
Longitudinal Analyses of Mutational Subclonal Architecture and Tumor Subtypes in Recurrent Bladder Cancer


Abstract
:1. Introduction
2. Results
2.1. Clinical History of Five Patients with Recurrent BC
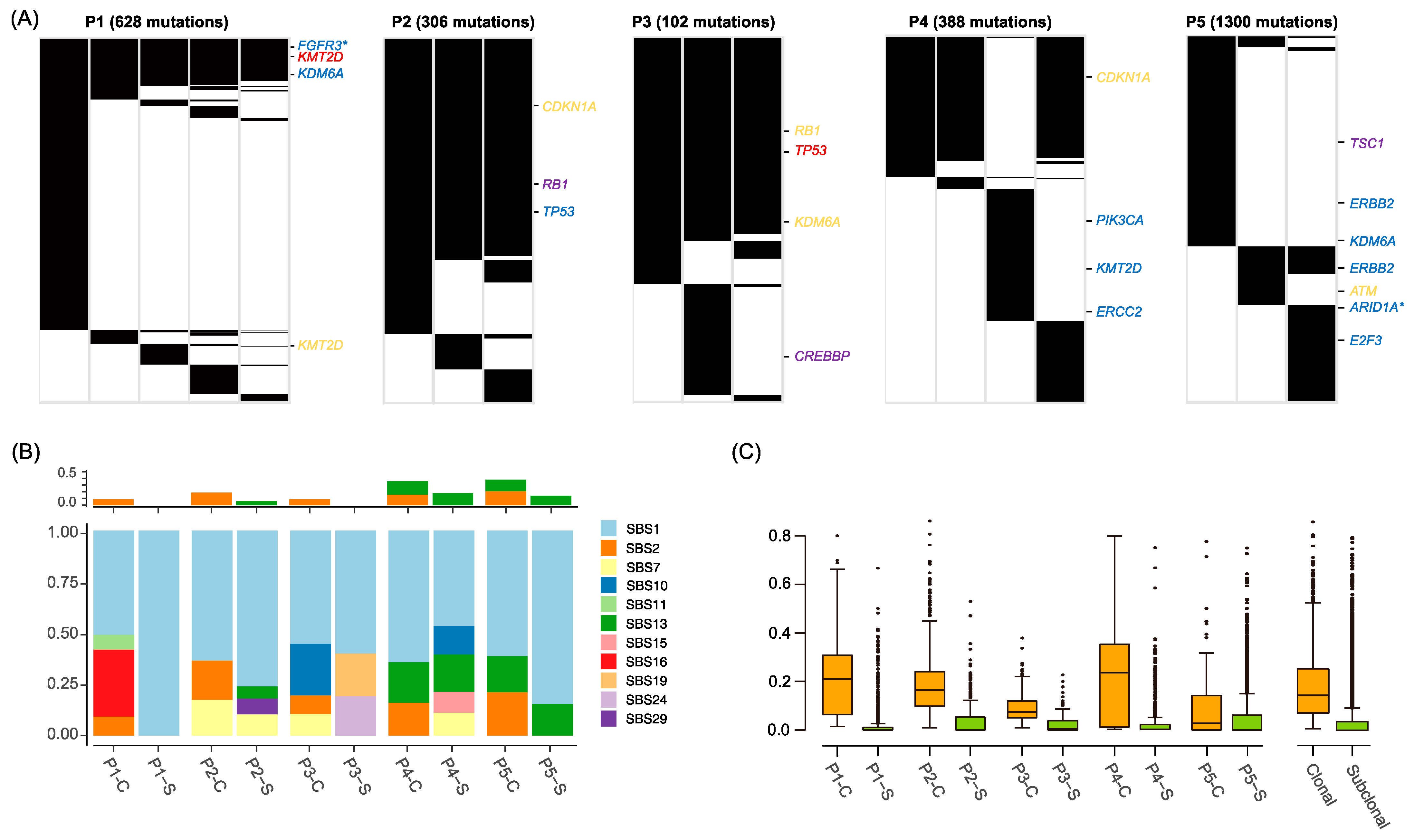
2.2. Mutation Landscape of BC Genomes
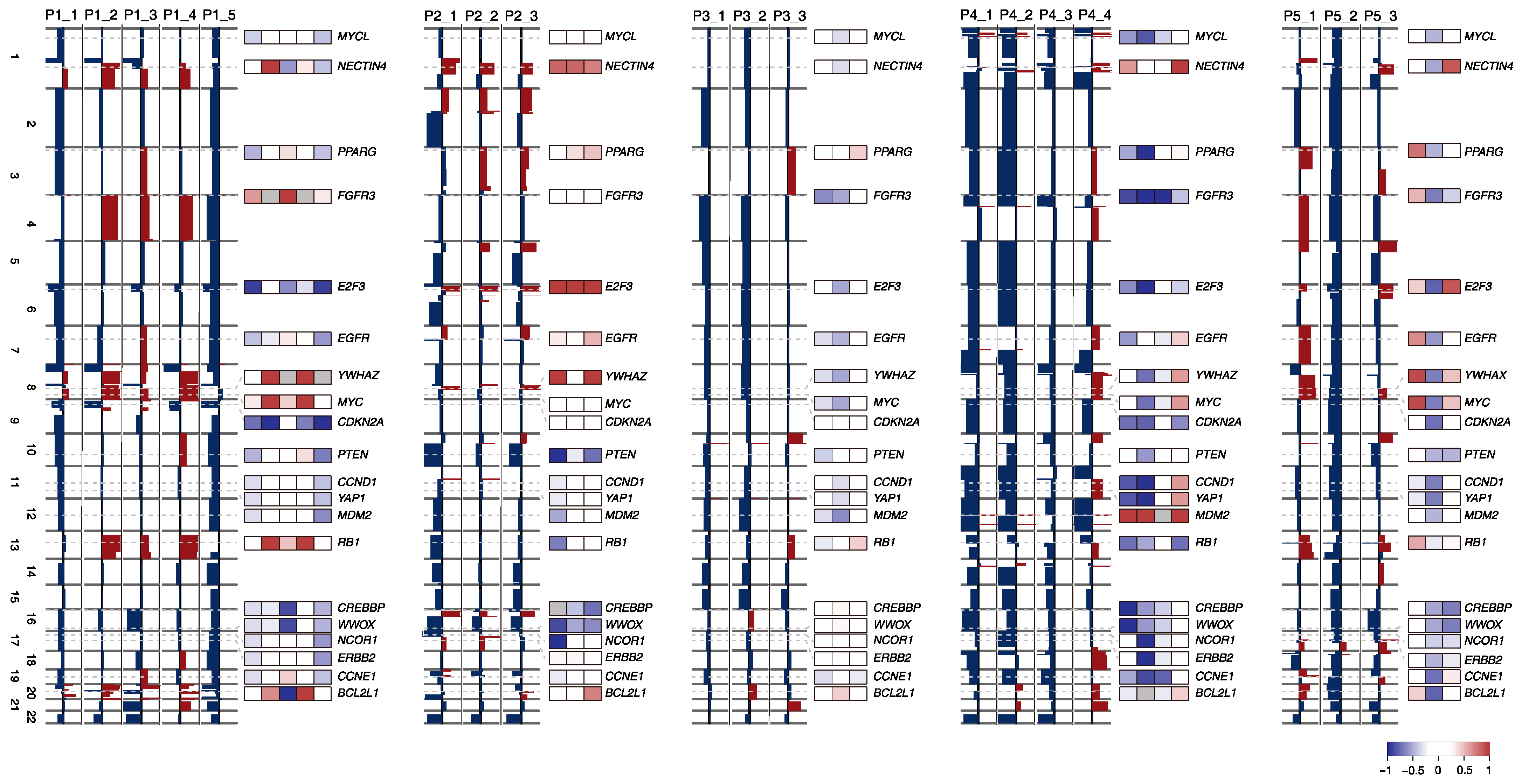
2.3. SCNA Profiles of BC Progression
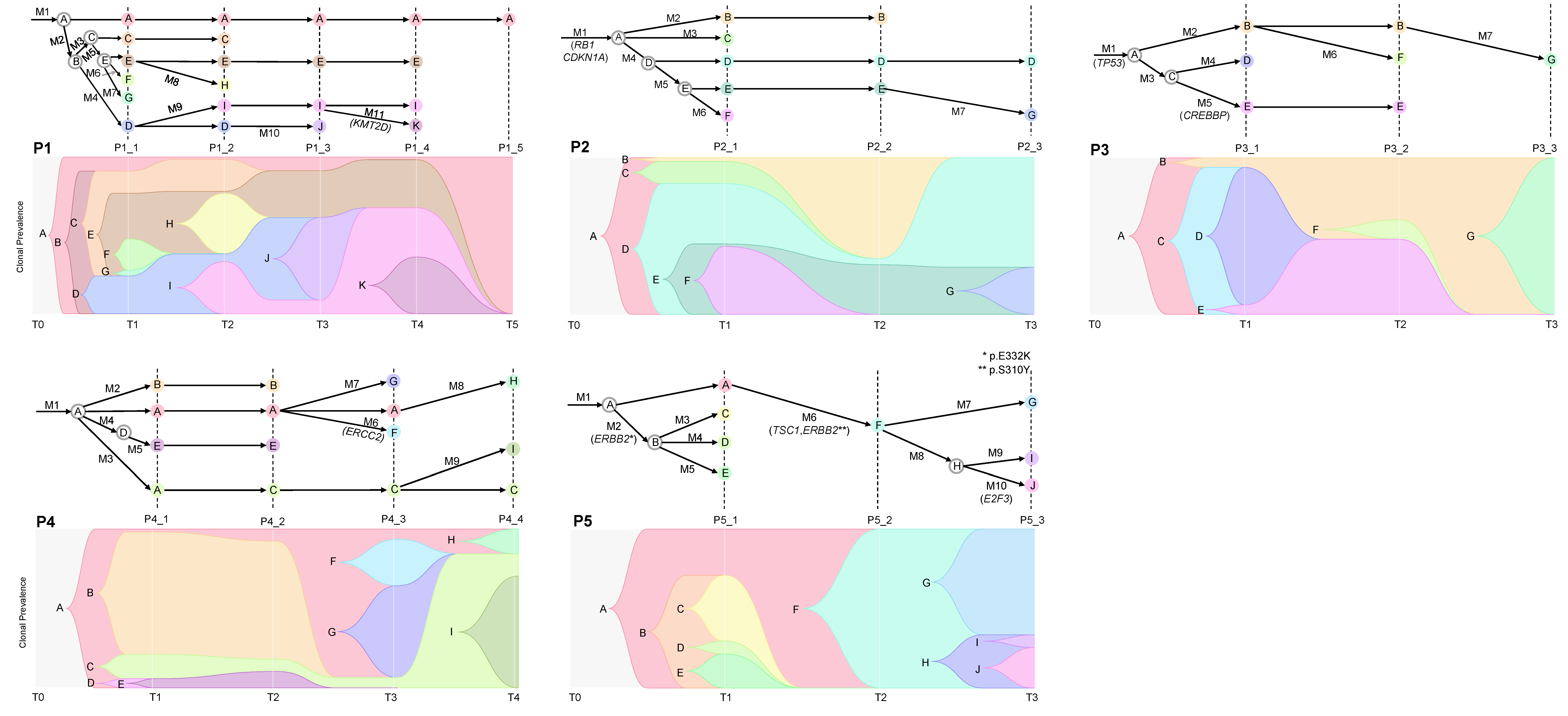
2.4. Cancer Evolution Inferred from the Multiregional Mutation Profiles
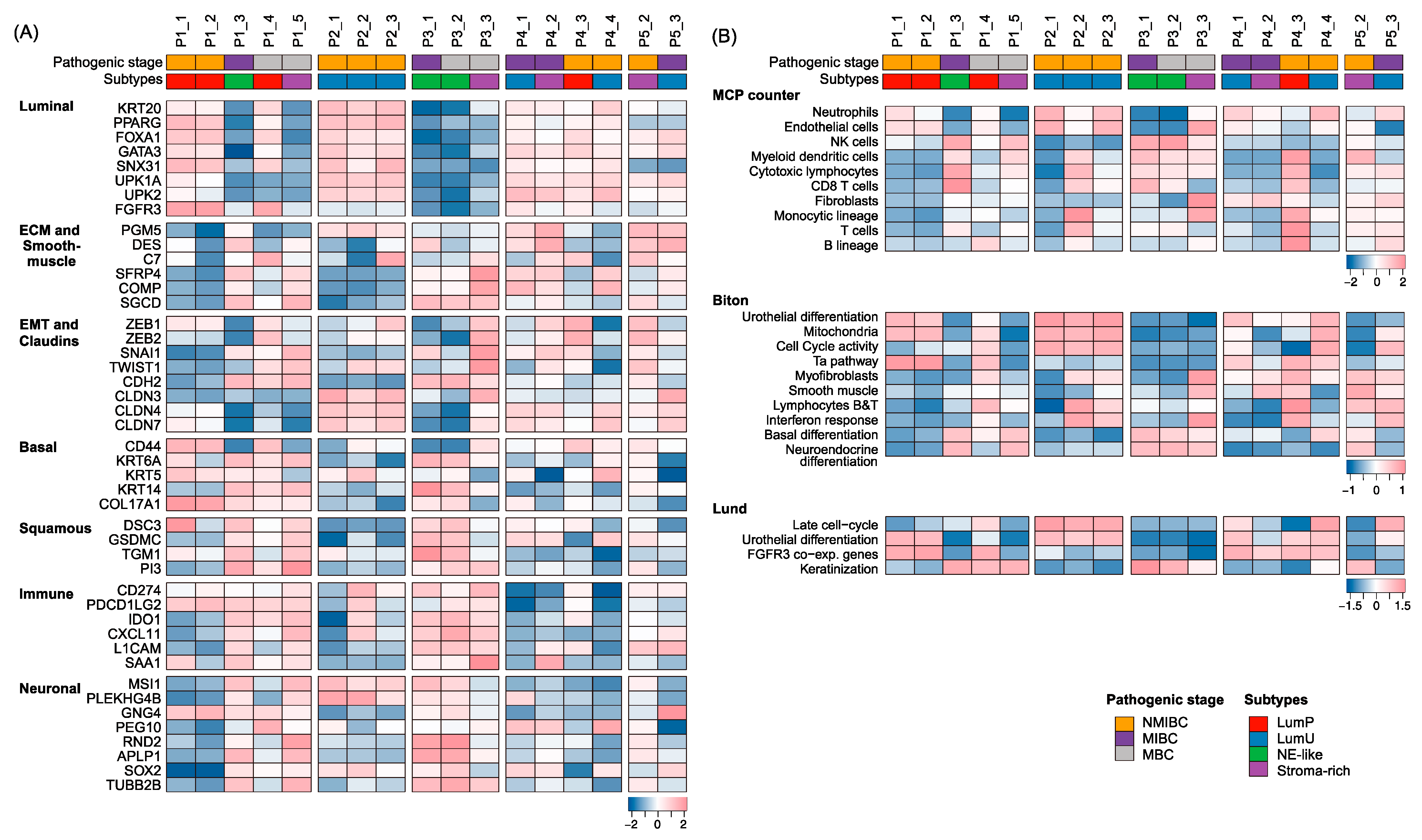
2.5. BC Subtypes along Tumor Progression
3. Materials and Methods
3.1. Patients
3.2. WES and RNA-Seq and Pre-Processing
3.3. Genomic Analyses
3.4. Transcriptomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Newhall, K.P.; Khani, F.; Barlow, L.; Nguyen, D.; Gu, L.; Eng, K.; Bhinder, B.; Uppal, M.; Récapet, C.; et al. The Cytidine Deaminase APOBEC3G Contributes to Cancer Mutagenesis and Clonal Evolution in Bladder Cancer. Cancer Res. 2023, 83, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Audisio, A.; Buttigliero, C.; Delcuratolo, M.D.; Parlagreco, E.; Audisio, M.; Ungaro, A.; Di Stefano, R.F.; Di Prima, L.; Turco, F.; Tucci, M. New Perspectives in the Medical Treatment of Non-Muscle-Invasive Bladder Cancer: Immune Checkpoint Inhibitors and Beyond. Cells 2022, 11, 357. [Google Scholar] [CrossRef]
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). SEER Cancer. In Book SEER Cancer Stat Facts: Bladder Cancer; National Cancer Institute (NCI): Bethesda, MD, USA, 2023. [Google Scholar]
- Faltas, B.M.; Prandi, D.; Tagawa, S.T.; Molina, A.M.; Nanus, D.M.; Sternberg, C.; Rosenberg, J.; Mosquera, J.M.; Robinson, B.; Elemento, O.; et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 2016, 48, 1490–1499. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [PubMed]
- Sjodahl, G.; Lauss, M.; Lovgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Ferno, M.; Ringner, M.; et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D. Mutation and epigenetic molecular clocks in cancer. Carcinogenesis 2011, 32, 123–128. [Google Scholar] [CrossRef]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.A.; Satas, G.; Raphael, B.J. CALDER: Inferring Phylogenetic Trees from Longitudinal Tumor Samples. Cell Syst. 2019, 8, 514–522.e15. [Google Scholar] [CrossRef]
- Kamoun, A.; de Reynies, A.; Allory, Y.; Sjodahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Eriksson, P.; Aine, M.; Veerla, S.; Liedberg, F.; Sjodahl, G.; Hoglund, M. Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Med. Genom. 2015, 8, 25. [Google Scholar] [CrossRef]
- Biton, A.; Bernard-Pierrot, I.; Lou, Y.; Krucker, C.; Chapeaublanc, E.; Rubio-Perez, C.; Lopez-Bigas, N.; Kamoun, A.; Neuzillet, Y.; Gestraud, P.; et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014, 9, 1235–1245. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; An, C.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Park, S.W.; Rhee, J.K.; Lee, S.H.; Chung, Y.J. Subclonal Genomic Architectures of Primary and Metastatic Colorectal Cancer Based on Intratumoral Genetic Heterogeneity. Clin. Cancer Res. 2015, 21, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; Baek, I.P.; Lee, S.H.; Choi, Y.J.; Lee, J.Y.; Chung, Y.J.; Lee, S.H. Regional biases in mutation screening due to intratumoural heterogeneity of prostate cancer. J. Pathol. 2014, 233, 425–435. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Al Bakir, M.; Huebner, A.; Martínez-Ruiz, C.; Grigoriadis, K.; Watkins, T.B.K.; Pich, O.; Moore, D.A.; Veeriah, S.; Ward, S.; Laycock, J.; et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature 2023, 616, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Abbosh, P.; Keliher, D.; Reardon, B.; Miao, D.; Mouw, K.; Weiner-Taylor, A.; Wankowicz, S.; Han, G.; Teo, M.Y.; et al. Mutational patterns in chemotherapy resistant muscle-invasive bladder cancer. Nat. Commun. 2017, 8, 2193. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.I.; Sjodahl, G.; Kaag, M.; Raman, J.D.; Merrill, S.; Shuman, L.; Chen, G.; Walter, V.; DeGraff, D.J. Intratumoral Heterogeneity of Bladder Cancer by Molecular Subtypes and Histologic Variants. Eur. Urol. 2019, 75, 18–22. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Meeks, J.J.; Al-Ahmadie, H.; Faltas, B.M.; Taylor, J.A., III; Flaig, T.W.; DeGraff, D.J.; Christensen, E.; Woolbright, B.L.; McConkey, D.J.; Dyrskjot, L. Genomic heterogeneity in bladder cancer: Challenges and possible solutions to improve outcomes. Nat. Rev. Urol. 2020, 17, 259–270. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtroder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients with Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1 | P2 | P3 | P4 | P5 | ||
|---|---|---|---|---|---|---|
| Age | 67 | 63 | 68 | 63 | 78 | |
| Sex | Male | Male | Male | Male | Male | |
| Number of samples | 5 | 3 | 3 | 4 | 3 (2 *) | |
| 1st | Tumor Stage | Ta | T1 | T3aN2/MBC (L) | T2 | Ta |
| Tumor Grade | Low | High | High | High | High | |
| 2nd | Tumor Stage | T1 | T1 | MBC (D) | T3 | T1 |
| Tumor Grade | High | High | High | High | High | |
| 3rd | Tumor Stage | T3bN+/MBC (L) | T1 | CIS + | T2 | |
| Tumor Grade | High | High | High | High | ||
| 4th | Tumor Stage | MBC (D) | T1 | |||
| Tumor Grade | High | High | ||||
| Treatment | ||||||
| BCG | 1 | 1 | 1 | 1 | ||
| Chemotherapy | 1 | 1 | 1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, D.; Kim, T.-M.; Lee, Y.-H.; Ha, U.-S. Longitudinal Analyses of Mutational Subclonal Architecture and Tumor Subtypes in Recurrent Bladder Cancer. Int. J. Mol. Sci. 2023, 24, 8418. https://doi.org/10.3390/ijms24098418
Ryu D, Kim T-M, Lee Y-H, Ha U-S. Longitudinal Analyses of Mutational Subclonal Architecture and Tumor Subtypes in Recurrent Bladder Cancer. International Journal of Molecular Sciences. 2023; 24(9):8418. https://doi.org/10.3390/ijms24098418
Chicago/Turabian StyleRyu, Daeun, Tae-Min Kim, Yun-Hee Lee, and U-Syn Ha. 2023. "Longitudinal Analyses of Mutational Subclonal Architecture and Tumor Subtypes in Recurrent Bladder Cancer" International Journal of Molecular Sciences 24, no. 9: 8418. https://doi.org/10.3390/ijms24098418