
BRCA Mutations and Fertility Preservation


Abstract
:1. Introduction
2. BRCA
2.1. Heritance of Breast Cancer
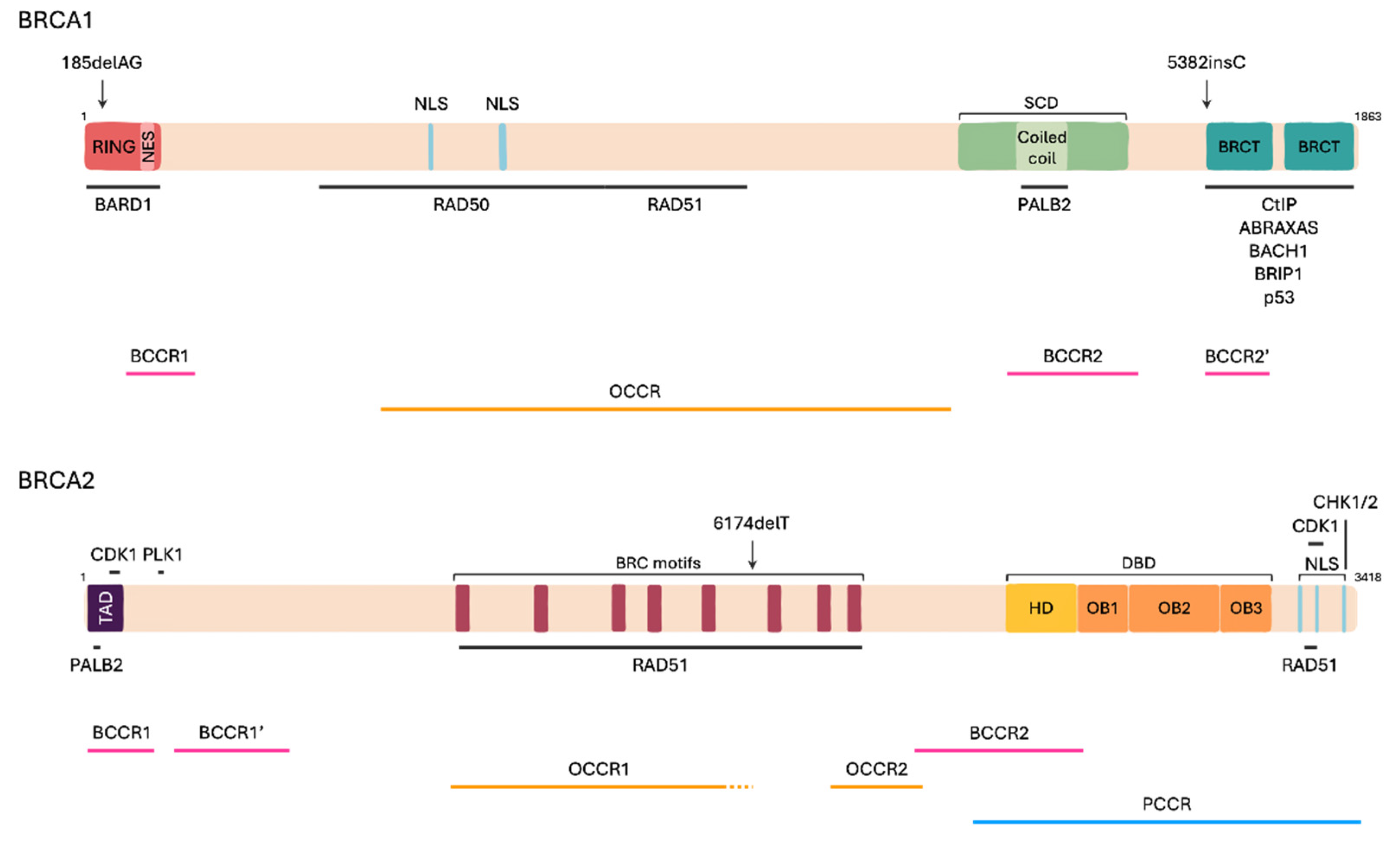
2.2. BRCA Genes Structure
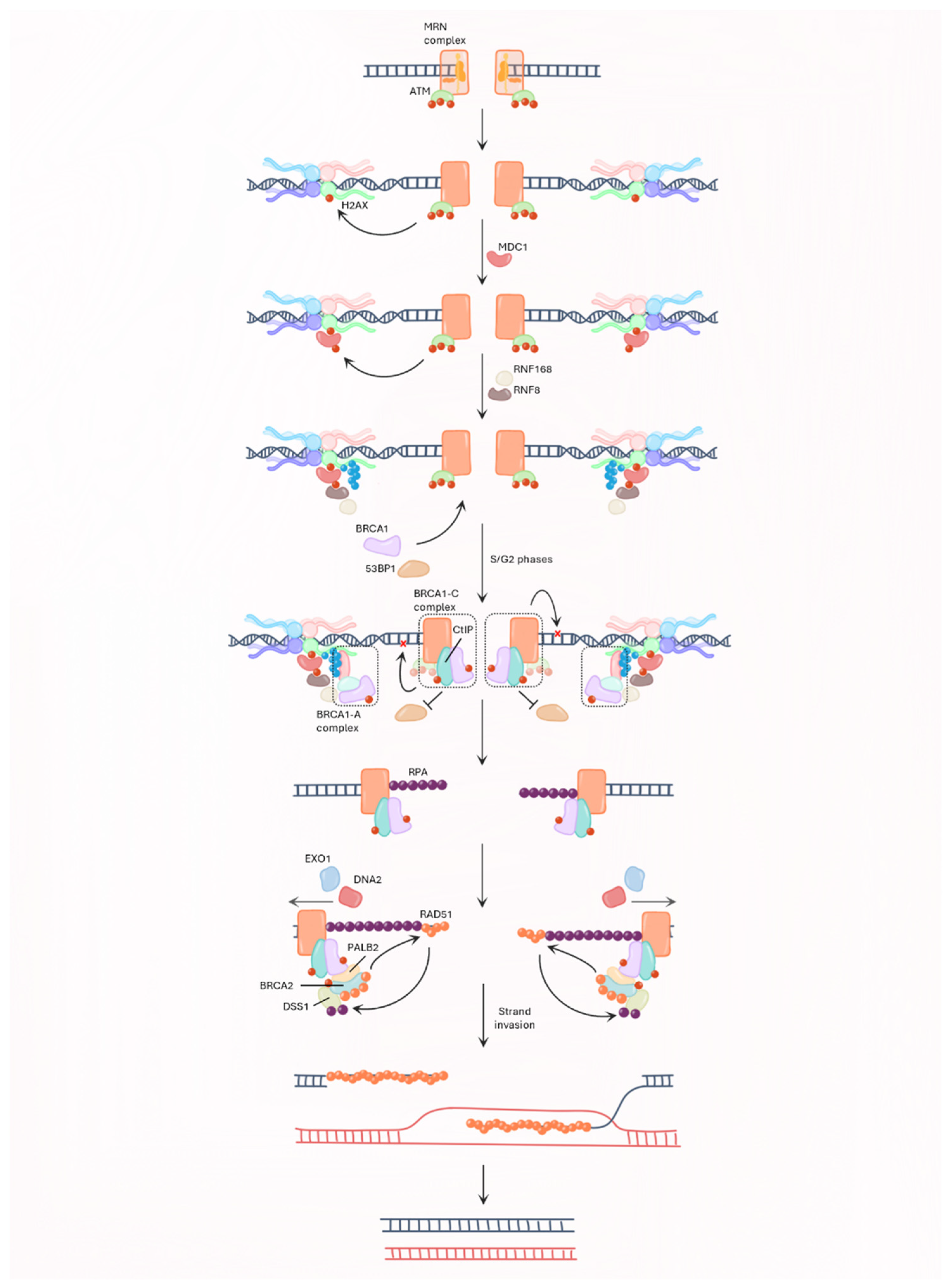
2.3. BRCA and Genome Integrity
2.4. Pathogenic Variants
3. BRCA Mutations and Fertility
3.1. Ovarian Ageing
3.2. Premature Ovarian Insufficiency
3.3. Female Fertility Preservation
3.4. BRCA Mutations and Male Infertility
4. Perspectives and Research Priorities
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer. Available online: https://www.europadonna.org/breast-cancer/ (accessed on 29 August 2023).
- Breast Cancer Facts and Statistics. 2023. Available online: https://www.breastcancer.org/facts-statistics (accessed on 29 August 2023).
- European Cancer Information System. European Commission Breast Cancer Burden in EU-27; European Commission: Brussel, Belgium, 2020. [Google Scholar]
- Breast Cancer in Young Women (Under 40). Available online: https://my.clevelandclinic.org/health/articles/16805-breast-cancer-in-young-women (accessed on 29 August 2023).
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 29 August 2023).
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primer 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.H.; Anders, C.K.; Litton, J.K.; Ruddy, K.J.; Bleyer, A. Breast Cancer in Adolescents and Young Adults. Pediatr. Blood Cancer 2018, 65, e27397. [Google Scholar] [CrossRef] [PubMed]
- Cathcart-Rake, E.J.; Ruddy, K.J.; Bleyer, A.; Johnson, R.H. Breast Cancer in Adolescent and Young Adult Women Under the Age of 40 Years. JCO Oncol. Pract. 2021, 17, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Bernstein-Molho, R.; Kaufman, B.; Wyld, L. Genetic syndromes and RT for breast cancer. In Breast Cancer Radiation Therapy: A Practical Guide for Technical Applications; Kaidar-Person, O., Meattini, I., Poortmans, P., Eds.; Springer: Cham, Switzerland, 2022; pp. 373–381. ISBN 978-3-030-91170-6. [Google Scholar]
- Buckley, K.H.; Niccum, B.A.; Maxwell, K.N.; Katona, B.W. Gastric Cancer Risk and Pathogenesis in BRCA1 and BRCA2 Carriers. Cancers 2022, 14, 5953. [Google Scholar] [CrossRef] [PubMed]
- Varol, U.; Kucukzeybek, Y.; Alacacioglu, A.; Somali, I.; Altun, Z.; Tarhan, M.O. BRCA Genes: BRCA 1 and BRCA 2. J. BUON 2018, 23, 862–866. [Google Scholar] [PubMed]
- Ricker, C. From families syndromes to genes… The first clinical and genetic characterizations of hereditary syndromes predisposing to cancer: What was the beginning? Rev. Méd. Clín. Condes 2017, 28, 482–490. [Google Scholar] [CrossRef]
- Newman, B.; Austin, M.A.; Lee, M.; King, M.C. Inheritance of Human Breast Cancer: Evidence for Autosomal Dominant Transmission in High-Risk Families. Proc. Natl. Acad. Sci. USA 1988, 85, 3044–3048. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- King, M.-C. “The Race” to Clone BRCA1. Science 2014, 343, 1462–1465. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancastert, J.; Swiftt, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the Breast Cancer Susceptibility Gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Tennen, R.I.; Laskey, S.B.; Koelsch, B.L.; McIntyre, M.H.; Tung, J.Y. Identifying Ashkenazi Jewish BRCA1/2 Founder Variants in Individuals Who Do Not Self-Report Jewish Ancestry. Sci. Rep. 2020, 10, 7669. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Lavoro, A.; Scalisi, A.; Candido, S.; Zanghì, G.; Rizzo, R.; Gattuso, G.; Caruso, G.; Libra, M.; Falzone, L. Identification of the Most Common BRCA Alterations through Analysis of Germline Mutation Databases: Is Droplet Digital PCR an Additional Strategy for the Assessment of Such Alterations in Breast and Ovarian Cancer Families? Int. J. Oncol. 2022, 60, 58. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.V.; Boehning, D. Structure-function of the tumor suppressor BRCA1. Comput. Struct. Biotechnol. J. 2012, 1, e201204005. [Google Scholar] [CrossRef] [PubMed]
- Paul, A. The Breast Cancer Susceptibility Genes (BRCA) in Breast and Ovarian Cancers. Front. Biosci. 2014, 19, 605. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Seo, J.; Wiek, C.; Hanenberg, H. Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes 2021, 12, 1034. [Google Scholar] [CrossRef]
- Lee, H. Cycling with BRCA2 from DNA Repair to Mitosis. Exp. Cell Res. 2014, 329, 78–84. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Sitz, J.; Grapton, D.; Orthwein, A. BRCA2 Functions: From DNA Repair to Replication Fork Stabilization. Endocr. Relat. Cancer 2016, 23, T1–T17. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/Ovarian Cancer Susceptibility Gene Products and Participants in DNA Double-Strand Break Repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Rothenberg, E. Preserving Genome Integrity in Human Cells via DNA Double-Strand Break Repair. Mol. Biol. Cell 2020, 31, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms Underlying Mutational Signatures in Human Cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘Complex’ Protein Involved in the Maintenance of Genomic Stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA End Resection and Its Role in DNA Replication and DSB Repair Choice in Mammalian Cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a Super Complex of BRCA1-Associated Proteins Involved in the Recognition and Repair of Aberrant DNA Structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [CrossRef]
- Ntemou, E.; Vidal, P.D.; Alexandri, C.; Van den Steen, G.; Lambertini, M.; Demeestere, I. Ovarian Toxicity of Carboplatin and Paclitaxel in Mouse Carriers of Mutation in BRIP1 Tumor Suppressor Gene. Sci. Rep. 2022, 12, 1658. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Y.; Li, W.; Yao, M.; Liu, C.; Zhang, Z.; Wang, C.; Wang, X.; Meng, K. Research Progress of the Fanconi Anemia Pathway and Premature Ovarian Insufficiency. Biol. Reprod. 2023, 109, 570–585. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 Gene: Function and Deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Leem, J.; Oh, J.S. Selective Utilization of Non-homologous End-joining and Homologous Recombination for DNA Repair during Meiotic Maturation in Mouse Oocytes. Cell Prolif. 2022, 56, e13384. [Google Scholar] [CrossRef] [PubMed]
- BRCA Exchange. Available online: https://brcaexchange.org/factsheet (accessed on 23 August 2023).
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, T.; Tischkowitz, M.; Antoniou, A.C. BRCA1 and BRCA2 Pathogenic Variants and Prostate Cancer Risk: Systematic Review and Meta-Analysis. Br. J. Cancer 2022, 126, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.L.; Busch, E.L.; Friebel, T.M.; Cronin, A.; Leslie, G.; McGuffog, L.; Adlard, J.; Agata, S.; Agnarsson, B.A.; Ahmed, M.; et al. Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res. 2020, 80, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, A. Regulation of Ovarian Follicular Development in Primates: Facts and Hypotheses. Endocr. Rev. 1996, 17, 121–155. [Google Scholar] [CrossRef]
- McGee, E.A.; Hsueh, A.J.W. Initial and Cyclic Recruitment of Ovarian Follicles*. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef]
- Broekmans, F.J.; Soules, M.R.; Fauser, B.C. Ovarian Aging: Mechanisms and Clinical Consequences. Endocr. Rev. 2009, 30, 465–493. [Google Scholar] [CrossRef]
- Vollenhoven, B.; Hunt, S. Ovarian Ageing and the Impact on Female Fertility. F1000 Res. 2018, 7, 1835. [Google Scholar] [CrossRef]
- ASRM. Committee Opinion No. 589. Female age-related fertility decline. Fertil. Steril. 2014, 101, 633–634. [Google Scholar] [CrossRef]
- Nelson, S.M.; Davis, S.R.; Kalantaridou, S.; Lumsden, M.A.; Panay, N.; Anderson, R.A. Anti-Müllerian Hormone for the Diagnosis and Prediction of Menopause: A Systematic Review. Hum. Reprod. Update 2023, 29, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Ménopause. Available online: https://www.who.int/fr/news-room/fact-sheets/detail/menopause (accessed on 11 September 2023).
- Melnick, A. Menopause. In Problem-Focused Reproductive Endocrinology and Infertility; Chung, P.H., Rosenwaks, Z., Eds.; Contemporary Endocrinology; Springer International Publishing: Cham, Switzerland, 2023; pp. 245–251. ISBN 978-3-031-19443-6. [Google Scholar]
- Kruszyńska, A.; Słowińska-Srzednicka, J. Anti-Müllerian Hormone (AMH) as a Good Predictor of Time of Menopause. Przegla̜d Menopauzalny Menopause Rev. 2017, 16, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Kunicki, M.; Kruszewska, J.; Skórska, J.; Laudy-Wiaderny, H.; Wrona, M.; Smolarczyk, R. Does the Value of FSH Predict Severity of Metabolic Complications in Females with POI? J. Clin. Med. 2022, 11, 2024. [Google Scholar] [CrossRef] [PubMed]
- Park, S.U.; Walsh, L.; Berkowitz, K.M. Mechanisms of Ovarian Aging. Reproduction 2021, 162, R19–R33. [Google Scholar] [CrossRef] [PubMed]
- Vanni, V.S.; Campo, G.; Cioffi, R.; Papaleo, E.; Salonia, A.; Viganò, P.; Lambertini, M.; Candiani, M.; Meirow, D.; Orvieto, R. The Neglected Members of the Family: Non- BRCA Mutations in the Fanconi Anemia/BRCA Pathway and Reproduction. Hum. Reprod. Update 2022, 28, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Winship, A.L.; Stringer, J.M.; Liew, S.H.; Hutt, K.J. The Importance of DNA Repair for Maintaining Oocyte Quality in Response to Anti-Cancer Treatments, Environmental Toxins and Maternal Ageing. Hum. Reprod. Update 2018, 24, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Turan, V.; Oktay, K. BRCA-Related ATM-Mediated DNA Double-Strand Break Repair and Ovarian Aging. Hum. Reprod. Update 2020, 26, 43–57. [Google Scholar] [CrossRef]
- Pailas, A.; Niaka, K.; Zorzompokou, C.; Marangos, P. The DNA Damage Response in Fully Grown Mammalian Oocytes. Cells 2022, 11, 798. [Google Scholar] [CrossRef]
- Titus, S.; Li, F.; Stobezki, R.; Akula, K.; Unsal, E.; Jeong, K.; Dickler, M.; Robson, M.; Moy, F.; Goswami, S.; et al. Impairment of BRCA1-Related DNA Double Strand Break Repair Leads to Ovarian Aging in Mice and Humans. Sci. Transl. Med. 2013, 5, 172ra21. [Google Scholar] [CrossRef]
- Govindaraj, V.; Keralapura Basavaraju, R.; Rao, A.J. Changes in the Expression of DNA Double Strand Break Repair Genes in Primordial Follicles from Immature and Aged Rats. Reprod. Biomed. Online 2015, 30, 303–310. [Google Scholar] [CrossRef]
- Govindaraj, V.; Krishnagiri, H.; Chakraborty, P.; Vasudevan, M.; Rao, A.J. Age-Related Changes in Gene Expression Patterns of Immature and Aged Rat Primordial Follicles. Syst. Biol. Reprod. Med. 2017, 63, 37–48. [Google Scholar] [CrossRef] [PubMed]
- He, D.-J.; Wang, L.; Zhang, Z.-B.; Guo, K.; Li, J.-Z.; He, X.-C.; Cui, Q.-H.; Zheng, P. Maternal Gene Ooep May Participate in Homologous Recombination-Mediated DNA Double-Strand Break Repair in Mouse Oocytes. Zool. Res. 2018, 39, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Shen, W.; Yan, W.; Zhou, S.; Cheng, J.; Pan, G.; Wu, M.; Ma, L.; Luo, A.; Wang, S. What Changed on the Folliculogenesis in the Process of Mouse Ovarian Aging? BioMed. Res. Int. 2019, 2019, 3842312. [Google Scholar] [CrossRef] [PubMed]
- Horta, F.; Catt, S.; Ramachandran, P.; Vollenhoven, B.; Temple-Smith, P. Female Ageing Affects the DNA Repair Capacity of Oocytes in IVF Using a Controlled Model of Sperm DNA Damage in Mice. Hum. Reprod. 2020, 35, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Horta, F.; Ravichandran, A.; Catt, S.; Vollenhoven, B.; Temple-Smith, P. Ageing and Ovarian Stimulation Modulate the Relative Levels of Transcript Abundance of Oocyte DNA Repair Genes during the Germinal Vesicle-Metaphase II Transition in Mice. J. Assist. Reprod. Genet. 2021, 38, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Bilotto, S.; Boni, R.; Russo, G.L.; Lioi, M.B. Meiosis Progression and Donor Age Affect Expression Profile of DNA Repair Genes in Bovine Oocytes. Zygote 2015, 23, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, X.; Yuan, J.; Li, L.; Zhang, D.; Guo, X.; Liu, L.; Zhang, S. Age-Specific Gene Expression Profiles of Rhesus Monkey Ovaries Detected by Microarray Analysis. BioMed. Res. Int. 2015, 2015, 625192. [Google Scholar] [CrossRef] [PubMed]
- Grøndahl, M.L.; Yding Andersen, C.; Bogstad, J.; Nielsen, F.C.; Meinertz, H.; Borup, R. Gene Expression Profiles of Single Human Mature Oocytes in Relation to Age. Hum. Reprod. 2010, 25, 957–968. [Google Scholar] [CrossRef]
- Bothun, A.M.; Gao, Y.; Takai, Y.; Ishihara, O.; Seki, H.; Karger, B.; Tilly, J.L.; Woods, D.C. Quantitative Proteomic Profiling of the Human Ovary from Early to Mid-Gestation Reveals Protein Expression Dynamics of Oogenesis and Folliculogenesis. Stem Cells Dev. 2018, 27, 723–735. [Google Scholar] [CrossRef]
- Yuan, L.; Yin, P.; Yan, H.; Zhong, X.; Ren, C.; Li, K.; Chin Heng, B.; Zhang, W.; Tong, G. Single-cell Transcriptome Analysis of Human Oocyte Ageing. J. Cell. Mol. Med. 2021, 25, 6289–6303. [Google Scholar] [CrossRef]
- Smits, M.A.J.; Janssens, G.E.; Goddijn, M.; Hamer, G.; Houtkooper, R.H.; Mastenbroek, S. Longevity Pathways Are Associated with Human Ovarian Ageing. Hum. Reprod. Open 2021, 2021, hoab020. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Ma, P.; Zhu, W.; Schultz, R.M. Age-Associated Increase in Aneuploidy and Changes in Gene Expression in Mouse Eggs. Dev. Biol. 2008, 316, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, V.; Krishnagiri, H.; Chauhan, M.S.; Rao, A.J. BRCA-1 Gene Expression and Comparative Proteomic Profile of Primordial Follicles from Young and Adult Buffalo (Bubalus Bubalis) Ovaries. Anim. Biotechnol. 2017, 28, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Titus, S.; Moy, F.; Ginsburg, E.S.; Oktay, K. Ovarian Aging in Women with BRCA Germline Mutations. J. Clin. Endocrinol. Metab. 2017, 102, 3839–3847. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.-L.; Wang, S.; Ye, X.; Zhang, D. Effects of BRCA Gene Mutation on Female Reproductive Potential: A Systematic Review. Maturitas 2020, 137, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Rzepka-Górska, I.; Tarnowski, B.; Chudecka-Głaz, A.; Górski, B.; Zielińska, D.; Tołoczko-Grabarek, A. Premature Menopause in Patients with BRCA1 Gene Mutation. Breast Cancer Res. Treat. 2006, 100, 59–63. [Google Scholar] [CrossRef]
- Rudnicka, E.; Kruszewska, J.; Klicka, K.; Kowalczyk, J.; Grymowicz, M.; Skórska, J.; Pięta, W.; Smolarczyk, R. Premature Ovarian Insufficiency—Aetiopathology, Epidemiology, and Diagnostic Evaluation. Przegla̜d Menopauzalny Menopause Rev. 2018, 17, 105–108. [Google Scholar] [CrossRef]
- Vujovic, S.; Ivovic, M.; Tancic Gajic, M.; Marina, L.; Dragojevic-Dikic, S. Premature ovarian insufficiency. In Amenorrhea; Genazzani, A.R., Hirschberg, A.L., Genazzani, A.D., Nappi, R., Vujovic, S., Eds.; ISGE Series; Springer: Cham, Switzerland, 2023; pp. 157–178. ISBN 978-3-031-22377-8. [Google Scholar]
- Committee on Adolescent Health Care. Primary Ovarian Insufficiency in Adolescents and Young Women; The American College of Obstetricians and Gynecologists: Washington, DC, USA, 2014. [Google Scholar]
- Bedoschi, G.; Navarro, P.A.; Oktay, K. Chemotherapy-Induced Damage to Ovary: Mechanisms and Clinical Impact. Future Oncol. 2016, 12, 2333–2344. [Google Scholar] [CrossRef]
- Meirow, D.; Biederman, H.; Anderson, R.A.; Wallace, W.H.B. Toxicity of Chemotherapy and Radiation on Female Reproduction. Clin. Obstet. Gynecol. 2010, 53, 727–739. [Google Scholar] [CrossRef]
- Spears, N.; Lopes, F.; Stefansdottir, A.; Rossi, V.; De Felici, M.; Anderson, R.A.; Klinger, F.G. Ovarian Damage from Chemotherapy and Current Approaches to Its Protection. Hum. Reprod. Update 2019, 25, 673–693. [Google Scholar] [CrossRef]
- Zhang, X.; Niu, J.; Che, T.; Zhu, Y.; Zhang, H.; Qu, J. Fertility Preservation in BRCA Mutation Carriers—Efficacy and Safety Issues: A Review. Reprod. Biol. Endocrinol. 2020, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Oktay, K.H.; Turan, V. Ovarian Stimulation and Oocyte Cryopreservation in Females with Cancer. Curr. Opin. Oncol. 2023, 35, 412–419. [Google Scholar] [CrossRef] [PubMed]
- El Moujahed, L.; Philis, R.; Grynberg, M.; Laot, L.; Mur, P.; Amsellem, N.; Mayeur, A.; Benoit, A.; Rakrouki, S.; Sifer, C.; et al. Response to Ovarian Stimulation for Urgent Fertility Preservation before Gonadotoxic Treatment in BRCA-Pathogenic-Variant-Positive Breast Cancer Patients. Cancers 2023, 15, 895. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, T.C.; Derks-Smeets, I.A.P.; Bos, A.M.E.; Oosterwijk, J.C.; van Golde, R.J.; de Die-Smulders, C.E.; van der Kolk, L.E.; van Zelst-Stams, W.A.G.; Velthuizen, M.E.; Hoek, A.; et al. Serum AMH Levels in Healthy Women from BRCA1/2 Mutated Families: Are They Reduced? Hum. Reprod. Oxf. Engl. 2016, 31, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Turan, V.; Lambertini, M.; Lee, D.-Y.; Wang, E.; Clatot, F.; Karlan, B.Y.; Demeestere, I.; Bang, H.; Oktay, K. Association of Germline BRCA Pathogenic Variants With Diminished Ovarian Reserve: A Meta-Analysis of Individual Patient-Level Data. J. Clin. Oncol. 2021, 39, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Di Micco, R.; Zuber, V.; Taghavi, K.; Bianchini, G.; Bellaminutti, S.; Meani, F.; Graffeo, R.; Candiani, M.; Mueller, M.D.; et al. Ovarian Reserve of Women with and without BRCA Pathogenic Variants: A Systematic Review and Meta-Analysis. Breast Off. J. Eur. Soc. Mastology 2021, 60, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Kim, T.H.; Han, J.Y.; Kim, S.K.; Lee, J.R.; Jee, U.C.; Suh, C.S.; Kim, S.H. Impact of BRCA Mutations and Hormone Receptor Status on Reproductive Potential in Breast Cancer Patients Undergoing Fertility Preservation. Gynecol. Endocrinol. 2022, 38, 227–230. [Google Scholar] [CrossRef]
- Drechsel Katja, C.E.; van Tilborg Theodora, C.; Eijkemans Marinus, J.C.; Lentjes Eef, G.W.M.; Homminga Irene Goddijn Mariette van Golde Ron, J.T.; Verpoest Willem Lichtenbelt Klaske, D.; Broekmans Frank, J.M.; Bos Anna, M.E. The Impact of BRCA1- and BRCA2 Mutations on Ovarian Reserve Status. Reprod. Sci. 2022, 30, 270–282. [Google Scholar] [CrossRef]
- Prokurotaite, E.; Condorelli, M.; Dechene, J.; Bouziotis, J.; Lambertini, M.; Demeestere, I. Impact of Breast Cancer and Germline BRCA Pathogenic Variants on Fertility Preservation in Young Women. Life 2023, 13, 930. [Google Scholar] [CrossRef]
- Michaelson-Cohen, R.; Mor, P.; Srebnik, N.; Beller, U.; Levy-Lahad, E.; Eldar-Geva, T. BRCA Mutation Carriers Do Not Have Compromised Ovarian Reserve. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2014, 24, 233–237. [Google Scholar] [CrossRef]
- Phillips, K.-A.; Collins, I.M.; Milne, R.L.; McLachlan, S.A.; Friedlander, M.; Hickey, M.; Stern, C.; Hopper, J.L.; Fisher, R.; Kannemeyer, G.; et al. Anti-Müllerian Hormone Serum Concentrations of Women with Germline BRCA1 or BRCA2 Mutations. Hum. Reprod. Oxf. Engl. 2016, 31, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Sammel, M.D.; Domchek, S.; Schanne, A.; Prewitt, M.; Gracia, C. Antimüllerian Hormone Levels Are Lower in BRCA2 Mutation Carriers. Fertil. Steril. 2017, 107, 1256–1265.e6. [Google Scholar] [CrossRef] [PubMed]
- Lambertini, M.; Goldrat, O.; Ferreira, A.R.; Dechene, J.; Azim Jr, H.A.; Desir, J.; Delbaere, A.; t’Kint De Roodenbeke, M.-D.; De Azambuja, E.; Ignatiadis, M.; et al. Reproductive Potential and Performance of Fertility Preservation Strategies in BRCA-Mutated Breast Cancer Patients. Ann. Oncol. 2018, 29, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, M.; Dagher Hayeck, B.; Papanikolaou, E.G.; Sifer, C.; Sermondade, N.; Sonigo, C. BRCA1/2 Gene Mutations Do Not Affect the Capacity of Oocytes from Breast Cancer Candidates for Fertility Preservation to Mature in Vitro. Hum. Reprod. 2019, 34, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Gunnala, V.; Fields, J.; Irani, M.; D’Angelo, D.; Xu, K.; Schattman, G.; Rosenwaks, Z. BRCA Carriers Have Similar Reproductive Potential at Baseline to Noncarriers: Comparisons in Cancer and Cancer-Free Cohorts Undergoing Fertility Preservation. Fertil. Steril. 2019, 111, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Son, K.-A.; Lee, D.-Y.; Choi, D. Association of BRCA Mutations and Anti-Müllerian Hormone Level in Young Breast Cancer Patients. Front. Endocrinol. 2019, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.; Fernandez-Gonzalez, S.; Calvo, I.; Climent, M.; Peñafiel, J.; Feliubadaló, L.; Teulé, A.; Lázaro, C.; Brunet, J.M.; Candás-Estébanez, B.; et al. Assessment of Ovarian Reserve and Reproductive Outcomes in BRCA1 or BRCA2 Mutation Carriers. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2020, 30, 83–88. [Google Scholar] [CrossRef]
- Pavone, M.E.; Hirshfeld-Cytron, J.; Tingen, C.; Thomas, C.; Thomas, J.; Lowe, M.P.; Schink, J.C.; Woodruff, T.K. Human Ovarian Tissue Cortex Surrounding Benign and Malignant Lesions. Reprod. Sci. 2014, 21, 582–589. [Google Scholar] [CrossRef]
- Fleming, R.; Seifer, D.B.; Frattarelli, J.L.; Ruman, J. Assessing Ovarian Response: Antral Follicle Count versus Anti-Müllerian Hormone. Reprod. Biomed. Online 2015, 31, 486–496. [Google Scholar] [CrossRef]
- Miller, K.D.; Fidler-Benaoudia, M.; Keegan, T.H.; Hipp, H.S.; Jemal, A.; Siegel, R.L. Cancer Statistics for Adolescents and Young Adults, 2020. CA Cancer J. Clin. 2020, 70, 443–459. [Google Scholar] [CrossRef]
- Lambertini, M.; Peccatori, F.A.; Demeestere, I.; Amant, F.; Wyns, C.; Stukenborg, J.-B.; Paluch-Shimon, S.; Halaska, M.J.; Uzan, C.; Meissner, J.; et al. Fertility Preservation and Post-Treatment Pregnancies in Post-Pubertal Cancer Patients: ESMO Clinical Practice Guidelines. Ann. Oncol. 2020, 31, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Paluch-Shimon, S.; Cardoso, F.; Partridge, A.H.; Abulkhair, O.; Azim, H.A.; Bianchi-Micheli, G.; Cardoso, M.-J.; Curigliano, G.; Gelmon, K.A.; Harbeck, N.; et al. ESO–ESMO 4th International Consensus Guidelines for Breast Cancer in Young Women (BCY4). Ann. Oncol. 2020, 31, 674–696. [Google Scholar] [CrossRef] [PubMed]
- Winship, A.L.; Griffiths, M.; Lliberos Requesens, C.; Sarma, U.; Phillips, K.-A.; Hutt, K.J. The PARP Inhibitor, Olaparib, Depletes the Ovarian Reserve in Mice: Implications for Fertility Preservation. Hum. Reprod. 2020, 35, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Takae, S.; Shiraishi, E.; Shinya, K.; Igualada, A.J.; Suzuki, N. Poly (ADP-Ribose) Polymerase Inhibitor Exposure Reduces Ovarian Reserve Followed by Dysfunction in Granulosa Cells. Sci. Rep. 2020, 10, 17058. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Amant, F.; Braat, D.; D’Angelo, A.; Chuva de Sousa Lopes, S.M.; Demeestere, I.; Dwek, S.; Frith, L.; Lambertini, M.; Maslin, C.; et al. ESHRE Guideline: Female Fertility Preservation. Hum. Reprod. Open 2020, 2020, hoaa052. [Google Scholar] [CrossRef] [PubMed]
- Cha, K.Y.; Chung, H.M.; Lim, J.M.; Ko, J.J.; Han, S.Y.; Choi, D.H.; Yoon, T.K. Freezing Immature Oocytes. Mol. Cell. Endocrinol. 2000, 169, 43–47. [Google Scholar] [CrossRef]
- Iussig, B.; Maggiulli, R.; Fabozzi, G.; Bertelle, S.; Vaiarelli, A.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. A Brief History of Oocyte Cryopreservation: Arguments and Facts. Acta Obstet. Gynecol. Scand. 2019, 98, 550–558. [Google Scholar] [CrossRef]
- Wong, K.M.; Mastenbroek, S.; Repping, S. Cryopreservation of Human Embryos and Its Contribution to in Vitro Fertilization Success Rates. Fertil. Steril. 2014, 102, 19–26. [Google Scholar] [CrossRef]
- Fraison, E.; Huberlant, S.; Labrune, E.; Cavalieri, M.; Montagut, M.; Brugnon, F.; Courbiere, B. Live Birth Rate after Female Fertility Preservation for Cancer or Haematopoietic Stem Cell Transplantation: A Systematic Review and Meta-Analysis of the Three Main Techniques; Embryo, Oocyte and Ovarian Tissue Cryopreservation. Hum. Reprod. Oxf. Engl. 2022, 38, 489–502. [Google Scholar] [CrossRef]
- Diaz, A.A.; Kubo, H.; Handa, N.; Hanna, M.; Laronda, M.M. A Systematic Review of Ovarian Tissue Transplantation Outcomes by Ovarian Tissue Processing Size for Cryopreservation. Front. Endocrinol. 2022, 13, 918899. [Google Scholar] [CrossRef]
- Corrado, G.; Marchetti, C.; Trozzi, R.; Scambia, G.; Fagotti, A. Fertility Preservation in Patients with BRCA Mutations or Lynch Syndrome. Int. J. Gynecol. Cancer 2021, 31, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, M.; Bruzzone, M.; Ceppi, M.; Ferrari, A.; Grinshpun, A.; Hamy, A.S.; de Azambuja, E.; Carrasco, E.; Peccatori, F.A.; Di Meglio, A.; et al. Safety of Assisted Reproductive Techniques in Young Women Harboring Germline Pathogenic Variants in BRCA1/2 with a Pregnancy after Prior History of Breast Cancer. ESMO Open 2021, 6, 100300. [Google Scholar] [CrossRef] [PubMed]
- Peccatori, F.A.; Mangili, G.; Bergamini, A.; Filippi, F.; Martinelli, F.; Ferrari, F.; Noli, S.; Rabaiotti, E.; Candiani, M.; Somigliana, E. Fertility Preservation in Women Harboring Deleterious BRCA Mutations: Ready for Prime Time? Hum. Reprod. 2018, 33, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Ghunaim, S.; Ghazeeri, G.; Khalife, D.; Azim, H.A. Fertility Preservation in Patients with BRCA Mutation. ecancermedicalscience 2020, 14, 1033. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Turan, V.; Oktay, K.H. Safety of Controlled Ovarian Stimulation with Gonadotropins and Letrozole in Women with BRCA Mutations. Fertil. Steril. 2015, 104, e265. [Google Scholar] [CrossRef]
- Bonardi, B.; Massarotti, C.; Bruzzone, M.; Goldrat, O.; Mangili, G.; Anserini, P.; Spinaci, S.; Arecco, L.; Del Mastro, L.; Ceppi, M.; et al. Efficacy and Safety of Controlled Ovarian Stimulation with or Without Letrozole Co-Administration for Fertility Preservation: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 574669. [Google Scholar] [CrossRef] [PubMed]
- Turan, V.; Bedoschi, G.; Emirdar, V.; Moy, F.; Oktay, K. Ovarian Stimulation in Patients with Cancer: Impact of Letrozole and BRCA Mutations on Fertility Preservation Cycle Outcomes. Reprod. Sci. 2018, 25, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Conduit, C.; Milne, R.L.; Friedlander, M.L.; Phillips, K.-A. Bilateral Salpingo-Oophorectomy and Breast Cancer Risk for BRCA1 and BRCA2 Mutation Carriers: Assessing the Evidence. Cancer Prev. Res. 2021, 14, 983–994. [Google Scholar] [CrossRef]
- Ibrahim, M.; Yadav, S.; Ogunleye, F.; Zakalik, D. Male BRCA Mutation Carriers: Clinical Characteristics and Cancer Spectrum. BMC Cancer 2018, 18, 179. [Google Scholar] [CrossRef]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J. Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, S.; Peterson, S.; Patel, D.S.; Huo, Y.; Cai, H.; Bowman-Colin, C.; Miller, S.; Ludwig, T.; Ganesan, S.; Bhaumik, M.; et al. Male Fertility Defect Associated with Disrupted BRCA1-PALB2 Interaction in Mice. J. Biol. Chem. 2014, 289, 24617–24629. [Google Scholar] [CrossRef] [PubMed]
- Kabartan, E.; Gunes, S.; Arslan, M.A.; Asci, R. Investigating the Relationship between BRCA1 and BRCA2 Genes Methylation Profile and Sperm DNA Fragmentation in Infertile Men. Andrologia 2019, 51, e13308. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, H.; Grievink, L.S.; Mano, R.; Ber, Y.; Ozalbo, R.; Tuval, S.; Baniel, J.; Margel, D. Is There a Difference in Testosterone Levels and Its Regulators in Men Carrying BRCA Mutations? Oncotarget 2017, 8, 103843–103850. [Google Scholar] [CrossRef]
- Dias, A.; Brook, M.N.; Bancroft, E.K.; Page, E.C.; Chamberlain, A.; Saya, S.; Amin, J.; Mikropoulos, C.; Taylor, N.; Myhill, K.; et al. Serum Testosterone and Prostate Cancer in Men with Germline BRCA1/2 Pathogenic Variants. BJUI Compass 2023, 4, 361–373. [Google Scholar] [CrossRef]
- Vakalopoulos, I.; Dimou, P.; Anagnostou, I.; Zeginiadou, T. Impact of Cancer and Cancer Treatment on Male Fertility. Hormones 2015, 14, 579–589. [Google Scholar] [CrossRef]
- Tharmalingam, M.D.; Matilionyte, G.; Wallace, W.H.B.; Stukenborg, J.-B.; Jahnukainen, K.; Oliver, E.; Goriely, A.; Lane, S.; Guo, J.; Cairns, B.; et al. Cisplatin and Carboplatin Result in Similar Gonadotoxicity in Immature Human Testis with Implications for Fertility Preservation in Childhood Cancer. BMC Med. 2020, 18, 374. [Google Scholar] [CrossRef]
- Tran, K.T.D.; Valli-Pulaski, H.; Colvin, A.; Orwig, K.E. Male Fertility Preservation and Restoration Strategies for Patients Undergoing Gonadotoxic Therapies. Biol. Reprod. 2022, 107, 382–405. [Google Scholar] [CrossRef]
- Welcsh, P.L.; King, M.-C. BRCA1 and BRCA2 and the Genetics of Breast and Ovarian Cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Gowen, L.C.; Johnson, B.L.; Latour, A.M.; Sulik, K.K.; Koller, B.H. Brca1 Deficiency Results in Early Embryonic Lethality Characterized by Neuroepithelial Abnormalities. Nat. Genet. 1996, 12, 191–194. [Google Scholar] [CrossRef]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic Lethality and Radiation Hypersensitivity Mediated by Rad51 in Mice Lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Samavat, H.; Kurzer, M.S. Estrogen Metabolism and Breast Cancer. Cancer Lett. 2015, 356, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA Polymerase II Pausing Mitigates BRCA1-Associated R-Loop Accumulation and Tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed]
- Winship, A.L.; Willson, C.; Hansen, K.R.; Hutt, K.J.; Hickey, M. Do BRCA1 and BRCA2 Gene Mutation Carriers Have a Reduced Ovarian Reserve? Protocol for a Prospective Observational Study. BMJ Open 2019, 9, e033810. [Google Scholar] [CrossRef] [PubMed]
- Picton, H.M. Activation of Follicle Development: The Primordial Follicle. Theriogenology 2001, 55, 1193–1210. [Google Scholar] [CrossRef] [PubMed]
- Monniaux, D.; Clément, F.; Dalbiès-Tran, R.; Estienne, A.; Fabre, S.; Mansanet, C.; Monget, P. The Ovarian Reserve of Primordial Follicles and the Dynamic Reserve of Antral Growing Follicles: What Is the Link?1. Biol. Reprod. 2014, 90, 85. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, T.C.; Broekmans, F.J.; Pijpe, A.; Schrijver, L.H.; Mooij, T.M.; Oosterwijk, J.C.; Verhoef, S.; Gómez Garcia, E.B.; van Zelst-Stams, W.A.; Adank, M.A.; et al. Do BRCA1/2 Mutation Carriers Have an Earlier Onset of Natural Menopause? Menopause 2016, 23, 903. [Google Scholar] [CrossRef]
- Kufel-Grabowska, J.; Podolak, A.; Maliszewski, D.; Bartoszkiewicz, M.; Ramlau, R.; Lukaszuk, K. Fertility Counseling in BRCA1/2-Mutated Women with Breast Cancer and Healthy Individuals. J. Clin. Med. 2022, 11, 3996. [Google Scholar] [CrossRef]
- Roti Roti, E.C.; Leisman, S.K.; Abbott, D.H.; Salih, S.M. Acute Doxorubicin Insult in the Mouse Ovary Is Cell- and Follicle-Type Dependent. PLoS ONE 2012, 7, e42293. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Zerafa, N.; Liew, S.H.; Findlay, J.K.; Hickey, M.; Hutt, K.J. Cisplatin- and Cyclophosphamide-Induced Primordial Follicle Depletion Is Caused by Direct Damage to Oocytes. Mol. Hum. Reprod. 2019, 25, 433–444. [Google Scholar] [CrossRef]
- Nguyen, Q.-N.; Zerafa, N.; Findlay, J.K.; Hickey, M.; Hutt, K.J. DNA Repair in Primordial Follicle Oocytes Following Cisplatin Treatment. J. Assist. Reprod. Genet. 2021, 38, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, R.; Heytens, E.; Darzynkiewicz, Z.; Oktay, K. Mechanisms of Chemotherapy-Induced Human Ovarian Aging: Double Strand DNA Breaks and Microvascular Compromise. Aging 2011, 3, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Oktay, K.H.; Bedoschi, G.; Goldfarb, S.B.; Taylan, E.; Titus, S.; Palomaki, G.E.; Cigler, T.; Robson, M.; Dickler, M.N. Increased Chemotherapy-Induced Ovarian Reserve Loss in Women with Germline BRCA Mutations Due to Oocyte Deoxyribonucleic Acid Double Strand Break Repair Deficiency. Fertil. Steril. 2020, 113, 1251–1260.e1. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Study | Design | Results |
|---|---|---|
| Anti-mullerian hormone (AMH) | ||
| Titus et al. (2013) [68] | 24 BRCA+ patients, 60 control patients | BRCA+ vs. control: lower AMH levels (1.22 ± 0.92 ng/mL vs. 2.23 ± 1.56 ng/mL; p < 0.0001) |
| Michaelson-Cohen et al. (2014) [101] | 41 BRCA+ patients, 324 control patients | BRCA+ vs. control: similar AMH levels (2.71 ± 0.59 ng/mL vs. 2.02 ± 0.12 ng/mL; p = 0.27) |
| Phillips et al. (2016) [102] | 172 BRCA1+ patients vs. 216 control patients for known BRCA1 mutation and 147 BRCA2+ patients vs. 158 control patients for known BRCA2 mutation | BRCA1+ vs. control: 25% lower AMH levels (exp(β) = 0.75; 95% CI = 0.59–0.95; p = 0.02) BRCA2+ vs. control: similar AMH levels (exp(β) = 0.99; 95% CI = 0.77–1.27; p = 0.94) |
| van Tilborg et al. (2016) [95] | 124 BRCA+ patients, 131 control patients | BRCA+ vs. control: similar AMH levels (1.90 [0.11–19.00] µg/L vs. 1.80 [0.11–10.00] µg/L; p = 0.34) |
| Johnson et al. (2017) [103] | 55 BRCA1+, 50 BRCA2+; 64 control patients | BRCA1+ vs. control: similar AMH levels (geometric mean ratio: 1.00; 95% CI 0.7–1.44; p = 0.999) BRCA2+ vs. control: 33% lower AMH levels (geometric mean ratio: 0.67; 95% CI 0.47–0.94; p = 0.037) |
| Lambertini et al. (2018) [104] | 25 BRCA+ patients, 60 control patients | BRCA+ vs. control: similar AMH levels (1.8 [1.0–2.7] µg/L vs. 2.6 [1.5–4.1] µg/L; p = 0.109) |
| Grynberg et al. (2019) [105] | 52 BRCA+ patients, 277 control patients | BRCA+ vs. control: similar AMH levels (3.6 ± 2.9 ng/mL vs. 4.1 ± 3.6 ng/mL; p = 0.3) |
| Gunnala et al. (2019) [106] | 38 BRCA+ BC, 53 control BC and 19 BRCA+, 600 control | BC-BRCA+ vs. control: similar AMH levels (2.6 ± 2.1 ng/mL vs. 2.4 ± 2.4 ng/mL; p = 0.915) Cancer-free-BRCA+ vs. control: similar AMH levels (3.2 ± 2.2 ng/mL vs. 2.3 ± 2.2 ng/mL; p = 0.403) |
| Son et al. (2019) [107] | 52 BRCA+ patients, 264 control patients | BRCA+ vs. control: lower AMH levels (2.60 ng/mL vs. 3.85 ng/mL; p = 0.004) |
| Ponce et al. (2020) [108] | 32 BRCA1+ patients, 37 BRCA2+, 66 control patients | BRCA1+ vs. BRCA2+ vs. control: similar AMH levels (3 ± 2.27 ng/mL vs. 2.54 ± 2.07 ng/mL vs. 2.27 ± 2.03 ng/mL; p = 0.28) but once adjusted by age showed lower AMH levels in BRCA2+ patients (20.2% vs. 23.5% BRCA1+ vs. 28.4% control) |
| Gasparri et al. (2021) [97] | 147 BRCA+ patients, 405 control patients (age under 42 years) | BRCA+ vs. control: lower AMH levels (odds ratio: −0.73 [−1.12, −0.35]; p = 0.10]; p = 0.0002) |
| Turan et al. (2021) [96] | 246 BRCA+ patients, 578 control patients | BRCA+ vs. control: lower AMH levels (23% lower; 95% CI, 4 to 38; p = 0.02) |
| Drechsel et al. (2022) [99] | 36 BRCA+ patients, 126 control patients | BRCA+ vs. control: similar AMH levels (2.40 [1.80–3.00] ng/mL vs. 2.15 [1.30–3.40] ng/mL; p = 0.45) |
| Kim et al. (2022) [98] | 39 BRCA+ patients, 20 control patients | BRCA+ vs. control: similar AMH levels (4.2 ± 3.6 ng/mL vs. 5.3 ± 3.5 ng/mL; p = 0.173) |
| El Moujahed et al. (2023) [94] | 57 BRCA+ patients, 254 control patients | BRCA+ vs. control: lower AMH levels (1.6 [0.8–2.9] ng/mL vs. 2.4 [1.4–3.7] ng/mL; p = 0.02) |
| Prokurotaite et al. (2023) [100] | 20 BRCA+ patients with BC, 10 BRCA+ without BC, 55 control patients | BRCA+ BC vs. BRCA+ without BC vs. control: similar AMH levels (1.7 [0.2–4.7] µg/L vs. 1.8 [0.5–8.3] µg/L vs. 2.3 [0.3–13] µg/L; p = 0.22) |
| Follicle density | ||
| Pavone et al. (2014) [109] | 35 risk-reducing surgery of which 15 BRCA+, 35 control patients (physiological findings) | BRCA+ vs. control: lower number of follicles per slide (15.4 vs. 23.3; p < 0.05) |
| Lin et al. (2017) [83] | 13 BRCA1+ patients, 5 BRCA2+ patients, 12 control patients | BRCA+ vs. control: lower number of PFs per mm3 (11.2 ± 6.7 vs. 44.18 ± 6.1; p = 0.0002) BRCA1+ and BRCA2+ vs. control: p = 0.0001 and p = 0.0003 |
| Lambertini et al. (2018) [104] | 19 BRCA+ patients, 53 control patients | BRCA+ vs. control: lower number of oocytes per mm2 (0.33 vs. 0.78; p = 0.153) |
| Antral follicle count (AFC) | ||
| Grynberg et al. (2019) [105] | 52 BRCA+ patients, 277 control patients | BRCA+ vs. control: similar AFC (3.6 ± 2.9 vs. 4.1 ± 3.6; p = 0.3) |
| Gunnala et al. (2019) [106] | 38 BRCA+ BC, 53 control BC and 19 BRCA+ cancer-free, 600 control cancer-free | BC-BRCA+ vs. control: similar AFC (15.2 ± 5.0 vs. 13.9 ± 6.3; p = 0.757) Cancer-free-BRCA+ vs. control: higher AFC (16.3 ± 3.9 vs. 12.2 ± 5.4; p = 0.025) |
| Drechsel et al. (2022) [99] | 30 BRCA+ patients, 122 control patients | BRCA+ vs. control: similar AFC (15.0 [10.8–20.3] vs. 14.5 [9.0–20.0]; p = 0.54) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias Nunes, J.; Demeestere, I.; Devos, M. BRCA Mutations and Fertility Preservation. Int. J. Mol. Sci. 2024, 25, 204. https://doi.org/10.3390/ijms25010204
Dias Nunes J, Demeestere I, Devos M. BRCA Mutations and Fertility Preservation. International Journal of Molecular Sciences. 2024; 25(1):204. https://doi.org/10.3390/ijms25010204
Chicago/Turabian StyleDias Nunes, Joana, Isabelle Demeestere, and Melody Devos. 2024. "BRCA Mutations and Fertility Preservation" International Journal of Molecular Sciences 25, no. 1: 204. https://doi.org/10.3390/ijms25010204
APA StyleDias Nunes, J., Demeestere, I., & Devos, M. (2024). BRCA Mutations and Fertility Preservation. International Journal of Molecular Sciences, 25(1), 204. https://doi.org/10.3390/ijms25010204