
Comprehensive Transcriptomic and Metabolic Profiling of Agrobacterium-tumefaciens-Infected Immature Wheat Embryos
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Agrobacterium Adsorbs into Holes on the Surface of Immature Embryos
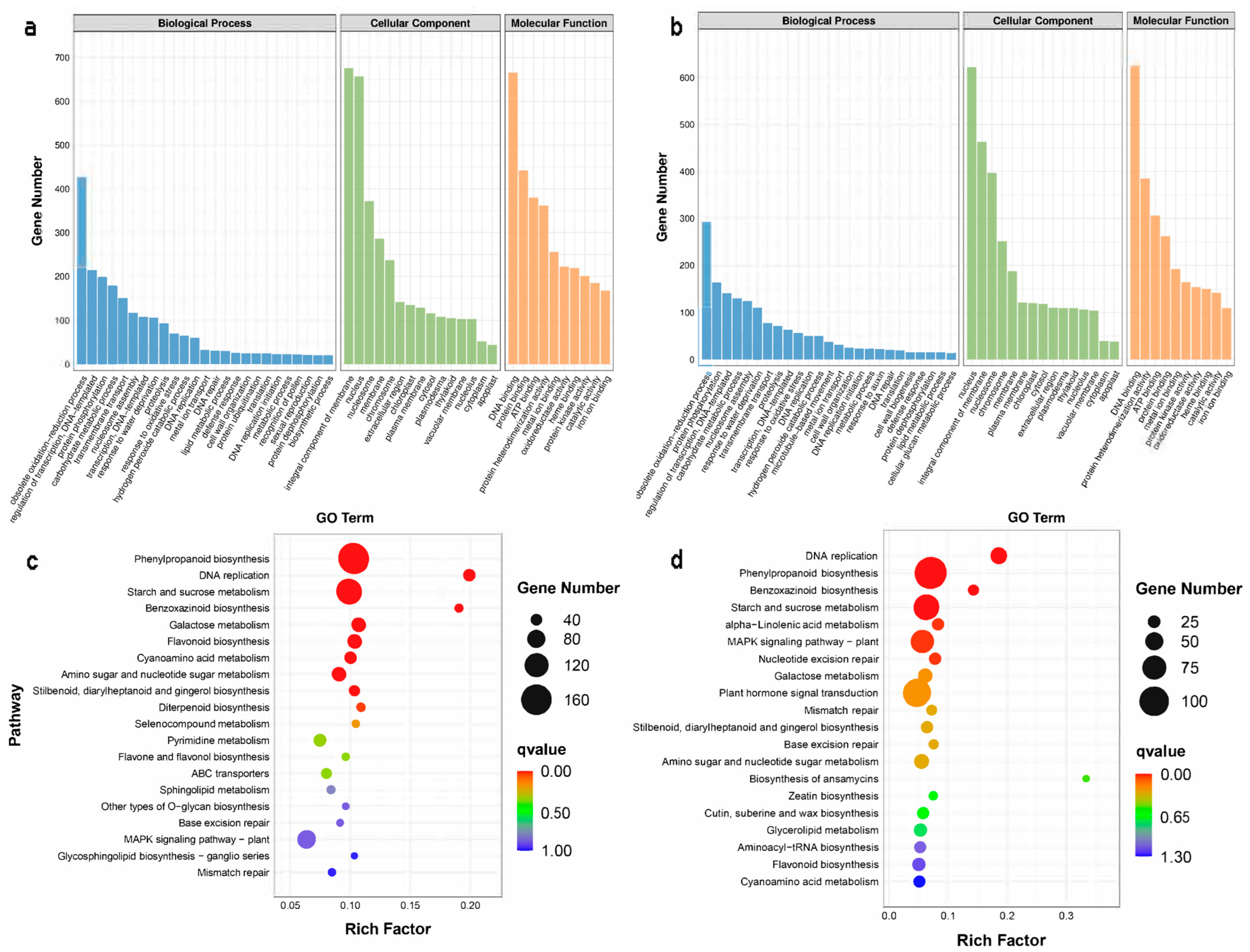
2.2. Transcriptomic Analysis of the Wheat Embryos Infected by Agrobacterium
2.3. Validation of RNA-Seq Data by Real-Time Quantitative Reverse-Transcription PCR (qRT-PCR)
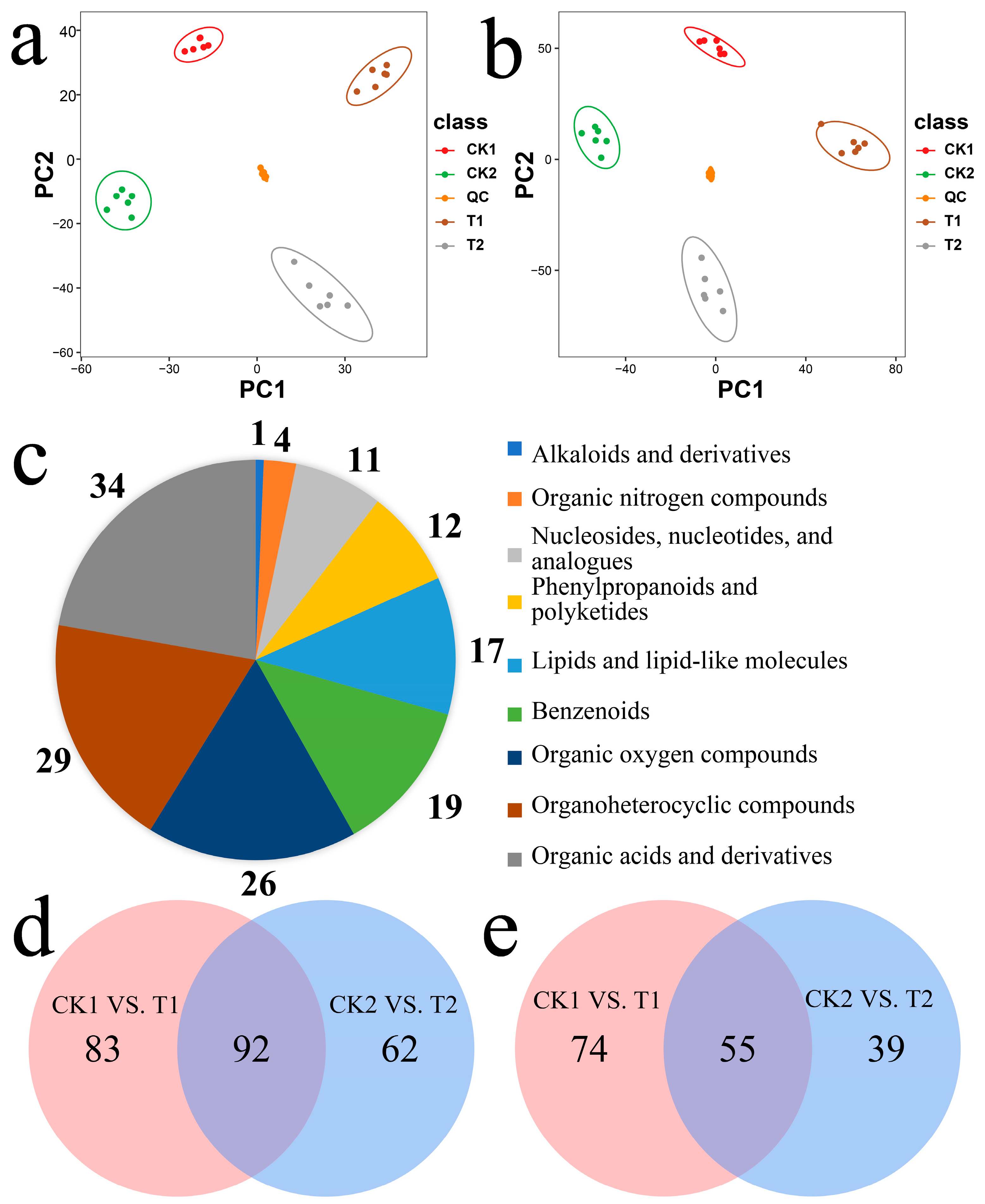
2.4. Metabolic Analysis of the Wheat Embryos Infected by Agrobacterium
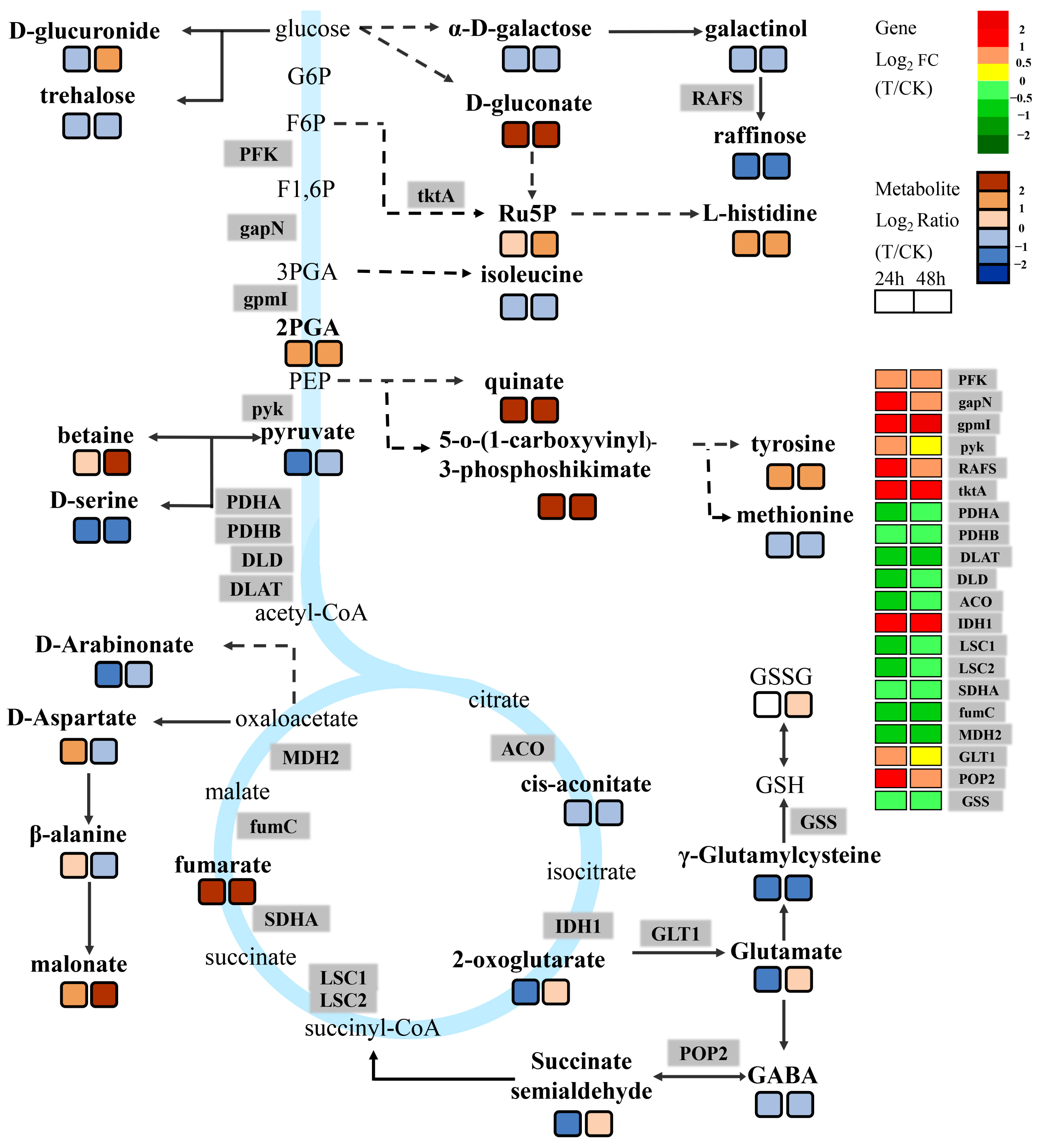
2.5. Association Analysis between DEGs and DAMs
3. Discussion
3.1. The Damage to Immature Embryos Promoted the Surface Colonization of Agrobacterium
3.2. Energy-Metabolism-Related Pathways Are Involved in the Regulation of Agrobacterium Infection of Immature Embryos
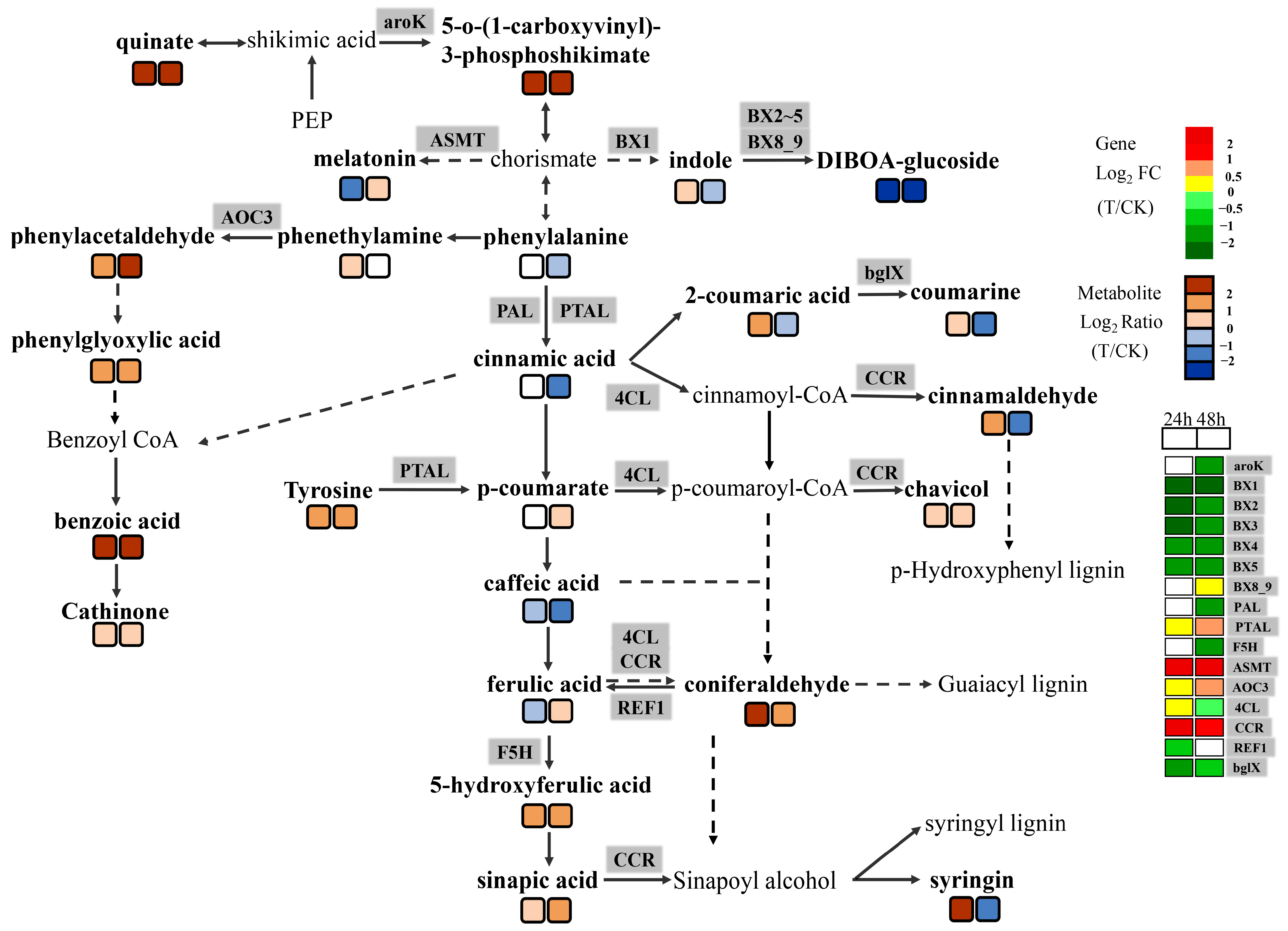
3.3. Wheat Embryos Infected by Agrobacterium tumefaciens Participated in Oxidative Stress and Biological Stress through the Phenylpropanoid Pathway
4. Materials and Methods
4.1. The Cultivation and Culture of cv. Fielder and the Acquisition of Immature Embryos
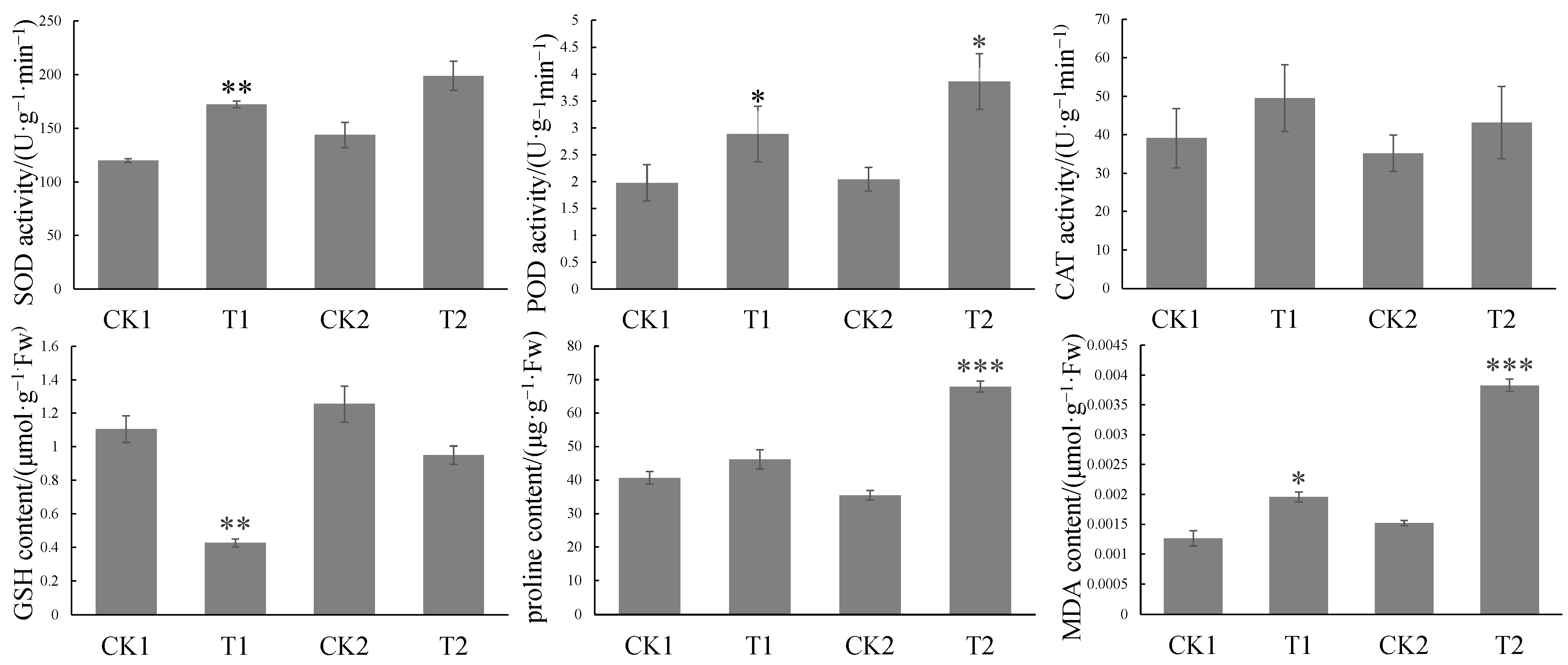
4.2. SEM Observation and Oxidation Index Determination
4.3. RNA Extraction, Quality Control, and RNA-Seq
4.4. Transcriptome Data Analysis
4.5. Validation of Transcriptomic Data by qRT-PCR
4.6. Metabolite Extraction and Metabolic Profiling Analysis
4.7. Combined Transcriptome and Metabolome Analyses
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.K.; Prasad, M. Advances in Agrobacterium tumefaciens-mediated genetic transformation of graminaceous crops. Protoplasma 2016, 253, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.X.; Long, D.F.; Lenk, I.; Nielsen, K.K. Comparative analysis of transgenic tall fescue (Festuca arundinacea Schreb.) plants obtained by Agrobacterium-mediated transformation and particle bombardment. Plant Cell Rep. 2008, 27, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gong, Q.; Ye, X.G. Recent developments and applications of genetic transformation and genome editing technologies in wheat. Theor. Appl. Genet. 2020, 133, 1603–1622. [Google Scholar] [CrossRef]
- Vasil, V.; Castillo, A.M.; Fromm, M.E.; Vasil, I.K. Herbicide resistant fertile transgenic wheat plants obtained by microprojectile bombardment of regenerable embryogenic callus. Bio/Technol. 1992, 10, 667–674. [Google Scholar] [CrossRef]
- Ishida, Y.; Tsunashima, M.; Hiei, Y.; Komari, T. Wheat (Triticum aestivum L.) transformation using immature embryos. Methods Mol. Biol. 2015, 1223, 189–198. [Google Scholar] [PubMed]
- Shrawat, A.K.; Lorz, H. Agrobacterium-mediated transformation of cereals: A promising approach crossing barriers. Plant Biotechnol. J. 2006, 4, 575–603. [Google Scholar] [CrossRef]
- Yang, J.J.; Lia, G.J.; Hua, S.Q.; Bishopp, A.; Heenatigala, P.P.M.; Kumar, S.; Duan, P.F.; Yao, L.G.; Hou, H.W. A protocol for efficient callus induction and stable transformation of Spirodela polyrhiza (L.) Schleiden using Agrobacterium tumefaciens. Aquat. Bot. 2018, 151, 80–86. [Google Scholar] [CrossRef]
- Shri, M.; Rai, A.; Verma, P.K.; Misra, P.; Dubey, S.; Kumar, S.; Verma, S.; Gautam, N.; Tripathi, R.D.; Trivedi, P.K.; et al. An improved Agrobacterium-mediated transformation of recalcitrant indica rice (Oryza sativa L.) cultivars. Protoplasma 2013, 250, 631–636. [Google Scholar] [CrossRef]
- Li, F.S.; Li, M.Y.; Zhan, C.; Wang, S.H. A reliable and high-efficiency Agrobacterium tumefaciens-mediated transformation system of Pogonatherum paniceum embryogenic callus using GFP as a reporter gene. Plant Cell Tissue Organ Cult. 2015, 120, 155–165. [Google Scholar] [CrossRef]
- Raza, A.; Razzaq, A.; Mehmood, S.S.; Hussain, M.A.; Su, W.; Huang, H.; Zaman, Q.U.; Zhang, X.; Cheng, Y.; Hasanuzamman, M. Omics: The way forward to enhance abiotic stress tolerance in Brassica napus L. Gm Crops Food-Biotechnol. Agric. Food Chain. 2021, 12, 434. [Google Scholar] [CrossRef]
- Qi, K.J.; Wu, X.; Gao, X.; Li, L.F.; Xie, Z.H.; Gu, C.; Zhang, S.L. Metabolome and transcriptome analyses unravel the inhibition of embryo germination by abscisic acid in pear. Sci. Hortic. 2022, 292, 110652. [Google Scholar]
- Yi, D.B.; Zhang, H.N.; Lai, B.; Liu, L.Q.; Pan, X.L.; Ma, Z.L.; Wang, Y.C.; Xie, J.H.; Shi, S.Y.; Wei, Y.Z. Integrative Analysis of the Coloring Mechanism of Red Longan Pericarp through Metabolome and Transcriptome Analyses. J. Agric. Food Chem. 2021, 69, 1806–1815. [Google Scholar] [CrossRef]
- Yang, F.; Li, C.H.; Das, D.; Zheng, Y.H.; Song, T.; Wang, L.X.; Chen, M.X.; Li, Q.Z.; Zhang, J.H. Comprehensive Transcriptome and Metabolic Profiling of Petal Color Development in Lycoris sprengeri. Front. Plant Sci. 2021, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, J.; Wang, B.K.; Huang, S.Y.; Hu, J.H.; Yang, T.; Asmutola, P.; Lan, H.Y.; Yu, Q.H. Identification of the Carbohydrate and Organic Acid Metabolism Genes Responsible for Brix in Tomato Fruit by Transcriptome and Metabolome Analysis. Front. Genet. 2021, 12, 1472. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yuan, G.; Zhang, Q.; Xuan, L.; Li, J.; Zhou, L.; Shi, H.; Wang, X.; Wang, C. Transcriptome and metabolome analyses reveal the pivotal role of hydrogen sulfide in promoting submergence tolerance in Arabidopsis. Environ. Exp. Bot. 2021, 183, 104365. [Google Scholar] [CrossRef]
- Liu, D.P.; Li, M.X.; Liu, Y.; Shi, L.X. Integration of the metabolome and transcriptome reveals the resistance mechanism to low nitrogen in wild soybean seedling roots. Environ. Exp. Bot. 2020, 175, 104043. [Google Scholar] [CrossRef]
- Xu, Z.S.; Chen, X.J.; Lu, X.P.; Zhao, B.P.; Yang, Y.M.; Liu, J.H. Integrative analysis of transcriptome and metabolome reveal mechanism of tolerance to salt stress in oat (Avena sativa L.). Plant Physiol. Biochem. 2021, 160, 315–328. [Google Scholar] [CrossRef]
- Xie, Z.Y.; Wang, J.; Wang, W.S.; Wang, Y.R.; Xu, J.L.; Li, Z.K.; Zhao, X.Q.; Fu, B.Y. Integrated Analysis of the Transcriptome and Metabolome Revealed the Molecular Mechanisms Underlying the Enhanced Salt Tolerance of Rice Due to the Application of Exogenous Melatonin. Front. Plant Sci. 2021, 11, 2277. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Xu, Y.F.; Zhang, M.; Zhu, F.D.; Sun, M.X.; Lian, X.Y.; Zhao, G.F.; Duan, D. Transcriptome and metabolome analysis of stress tolerance to aluminium in Vitis quinquangularis. Planta 2021, 254, 105. [Google Scholar] [CrossRef]
- Raza, A.; Su, W.; Hussain, M.A.; Mehmood, S.S.; Zhang, X.K.; Cheng, Y.; Zou, X.L.; Lv, Y. Integrated Analysis of Metabolome and Transcriptome Reveals Insights for Cold Tolerance in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 12, 1796. [Google Scholar] [CrossRef]
- Krugman, T.; Peleg, Z.; Quansah, L.; Chague, V.; Korol, A.B.; Nevo, E.; Saranga, Y.; Fait, A.; Chalhoub, B.; Fahima, T. Alteration in expression of hormone-related genes in wild emmer wheat roots associated with drought adaptation mechanisms. Funct. Integr. Genom. 2011, 11, 565–583. [Google Scholar] [CrossRef]
- Sun, T.T.; Zhang, J.K.; Zhang, Q.; Li, X.L.; Li, M.J.; Yang, Y.Z.; Zhou, J.; Wei, Q.P.; Zhou, B.B. Transcriptome and metabolome analyses revealed the response mechanism of apple to different phosphorus stresses. Plant Physiol. Biochem. 2021, 167, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.T.; Zhang, J.K.; Zhang, Q.; Li, X.L.; Li, M.J.; Yang, Y.Z.; Zhou, J.; Wei, Q.P.; Zhou, B.B. Integrative physiological, transcriptome, and metabolome analysis reveals the effects of nitrogen sufficiency and deficiency conditions in apple leaves and roots. Environ. Exp. Bot. 2021, 192, 104633. [Google Scholar] [CrossRef]
- Pei, L.M.; Liu, J.J.; Zhou, Y.Y.; Jiang, Y.H.; Li, H. Transcriptomic and metabolomic profiling reveals the protective role of anthocyanins in alleviating low phosphate stress in maize. Physiol. Mol. Biol. Plants 2021, 27, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Liu, J.X.; Zong, J.Q.; Guo, H.L.; Li, J.J.; Wang, J.J.; Wang, H.R.; Li, L.; Chen, J.B. Integration of the metabolome and transcriptome reveals the mechanism of resistance to low nitrogen supply in wild bermudagrass (Cynodon dactylon (L.) Pers.) roots. BMC Plant Biol. 2021, 21, 480. [Google Scholar] [CrossRef]
- Nussbaumer, T.; Warth, B.; Sharma, S.; Ametz, C.; Bueschl, C.; Parich, A.; Pfeifer, M.; Siegwart, G.; Steiner, B.; Lemmens, M.; et al. Joint Transcriptomic and Metabolomic Analyses Reveal Changes in the Primary Metabolism and Imbalances in the Subgenome Orchestration in the Bread Wheat Molecular Response to Fusarium graminearum. G3—Genes Genomes Genet. 2015, 5, 2579–2592. [Google Scholar] [CrossRef]
- Lang, J.; Fu, Y.X.; Zhou, Y.; Cheng, M.P.; Deng, M.; Li, M.L.; Zhu, T.T.; Yang, J.; Guo, X.J.; Gui, L.X.; et al. Myb10-D confers PHS-3D resistance to pre-harvest sprouting by regulating NCED in ABA biosynthesis pathway of wheat. New Phytol. 2021, 230, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Muthamilarasan, M.; Theriappan, P.; Prasad, M. Recent advances in crop genomics for ensuring food security. Curr. Sci. 2013, 105, 155–158. [Google Scholar]
- Ge, F.; Luo, X.; Huang, X.; Zhang, Y.L.; He, X.J.; Liu, M.; Lin, H.J.; Peng, H.W.; Li, L.J.; Zhang, Z.M.; et al. Genome-wide analysis of transcription factors involved in maize embryonic callus formation. Physiol. Plant. 2016, 158, 452–462. [Google Scholar] [CrossRef]
- Akram, M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Zhou, X.H.; Wang, K.; Lv, D.W.; Wu, C.J.; Li, J.R.; Zhao, P.; Lin, Z.S.; Du, L.P.; Yan, Y.M.; Ye, X.G. Global Analysis of Differentially Expressed Genes and Proteins in the Wheat Callus Infected by Agrobacterium tumefaciens. PLoS ONE 2013, 8, e79390. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Hu, H.M.; Huang, X.; Zhang, Y.L.; Wang, Y.L.; Li, Z.L.; Zou, C.Y.; Peng, H.W.; Li, L.J.; Gao, S.B.; et al. Metabolomic and Proteomic Analysis of Maize Embryonic Callus induced from immature embryo. Sci. Rep. 2017, 7, 1004. [Google Scholar] [CrossRef]
- Chu, Z.L.; Chen, J.Y.; Sun, J.Y.; Dong, Z.D.; Yang, X.; Wang, Y.; Xu, H.X.; Zhang, X.K.; Chen, F.; Cui, D.Q. De novo assembly and comparative analysis of the transcriptome of embryogenic callus formation in bread wheat (Triticum aestivum L.). BMC Plant Biol. 2017, 17, 244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, S.J.; Zhou, H.L.; Yuan, Z.; Zhou, T.; Zhang, Y.H.; Xiang, S.; Yang, F.; Shen, X.L.; Zhang, D.C. Transcriptome sequencing analysis of sorghum callus with various regeneration capacities. Planta 2021, 254, 33. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.P.; Zhang, C.J.; Liu, Q.H.; Zhang, Z.; Zheng, B.; Bao, M.Z. De novo comparative transcriptome analysis provides new insights into sucrose induced somatic embryogenesis in camphor tree (Cinnamomum camphora L.). BMC Genom. 2016, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gao, Y.; Wang, J.B. Transcriptomic Analysis of Rice (Oryza sativa) Developing Embryos Using the RNA-Seq Technique. PLoS ONE 2012, 7, e30646. [Google Scholar] [CrossRef]
- Muhammad, M.H.; Idris, A.L.; Fan, X.; Guo, Y.C.; Yu, Y.Y.; Jin, X.; Qiu, J.Z.; Guan, X.; Huang, T.P. Beyond Risk: Bacterial Biofilms and Their Regulating Approaches. Front. Microbiol. 2020, 11, 928. [Google Scholar] [CrossRef]
- Merritt, P.M.; Danhorn, T.; Fuqua, C. Motility and chemotaxis in Agrobacterium tumefaciens surface attachment and Biofilm formation. J. Bacteriol. 2007, 189, 8005–8014. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Liu, Z.-H.; Yang, R.-J.; Li, G.-L.; Guo, X.-L.; Zhang, H.-N.; Zhang, H.-M.; Di, R.; Zhao, Q.-S.; Zhang, M.-C. Improvement of soybean transformation via Agrobacterium tumefaciens methods involving alpha-aminooxyacetic acid and sonication treatments enlightened by gene expression profile analysis. Plant Cell Rep. 2016, 35, 1259–1271. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Sood, P.; Citovsky, V. The roles of plant phenolics in defence and communication during Agrobacterium and Rhizobium infection. Mol. Plant Pathol. 2010, 11, 705–719. [Google Scholar] [CrossRef]
- Cheng, M.; Hu, T.C.; Layton, J.; Liu, C.N.; Fry, J.E. Desiccation of plant tissues post-Agrobacterium infection enhances T-DNA delivery and increases stable transformation efficiency in wheat. Vitr. Cell. Dev. Biol.-Plant 2003, 39, 595–604. [Google Scholar] [CrossRef]
- Norelli, J.; Mills, J.; Aldwinckle, H. Leaf wounding increases efficiency of Agrobacterium-mediated transformation of apple. HortScience 1996, 31, 1026–1027. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, A.; Prasad, B.C.N.; Gururaj, H.B.; Ravishankar, G.A. Agrobacterium rhizogenes mediated genetic transformation resulting in hairy root formation is enhanced by ultrasonication and acetosyringone treatment. Electron. J. Biotechnol. 2006, 9, 349–357. [Google Scholar] [CrossRef]
- Mannervik, B. The enzymes of glutathione metabolism: An overview. Biochem. Soc. Trans. 1987, 15, 717–718. [Google Scholar] [CrossRef]
- Guo, J.; Carrington, Y.; Alber, A.; Ehlting, J. Molecular Characterization of Quinate and Shikimate Metabolism in Populus trichocarpa. J. Biol. Chem. 2014, 289, 23846–23858. [Google Scholar] [CrossRef]
- Le Roy, J.; Huss, B.; Creach, A.; Hawkins, S.; Neutelings, G. Glycosylation Is a Major Regulator of Phenylpropanoid Availability and Biological Activity in Plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Saito, K. Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Plant Biol. 2015, 24, 10–16. [Google Scholar] [CrossRef]
- Ohl, S.; Hedrick, S.A.; Chory, J.; Lamb, C.J. Functional properties of a phenylalanine ammonia-lyase promoter from Arabidopsis. Plant Cell 1990, 2, 837–848. [Google Scholar]
- Fritz, R.R.; Hodgins, D.S.; Abell, C.W. Phenylalanine ammonia-lyase. Induction and purification from yeast and clearance in mammals. J. Biol. Chem. 1976, 251, 4646–4650. [Google Scholar] [CrossRef]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Wang, Y.; Reiter, R.J.; Chan, Z. Phytomelatonin: A universal abiotic stress regulator. J. Exp. Bot. 2018, 69, 963–974. [Google Scholar] [CrossRef]
- Cao, Q.; Li, G.; Cui, Z.; Yang, F.; Jiang, X.; Diallo, L.; Kong, F. Seed Priming with Melatonin Improves the Seed Germination of Waxy Maize under Chilling Stress via Promoting the Antioxidant System and Starch Metabolism. Sci. Rep. 2019, 9, 15044. [Google Scholar] [CrossRef]
- Byeon, Y.; Lee, H.-J.; Lee, H.Y.; Back, K. Cloning and functional characterization of the Arabidopsis N-acetylserotonin O-methyltransferase responsible for melatonin synthesis. J. Pineal Res. 2016, 60, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Steinmacher, D.A.; Guerra, M.P.; Saare-Surminski, K.; Lieberei, R. A temporary immersion system improves in vitro regeneration of peach palm through secondary somatic embryogenesis. Ann. Bot. 2011, 108, 1463–1475. [Google Scholar] [CrossRef]
- Razzaq, K.; Khan, A.S.; Malik, A.U.; Shahid, M.; Ullah, S. Role of putrescine in regulating fruit softening and antioxidative enzyme systems in ‘Samar Bahisht Chaunsa’ mango. Postharvest Biol. Technol. 2014, 96, 23–32. [Google Scholar] [CrossRef]
- Hemalatha, M.S.; Manu, B.T.; Bhagwat, S.G.; Leelavathi, K.; Rao, U.J.S.P. Protein characteristics and peroxidase activities of different Indian wheat varieties and their relationship to chapati-making quality. Eur. Food Res. Technol. 2007, 225, 463–471. [Google Scholar] [CrossRef]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts. I. Kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys. 1968, 125, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Fahey, R.C.; Di Stefano, D.L.; Meier, G.P.; Bryan, R.N. Role of hydration state and thiol-disulfide status in the control of thermal stability and protein synthesis in wheat embryo. Plant Physiol. 1980, 65, 1062–1066. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Guo, J.; Ma, J.; Wang, Z.; Zhang, L.; Wang, Z.; Meng, M.; Zhang, C.; Sun, F.; Xi, Y. Comprehensive Transcriptomic and Metabolic Profiling of Agrobacterium-tumefaciens-Infected Immature Wheat Embryos. Int. J. Mol. Sci. 2023, 24, 8449. https://doi.org/10.3390/ijms24098449
Wang W, Guo J, Ma J, Wang Z, Zhang L, Wang Z, Meng M, Zhang C, Sun F, Xi Y. Comprehensive Transcriptomic and Metabolic Profiling of Agrobacterium-tumefaciens-Infected Immature Wheat Embryos. International Journal of Molecular Sciences. 2023; 24(9):8449. https://doi.org/10.3390/ijms24098449
Chicago/Turabian StyleWang, Weiwei, Jinliang Guo, Jiayang Ma, Zhulin Wang, Lining Zhang, Zixu Wang, Min Meng, Chao Zhang, Fengli Sun, and Yajun Xi. 2023. "Comprehensive Transcriptomic and Metabolic Profiling of Agrobacterium-tumefaciens-Infected Immature Wheat Embryos" International Journal of Molecular Sciences 24, no. 9: 8449. https://doi.org/10.3390/ijms24098449