
Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile


Abstract
:1. Introduction
2. Results
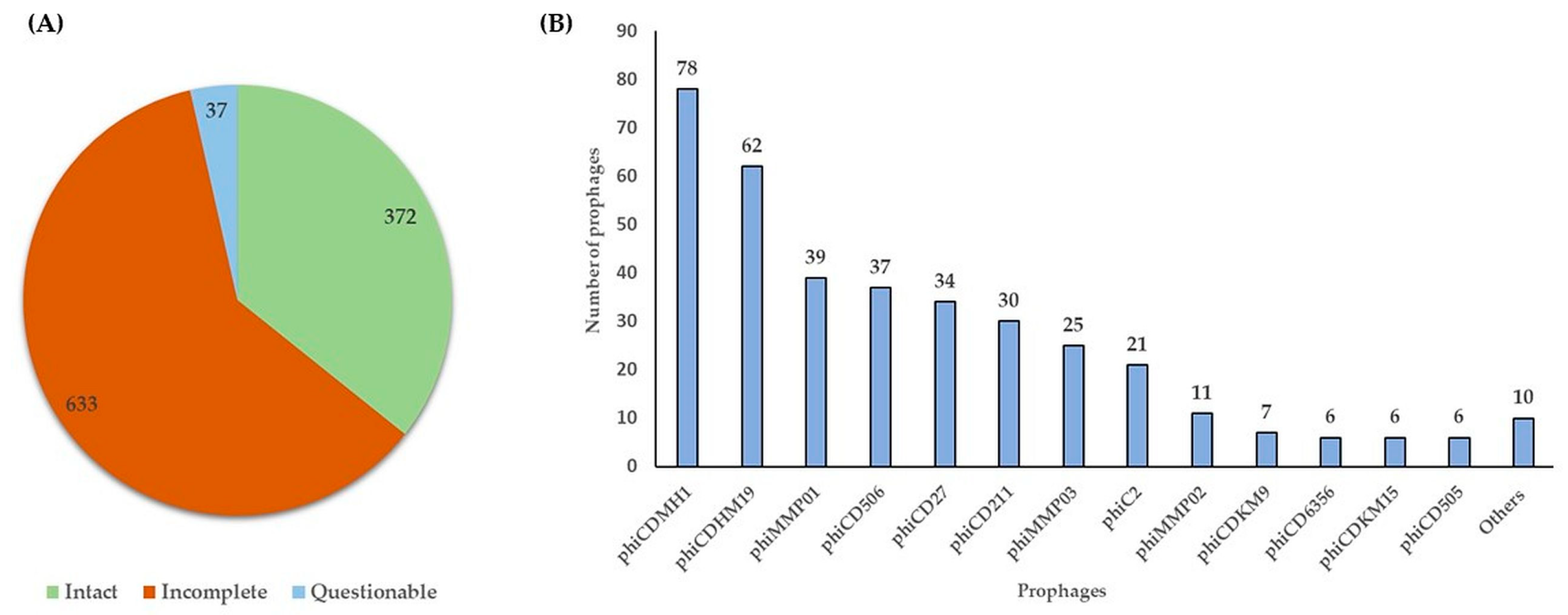
2.1. Prevalence of Prophages in the Genomes of Environmental C. difficile Isolates
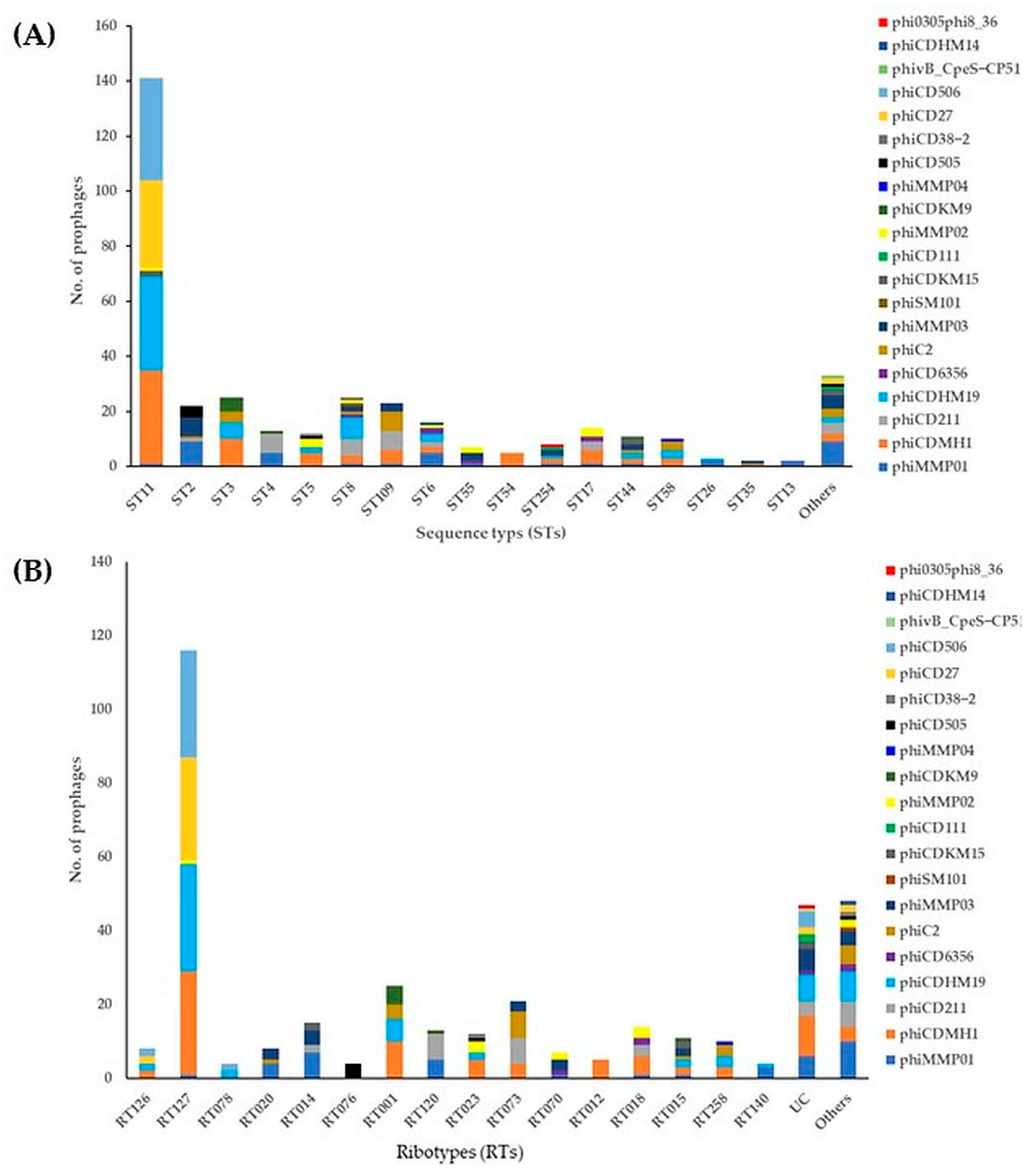
2.2. Prevalence and Diversity of Intact Prophages in STs/RTs of Environmental C. difficile Isolates
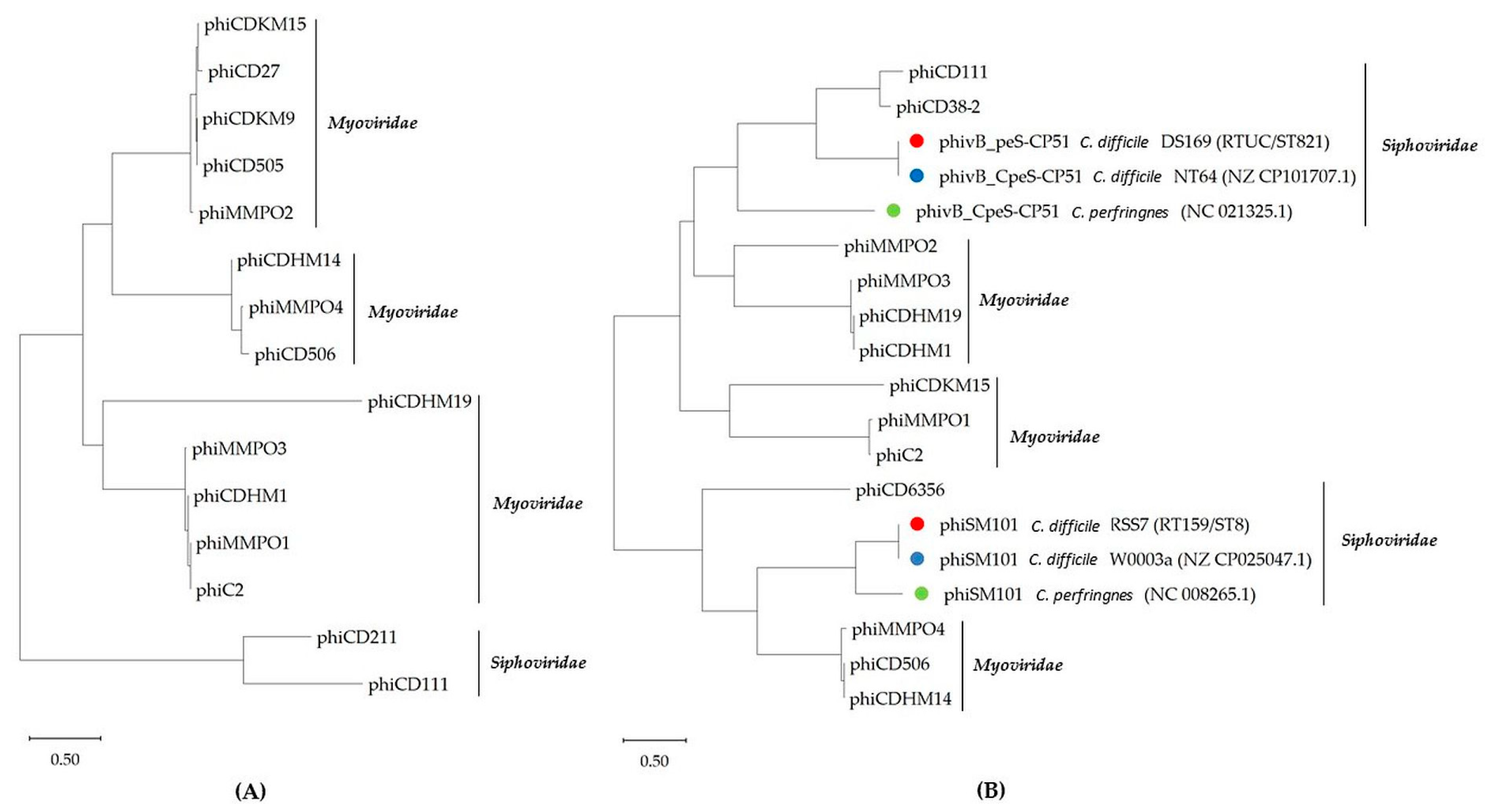
2.3. Phylogenetic Analysis of Terminase Large Subunits (TerL) and LysM Proteins of C. difficile Prophages
2.4. Phage-Unrelated Genes of Intact Prophages Identified in C. difficile Genomes
2.5. The Genome Features of Newly Discovered phivB_CpeS-CP51- and phiSM101- Clostridium Phages Identified in C. difficile Genomes
2.6. The CRISPR Arrays and Cas-Systems in Prophages Identified in C. difficile Genomes
3. Discussion
4. Materials and Methods
4.1. Whole Genome Sequencing and Data Analysis
4.2. Phylogenetic Analysis
4.3. Features of Newly Identified Prophages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef] [PubMed]
- Lawson, P.A.; Citron, D.M.; Tyrrell, K.L.; Finegold, S.M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 2016, 40, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Usacheva, E.A.; Jin, J.-P.; Peterson, L.R. Host response to Clostridium difficile infection: Diagnostics and detection. J. Glob. Antimicrob. Resist. 2016, 7, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Leffler, D.A.; Lamont, J.T. Clostridium difficile Infection. N. Engl. J. Med. 2015, 373, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Chipman, P.R.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell 2004, 118, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Plisson, C.; White, H.E.; Auzat, I.; Zafarani, A.; São-José, C.; Lhuillier, S.; Tavares, P.; Orlova, E.V. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 2007, 26, 3720–3728. [Google Scholar] [CrossRef]
- Heuler, J.; Fortier, L.-C.; Sun, X. Clostridioides difficile phage biology and application. FEMS Microbiol. Rev. 2021, 45, fuab012. [Google Scholar] [CrossRef]
- Meader, E.; Mayer, M.J.; Gasson, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe 2010, 16, 549–554. [Google Scholar] [CrossRef]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef]
- Umansky, A.A.; Fortier, L.C. The long and sinuous road to phage-based therapy of Clostridioides difficile infections. Front. Med. 2023, 10, 1259427. [Google Scholar] [CrossRef]
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeño-Tárraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786. [Google Scholar] [CrossRef]
- Goh, S.; Hussain, H.; Chang, B.J.; Emmett, W.; Riley, T.V.; Mullany, P. Phage ϕC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. mBio 2013, 4, e00840-13. [Google Scholar] [CrossRef]
- Govind, R.; Vediyappan, G.; Rolfe, R.D.; Dupuy, B.; Fralick, J.A. Bacteriophage-mediated toxin gene regulation in Clostridium difficile. J. Virol. 2009, 83, 12037–12045. [Google Scholar] [CrossRef]
- Sekulovic, O.; Meessen-Pinard, M.; Fortier, L.-C. Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J. Bacteriol. 2011, 193, 2726–2734. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. What does the talking?: Quorum sensing signalling genes discovered in a bacteriophage genome. PLoS ONE 2014, 9, e85131. [Google Scholar] [CrossRef]
- Garneau, J.R.; Sekulovic, O.; Dupuy, B.; Soutourina, O.; Monot, M.; Fortier, L.-C. High Prevalence and Genetic Diversity of Large phiCD211 (phiCDIF1296T)-Like Prophages in Clostridioides difficile. Appl. Environ. Microbiol. 2018, 84, e02164-17. [Google Scholar] [CrossRef]
- Brouwer, M.S.M.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef]
- Nale, J.Y.; Thanki, A.M.; Rashid, S.J.; Shan, J.; Vinner, G.K.; Dowah, A.S.A.; Cheng, J.K.J.; Sicheritz-Pontén, T.; Clokie, M.R.J. Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages. Viruses 2022, 14, 2772. [Google Scholar] [CrossRef]
- Boudry, P.; Semenova, E.; Monot, M.; Datsenko, K.A.; Lopatina, A.; Sekulovic, O.; Ospina-Bedoya, M.; Fortier, L.C.; Severinov, K.; Dupuy, B.; et al. Function of the CRISPR-Cas System of the Human Pathogen Clostridium difficile. mBio 2015, 6, 10–1128. [Google Scholar] [CrossRef]
- Blau, K.; Berger, F.K.; Mellmann, A.; Gallert, C. Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective. Microorganisms 2023, 11, 497. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Myers, G.S.A.; Rasko, D.A.; Cheung, J.K.; Ravel, J.; Seshadri, R.; DeBoy, R.T.; Ren, Q.; Varga, J.; Awad, M.M.; Brinkac, L.M.; et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006, 16, 1031–1040. [Google Scholar] [CrossRef]
- Thomas, J.A.; Hardies, S.C.; Rolando, M.; Hayes, S.J.; Lieman, K.; Carroll, C.A.; Weintraub, S.T.; Serwer, P. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305ϕ8–36. Virology 2007, 368, 405–421. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Gervasi, T.; Lo Curto, R.; Narbad, A.; Mayer, M.J. Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch. Virol. 2013, 158, 2015–2017. [Google Scholar] [CrossRef]
- Frey, J.; Falquet, L. Patho-genetics of Clostridium chauvoei. Res. Microbiol. 2015, 166, 384–392. [Google Scholar] [CrossRef]
- Thomas, P.; Abdel-Glil, M.Y.; Subbaiyan, A.; Busch, A.; Eichhorn, I.; Wieler, L.H.; Neubauer, H.; Pletz, M.; Seyboldt, C. First Comparative Analysis of Clostridium septicum Genomes Provides Insights into the Taxonomy, Species Genetic Diversity, and Virulence Related to Gas Gangrene. Front. Microbiol. 2021, 12, 771945. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, C.; Li, W.-G.; Xu, J.-L.; Zhang, W.-Z.; Dai, Y.-F.; Lu, J.-X. Independent Microevolution Mediated by Mobile Genetic Elements of Individual Clostridium difficile Isolates from Clade 4 Revealed by Whole-Genome Sequencing. mSystems 2019, 4, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.-C. Bacteriophages Contribute to Shaping Clostridioides (Clostridium) difficile Species. Front. Microbiol. 2018, 9, 2033. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Vargas, G.; Goh, S.; Rodríguez, C. The Novel Phages phiCD5763 and phiCD2955 Represent Two Groups of Big Plasmidial Siphoviridae Phages of Clostridium difficile. Front. Microbiol. 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, J.; Riedel, T.; Bunk, B.; Spröer, C.; Gronow, S.; Overmann, J. Complete Genome Sequence of the Novel Temperate Clostridium difficile Phage phiCDIF1296T. Genome Announc. 2015, 3, e00839-15. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Horgan, M.; O’Sullivan, O.; Coffey, A.; Fitzgerald, G.F.; van Sinderen, D.; McAuliffe, O.; Ross, R.P. Genome analysis of the Clostridium difficile phage PhiCD6356, a temperate phage of the Siphoviridae family. Gene 2010, 462, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Wittmann, J.; Bunk, B.; Schober, I.; Spröer, C.; Gronow, S.; Overmann, J. A Clostridioides difficile bacteriophage genome encodes functional binary toxin-associated genes. J. Biotechnol. 2017, 250, 23–28. [Google Scholar] [CrossRef]
- Novick, R.P.; Geisinger, E. Quorum sensing in staphylococci. Annu. Rev. Genet. 2008, 42, 541–564. [Google Scholar] [CrossRef]
- Goh, S.; Ong, P.F.; Song, K.P.; Riley, T.V.; Chang, B.J. The complete genome sequence of Clostridium difficile phage phiC2 and comparisons to phiCD119 and inducible prophages of CD630. Microbiology 2007, 153, 676–685. [Google Scholar] [CrossRef]
- Paredes-Sabja, D.; Shen, A.; Sorg, J.A. Clostridium difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef]
- Garcia-Pino, A.; Christensen-Dalsgaard, M.; Wyns, L.; Yarmolinsky, M.; Magnuson, R.D.; Gerdes, K.; Loris, R. Doc of prophage P1 is inhibited by its antitoxin partner Phd through fold complementation. J. Biol. Chem. 2008, 283, 30821–30827. [Google Scholar] [CrossRef]
- Rashid, S.J.; Barylski, J.; Hargreaves, K.R.; Millard, A.A.; Vinner, G.K.; Clokie, M.R.J. Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology. Viruses 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Flores, C.O.; Lawley, T.D.; Clokie, M.R.J. Abundant and diverse clustered regularly interspaced short palindromic repeat spacers in Clostridium difficile strains and prophages target multiple phage types within this pathogen. mBio 2014, 5, e01045-13. [Google Scholar] [CrossRef] [PubMed]
- Hatoum-Aslan, A.; Samai, P.; Maniv, I.; Jiang, W.; Marraffini, L.A. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J. Biol. Chem. 2013, 288, 27888–27897. [Google Scholar] [CrossRef] [PubMed]
- Lemriss, H.; Lemriss, S.; Butin, M.; Ibrahimi, A.; El Kabbaj, S.; Rasigade, J.; Laurent, F. Non-contiguous finished genome sequence of Staphylococcus capitis CR01 (pulsetype NRCS-A). Stand. Genomic Sci. 2014, 9, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Golding, G.R.; Bryden, L.; Levett, P.N.; McDonald, R.R.; Wong, A.; Graham, M.R.; Tyler, S.; van Domselaar, G.; Mabon, P.; Kent, H.; et al. whole-genome sequence of livestock-associated st398 methicillin-resistant Staphylococcus aureus Isolated from Humans in Canada. J. Bacteriol. 2012, 194, 6627–6628. [Google Scholar] [CrossRef]
- Blau, K.; Gallert, C. Prevalence, Antimicrobial Resistance and Toxin-Encoding Genes of Clostridioides difficile from Environmental Sources Contaminated by Feces. Antibiotics 2023, 12, 162. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | No. of Prophages (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| phiMMP01 | phiMMP02 | phiMMP03 | phiC2 | phiCD211 | phiCDHM1 | phiCDHM14 | phiCD6356 | |
| Agr-QS systems | agrC, agrD (1/39, 3%) | agrC, agrD (1/11, 9%) | AgrC, agrD (1/25, 4%) | - | - | agrC, agrB, agrD (4/78, 5%) | - | - |
| Spore protease YyaC | - | - | - | - | 19/30 (63%) | - | - | - |
| Abi systems | 19/39 (49%) | - | - | 6/21 (29%) | - | - | - | - |
| Death-on-curing (DOC) protein | - | - | - | - | 15/30 (50%) | - | - | - |
| parA | - | - | - | - | - | - | 1/1 (100%) | 2/6 (33%) |
| RT/ST 1 | Prophages | Family | CRISPR No. | Spacer No. | DR No. | DR ID 2 |
|---|---|---|---|---|---|---|
| RT005/ST6 | phiMMP01 | Myoviridae | 2 | 14, 3 | 21, 2 | R7326, R8849 |
| phiMMP01 | 4 | 6, 17, 3, 2 | 19, 0 | R7360, UN | ||
| phiCDHM19 | 1 | 1 | 0 | UN | ||
| RT090/ST1073 | phiMMP01 | Myoviridae | 2 | 2 | 9 | R1411 |
| RT011/ST36 | phiC2 | Myoviridae | 3 | 2, 17, 3, 19 | 21, 2 | R7326, R8849 |
| UC/ST2 | phiMMP01 | Myoviridae | 4 | 2, 3, 17, 6 | 0 | UN |
| phiMMP01 | 2 | 3, 14 | 0, 9 | UN, R1411 | ||
| phiCD6356 | Siphoviridae | 1 | 1 | 0 | UN | |
| RT020/ST2 | phiC2 | Myoviridae | 2 | 5, 8 | 1 | R7327, R6417 |
| phiMMP01 | 3 | 5, 4, 12, 6, 14 | 21, 0, 19, 9 | R7326, R7360, R1411, UN | ||
| RT070/ST55 | phiMMP02 | Myoviridae | 1, 2 | 3, 4, 6 | 2, 9 | R1411, R3412 |
| RT159/ST8 | phiCD211 | Siphoviridae | 1 | 1 | 0 | UN |
| phiC2 | Myoviridae | 2 | 4, 13 | 0 | UN | |
| RT015/ST44 | phiCDKM15 | Myoviridae | 2 | 6 | 0 | UN |
| phiMMP03 | 2 | 7, 10 | 0, 1 | R7327, UN | ||
| phiCDHM19 | 1 | 1 | 0 | UN | ||
| phiMMP01 | 1 | 3 | 0 | UN | ||
| RTUC/ST254 | phiMMP03 | Myoviridae | 1, 2 | 5 | 0, 19 | R7360, UN |
| phiMMP01 | 4 | 5, 7, 10, 14 | 0, 9 | R1411, UN | ||
| phiCDHM1 | 2 | 5 | 0, 1 | R6417, UN | ||
| RT010/ST15 | phiMMP01 | Myoviridae | 2 | 8, 13 | 21 | R7326 |
| RT140/ST26 | phiMMP01 | Myoviridae | 2 | 2, 13, 14 | 1, 21 | R7326, R7327 |
| RT140/ST515 | phiMMP01 | Myoviridae | 2 | 2, 14 | 21 | R7326 |
| RT023/ST5 | phiMMP02 | Myoviridae | 2 | 3, 7 | 0, 9, 21 | R7326, R1411, UN |
| phiCDHM1 | 1 | 3, 14 | 9, 21 | R1411, R7326 | ||
| phiCD505 | 1 | 13 | 21 | R7326 | ||
| RT014/ST14 | phiMMP01 | Myoviridae | 3 | 4, 6, 14 | 0, 9 | R1411, UN |
| phiCDKM15 | 2 | 3, 5 | 0, 9 | R1411, UN | ||
| RT014/ST2 | phiMMP03 | Myoviridae | 1, 2 | 5, 7, 8 | 0, 1 | R7327, R6417, UN |
| phiMMP01 | 3 | 4, 5, 6, 11, 12 | 0, 9, 19, 21 | R7326, R7360, R1411, UN | ||
| RT014/ST13 | phiMMP01 | Myoviridae | 3 | 4, 6, 14 | 0, 19, 21 | R7326, R7360, UN |
| RT014/ST49 | phiCDKM15 | Myoviridae | 1 | 6 | 0 | UN |
| phiMMP01 | 3 | 4, 6, 14 | 0, 19, 21 | R7326, R7360, UN | ||
| RT018/ST17 | phiCDHM1 | Myoviridae | 4 | 3, 5, 6 | 0, 2, 9, 21 | R1411, R7326, R8849, UN |
| phiMMP02 | 1 | 17 | 9 | R1411 | ||
| phiMMP01 | 1 | 5 | 0 | UN | ||
| RT001/ST3 | phiC2 | Myoviridae | 4 | 1, 3, 7, 13 | 0, 9 | R1411, UN |
| phiCDHM1 | 3, 4 | 2, 3, 4, 5, 6 | 0, 1, 9, 19, 21 | R7360, R7326, R1411, R8402, UN | ||
| phiCDKM9 | 1 | 2 | 0 | UN | ||
| phiCDHM19 | 1 | 1 | 0 | UN | ||
| RTUC/ST821 | phiMMP03 | Myoviridae | 2 | 8, 11 | 19 | R7360 |
| RTUC/ST917 | phiMMP01 | Myoviridae | 2 | 1, 5 | 0 | UN |
| RT126/ST11 | phiCDHM19 | Myoviridae | 1 | 3 | 9, 21 | R7326 |
| phiCD27 | 1 | 4 | 0 | UN | ||
| RT031/ST26 | phiMMP03 | Myoviridae | 2 | 4, 5 | 0 | UN |
| RT017/ST37 | phiMMP01 | Myoviridae | 2 | 3, 4 | 0, 1 | R8649, UN |
| phiCDHM19 | 1 | 1 | 0 | UN | ||
| RT002/ST8 | phiMMP01 | Myoviridae | 2 | 4 | 0, 19 | R7360, UN |
| phiMMP03 | 1 | 1 | 0 | UN | ||
| RT127/ST11 | phiCD27 | Myoviridae | 1 | 4 | 0 | UN |
| phiCDHM19 | 1 | 3 | 9, 21 | R1411, R7326 | ||
| phiMMP02 | 1 | 4 | 0 | UN | ||
| RTUC/ST11 | phiCDHM1 | Myoviridae | 1 | 5 | 0 | UN |
| phiCDKM15 | 1 | 8 | 0 | UN | ||
| phiCD27 | 1 | 4, 8 | 0 | UN | ||
| RTUC/ST11 | phiCDHM19 | Myoviridae | 1 | 3 | 21 | UN |
| RT095/ST2 | phiMMP01 | Myoviridae | 3 | 4, 6, 14 | 0, 9 | R1411, UN |
| RT077/ST13 | phiMMP01 | Myoviridae | 3 | 4, 6, 14 | 0, 9, 21 | R7326, R7360, UN |
| RT120/ST4 | phiMMP01 | Myoviridae | 1 | 5 | 0 | UN |
| phiCDKM9 | 1 | 5 | 0 | UN | ||
| RT328/ST35 | phiMMP03 | Myoviridae | 2 | 8, 9 | 0, 9 | R1411, UN |
| phiC2 | 2 | 8, 9 | 0, 9 | R1411, UN | ||
| RT103/ST53 | phiCD27 | Myoviridae | 3 | 1, 3, 5 | 0 | UN |
| phiMMP01 | 2 | 5, 6 | 21 | R7326 | ||
| RT073/ST109 | phiC2 | Myoviridae | 3 | 5, 9 | 0, 9, 21 | R7326, R1411, UN |
| phiCDHM1 | 1 | 1 | 0 | UN | ||
| RT085/ST39 | phiC2 | Myoviridae | 3 | 5, 8, 12 | 0, 21 | R7326, UN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blau, K.; Gallert, C. Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile. Int. J. Mol. Sci. 2024, 25, 2. https://doi.org/10.3390/ijms25010002
Blau K, Gallert C. Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile. International Journal of Molecular Sciences. 2024; 25(1):2. https://doi.org/10.3390/ijms25010002
Chicago/Turabian StyleBlau, Khald, and Claudia Gallert. 2024. "Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile" International Journal of Molecular Sciences 25, no. 1: 2. https://doi.org/10.3390/ijms25010002
APA StyleBlau, K., & Gallert, C. (2024). Prophage Carriage and Genetic Diversity within Environmental Isolates of Clostridioides difficile. International Journal of Molecular Sciences, 25(1), 2. https://doi.org/10.3390/ijms25010002