1A1, EEF1A2, EEF1B2, EEF1B2P2, EEF1D, EEF1G, EEF2, DPH1, DPH2, DPH3, DPH5, DPH6, DPH7, EEFSEC, EIF5A, EIF5A2, EIF5AL1, DHPS, DOHH |
| TERMINATION | ETF1, GSPT1, GSPT2 |
| TERMINATION+ | ETF1, GSPT1, GSPT2, PABPC1, ABCE1, EIF2D, MCTS1, DENR, MCTS2P, HBS1L, PELO |
| PABPC paralogs | PABPC1L, PABPC1L2A, PABPC1L2B, PABPC3, PABPC4, PABPC4L, PABPC5, PABPC1 |
| ARSases | AIMP1, AIMP2, EEF1E1, NARS, RARS, EPRS, MARS, QARS, IARS, KARS, LARS, YARS, VARS, AARS, CARS, DARS, FARSA, FARSB, GARS, HARS, HARS2, SARS, SARS2, TARS, TARSL2, WARS, LARS2, RARS2, VARS2, AARS2, TARS2, YARS2, WARS2, NARS2, PARS2, MARS2, IARS2, FARS2, EARS2, DARS2, CARS2 |
| ARSase COMPLEX | AIMP1, AIMP2, EEF1E1, NARS, RARS, EPRS, MARS, QARS, IARS, KARS, LARS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anisimova, A.S.; Kolyupanova, N.M.; Makarova, N.E.; Egorov, A.A.; Kulakovskiy, I.V.; Dmitriev, S.E. Human Tissues Exhibit Diverse Composition of Translation Machinery. Int. J. Mol. Sci. 2023, 24, 8361. https://doi.org/10.3390/ijms24098361
Anisimova AS, Kolyupanova NM, Makarova NE, Egorov AA, Kulakovskiy IV, Dmitriev SE. Human Tissues Exhibit Diverse Composition of Translation Machinery. International Journal of Molecular Sciences. 2023; 24(9):8361. https://doi.org/10.3390/ijms24098361
Chicago/Turabian StyleAnisimova, Aleksandra S., Natalia M. Kolyupanova, Nadezhda E. Makarova, Artyom A. Egorov, Ivan V. Kulakovskiy, and Sergey E. Dmitriev. 2023. "Human Tissues Exhibit Diverse Composition of Translation Machinery" International Journal of Molecular Sciences 24, no. 9: 8361. https://doi.org/10.3390/ijms24098361
APA StyleAnisimova, A. S., Kolyupanova, N. M., Makarova, N. E., Egorov, A. A., Kulakovskiy, I. V., & Dmitriev, S. E. (2023). Human Tissues Exhibit Diverse Composition of Translation Machinery. International Journal of Molecular Sciences, 24(9), 8361. https://doi.org/10.3390/ijms24098361