
Mitochondrial Reactive Oxygen Species in TRIF-Dependent Toll-like Receptor 3 Signaling in Bronchial Epithelial Cells against Viral Infection


Abstract
:1. Introduction
2. Results
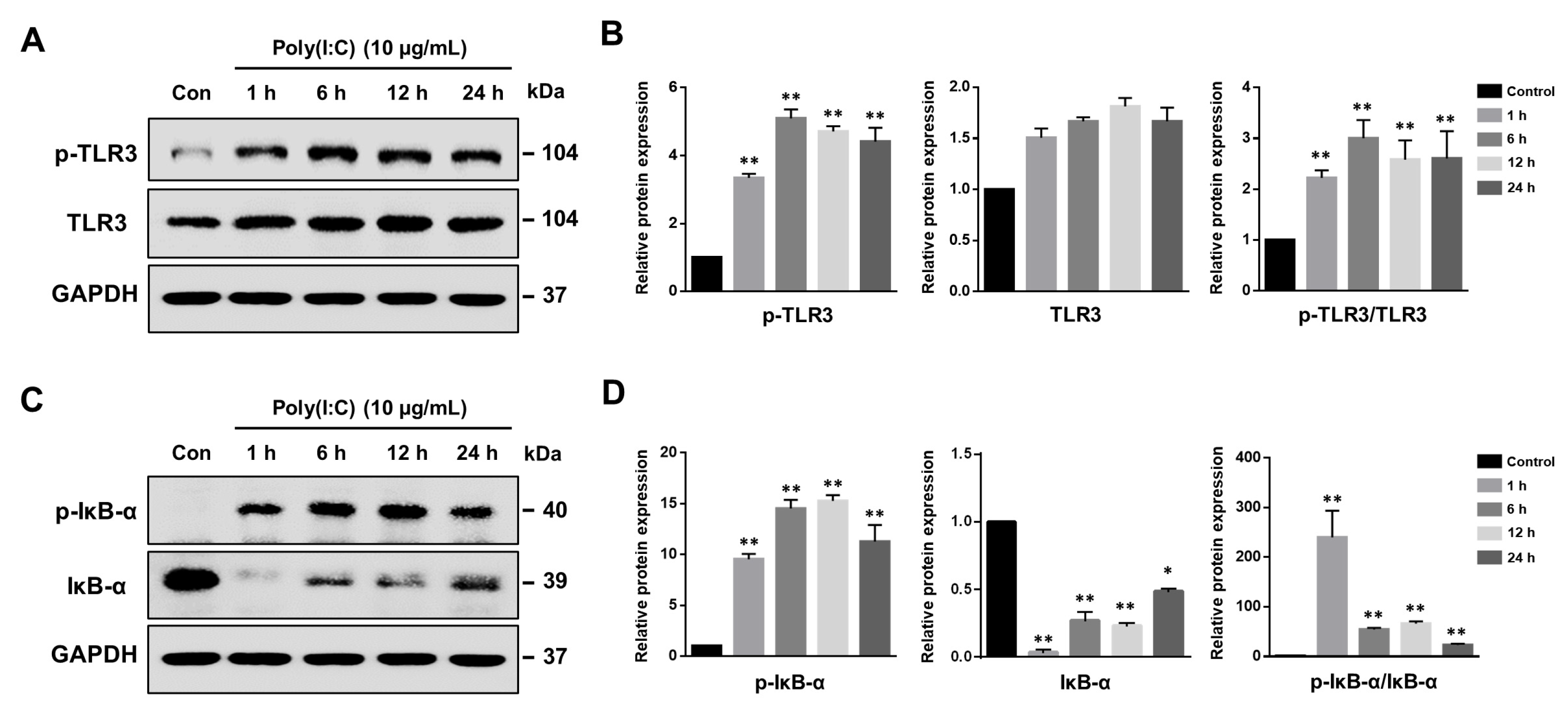
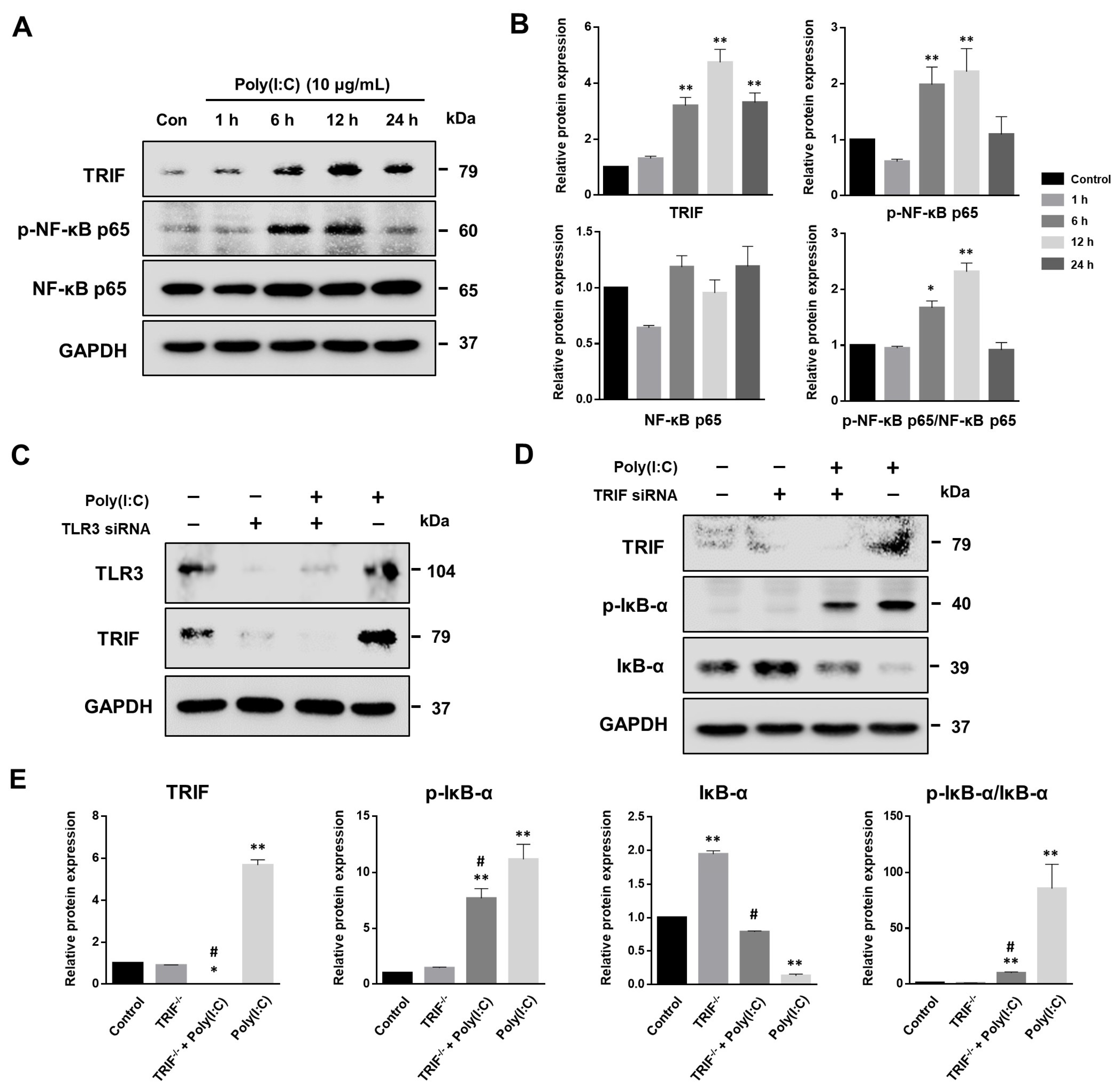
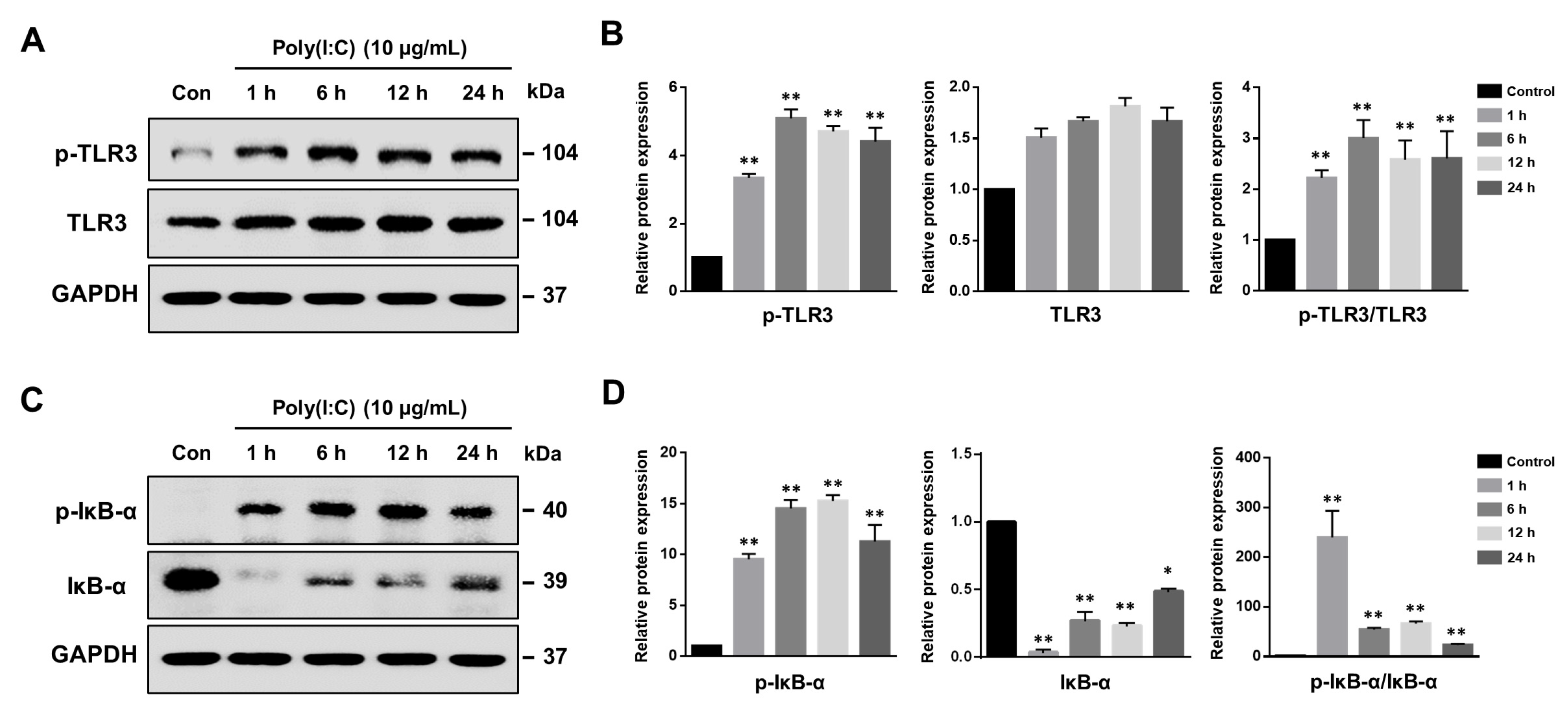
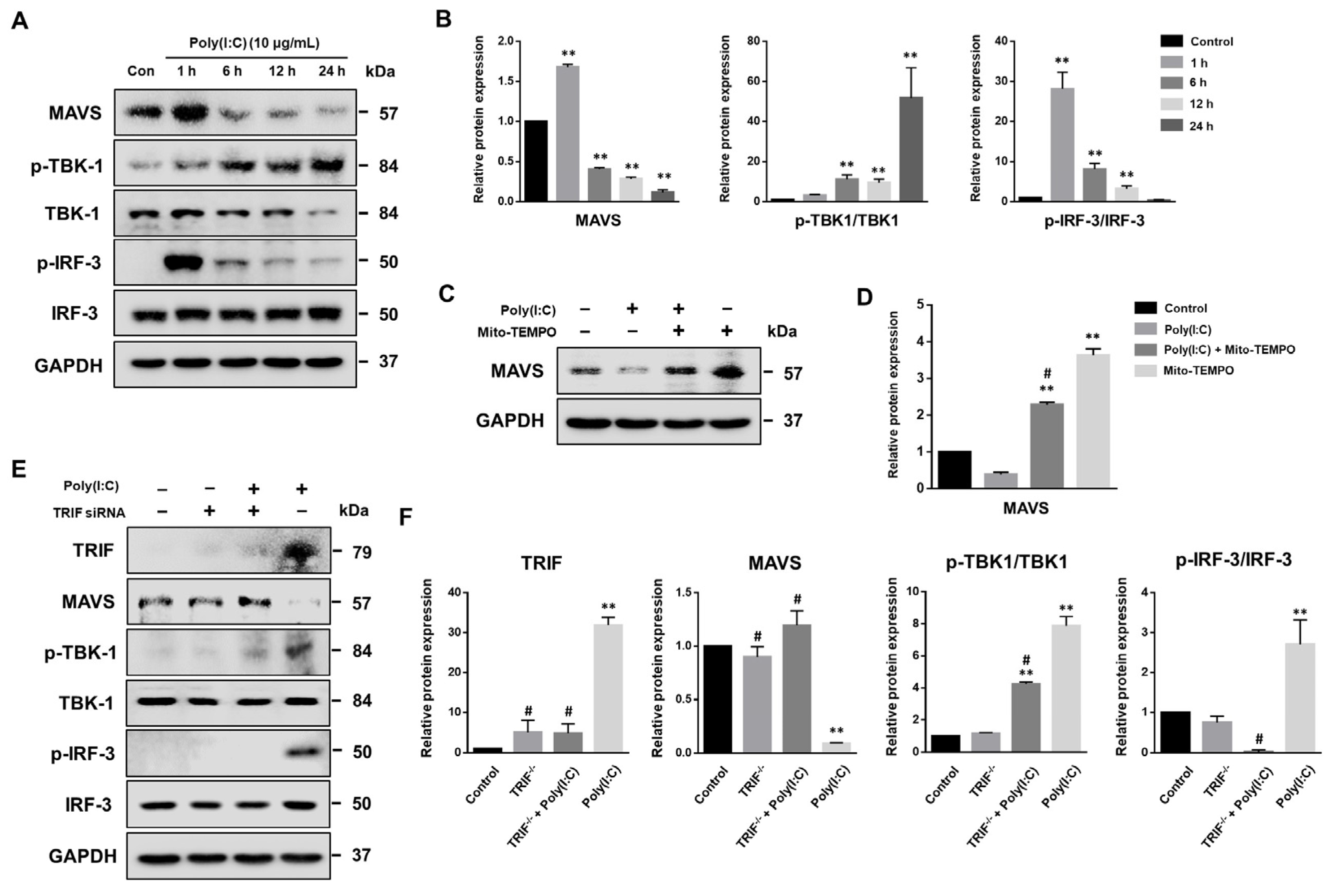
2.1. Poly(I:C)-Induced TLR3 Activation and NF-κB Signaling Pathway Initiation in Bronchial Epithelial Cells
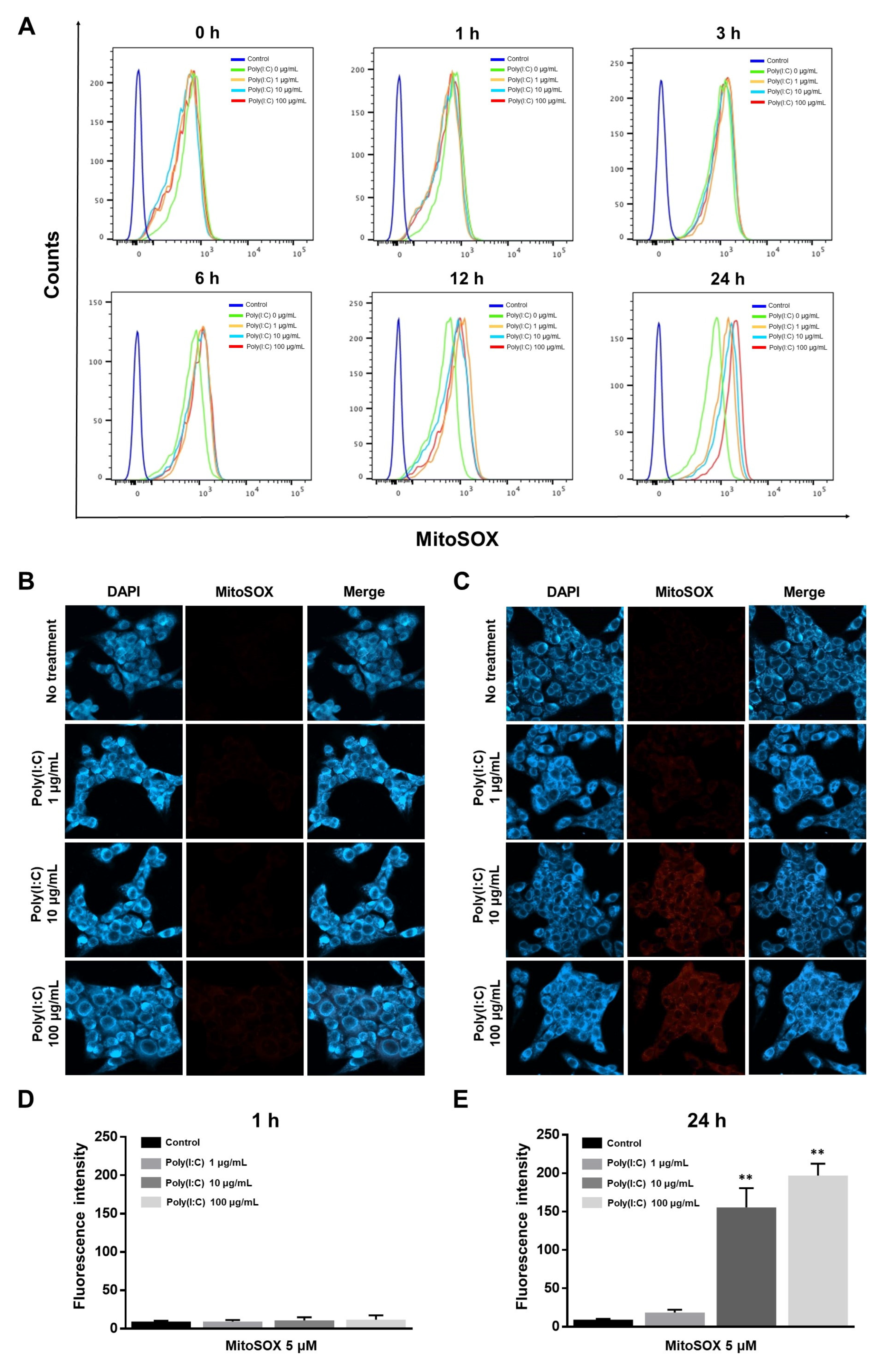
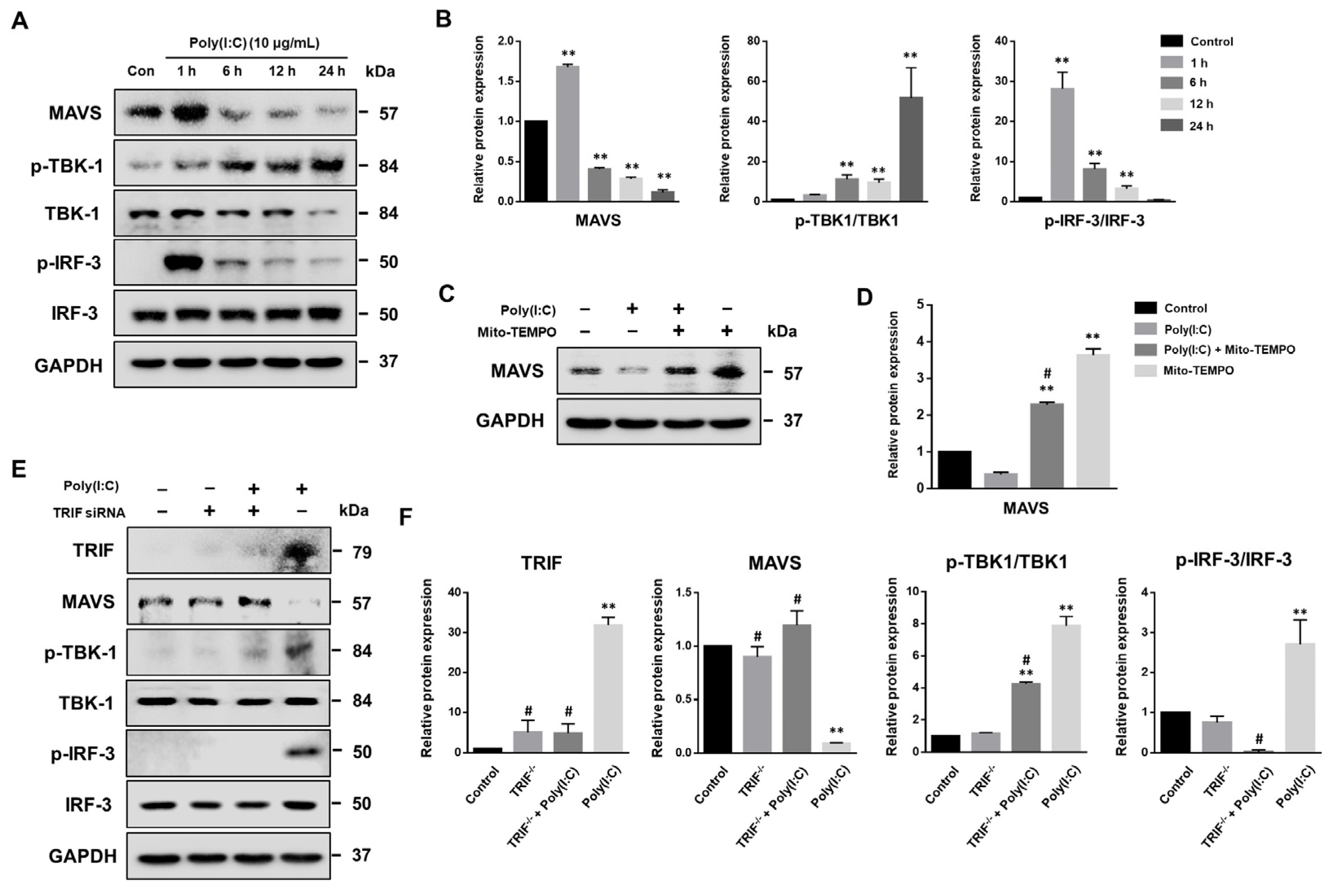
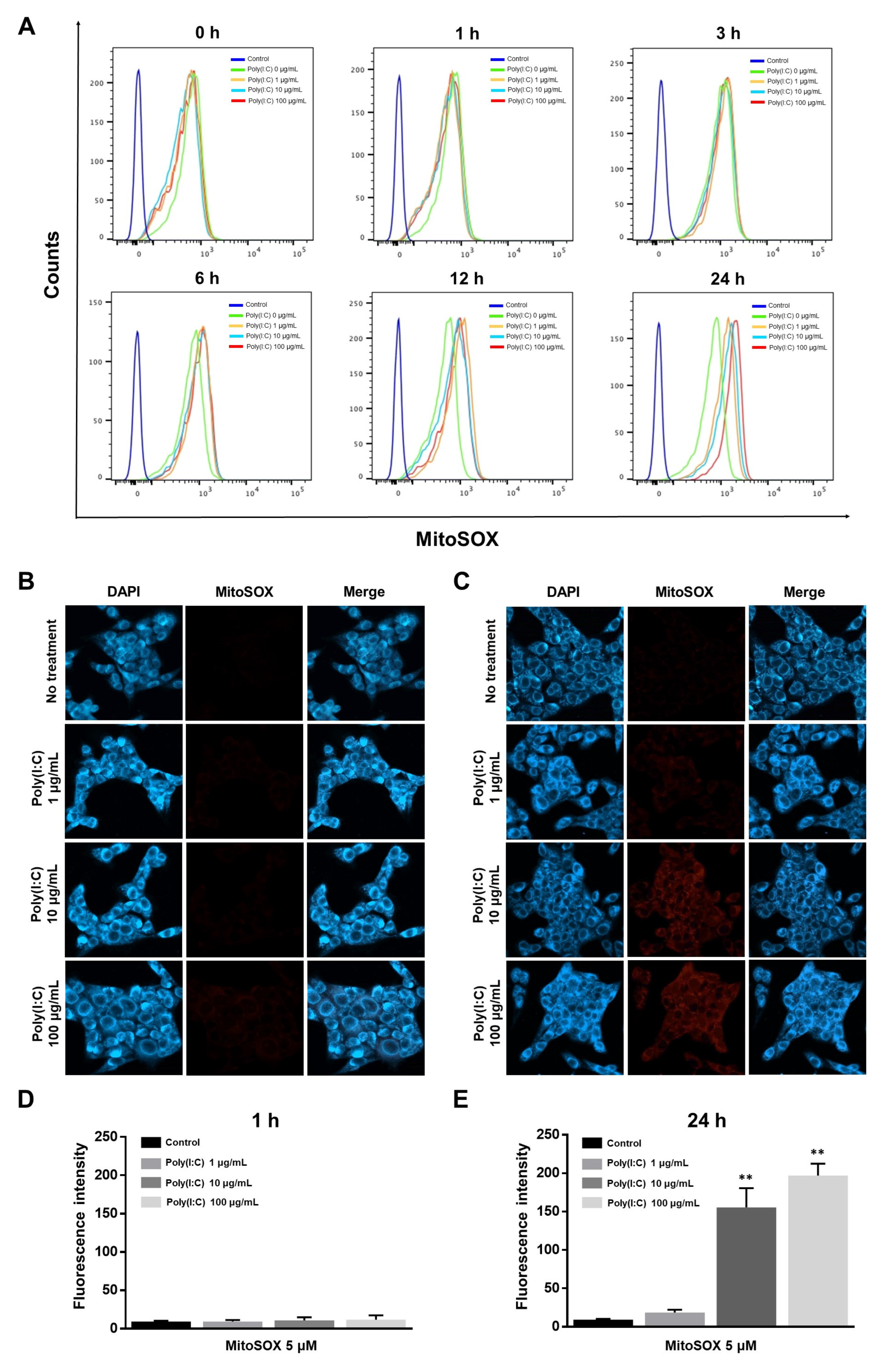
2.2. mtROS Generation Induced in TLR3 Signaling after 6 h of Poly(I:C) Treatment in Bronchial Epithelial Cells
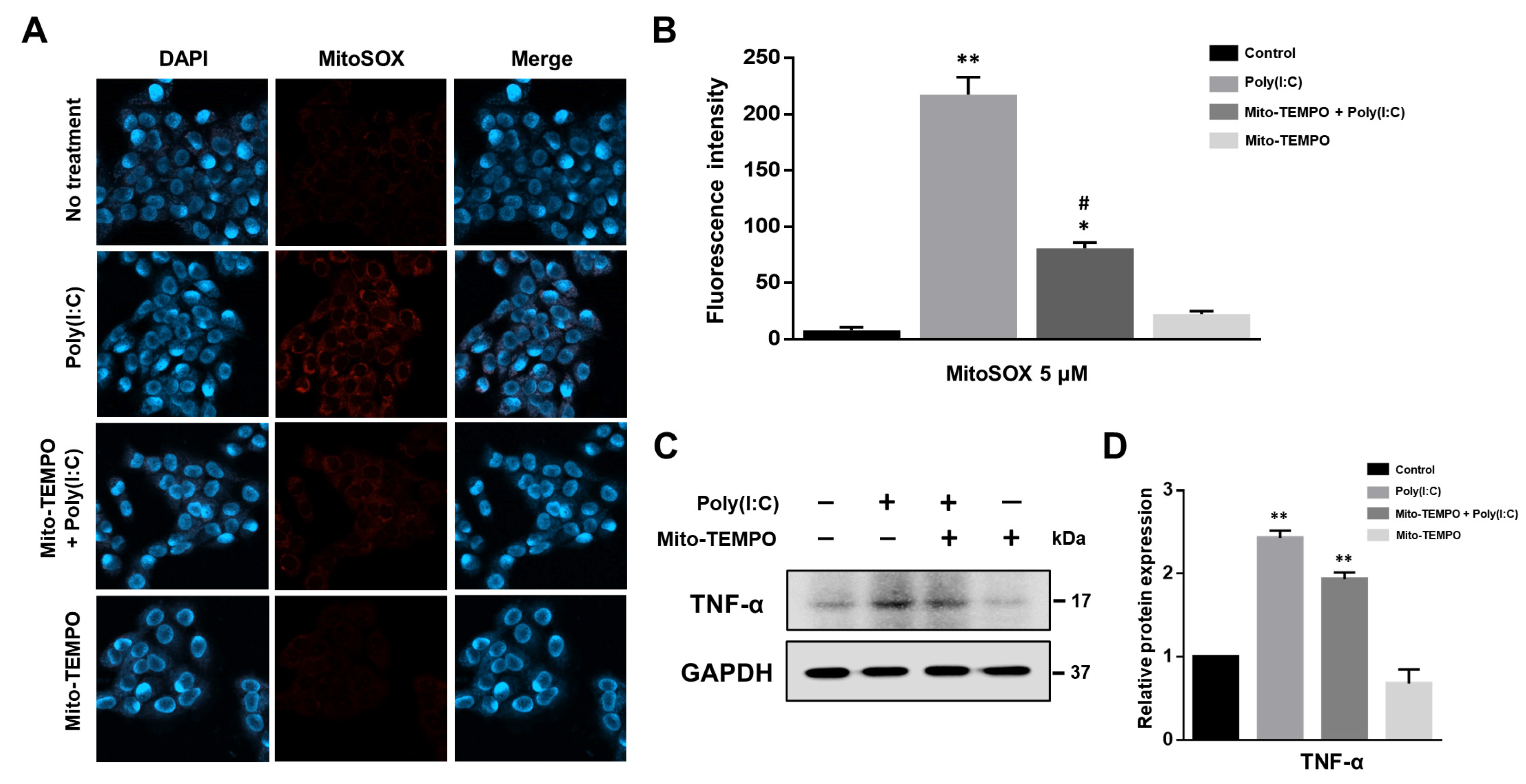
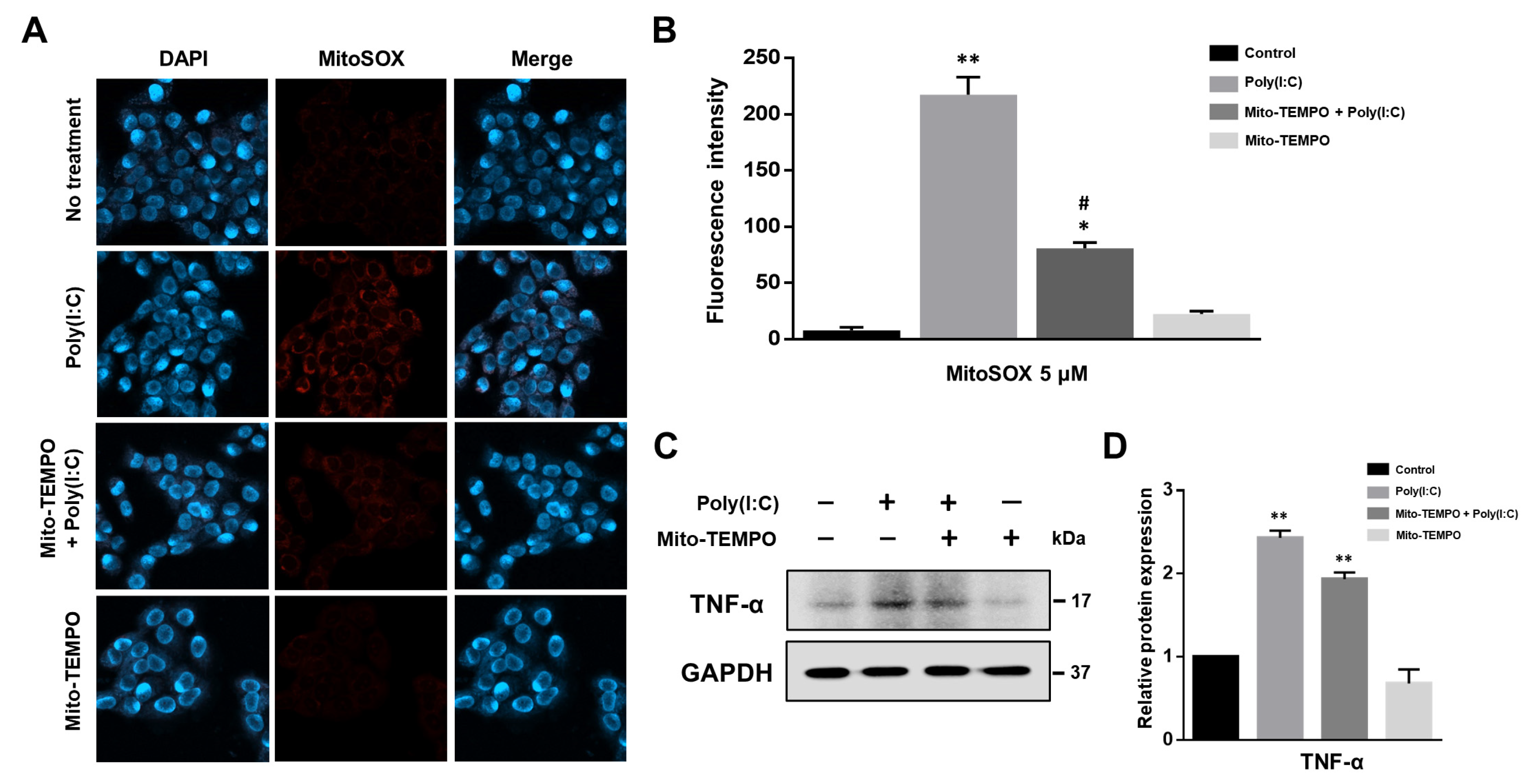
2.3. Cytokine Production Affected by Poly(I:C)-Induced mtROS Generation in Bronchial Epithelial Cells
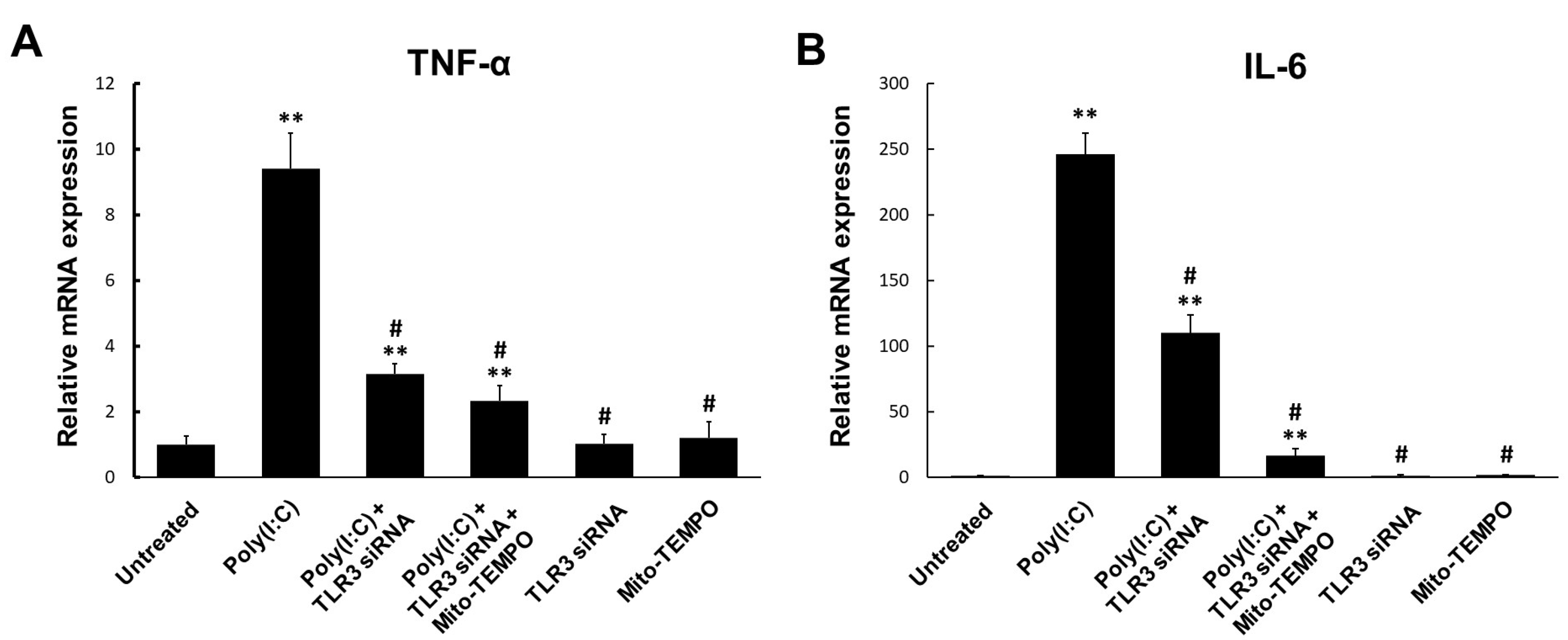
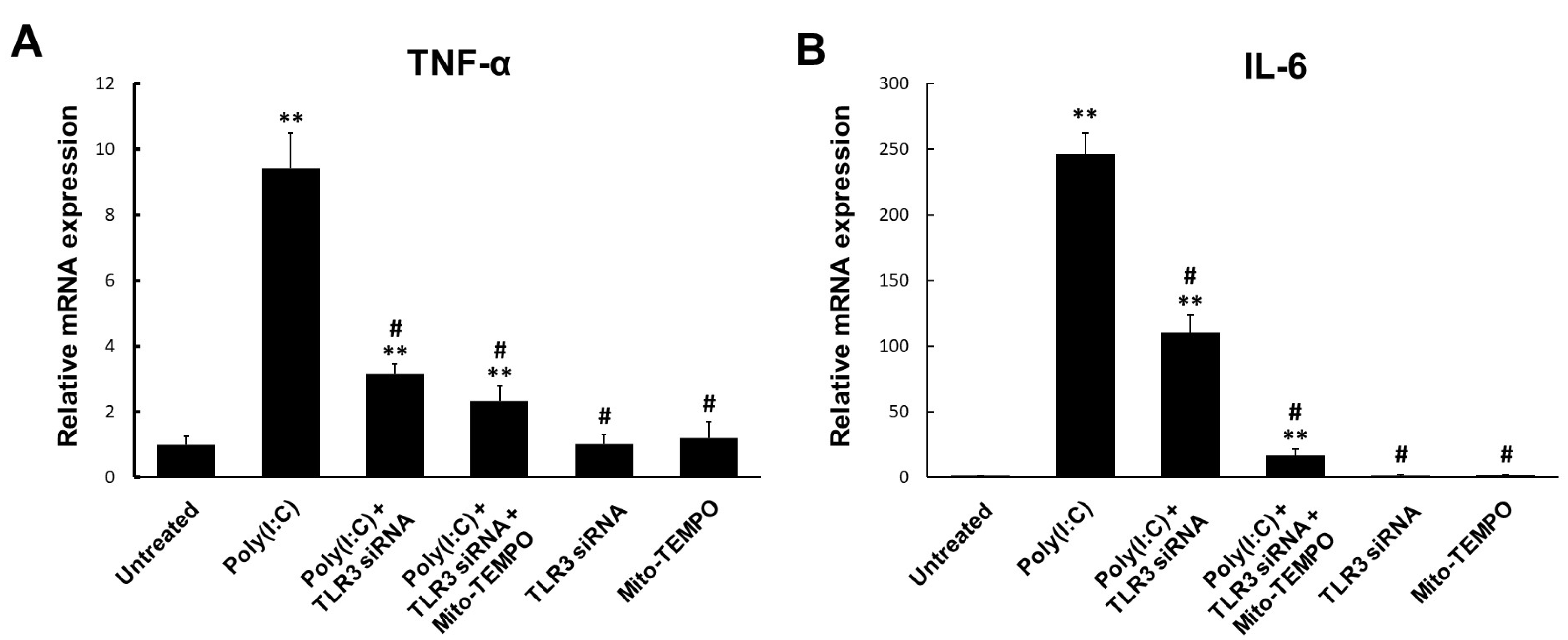
2.4. TLR3-Triggered mtROS Contribute to Inflammatory Cytokine Production in Bronchial Epithelial Cells
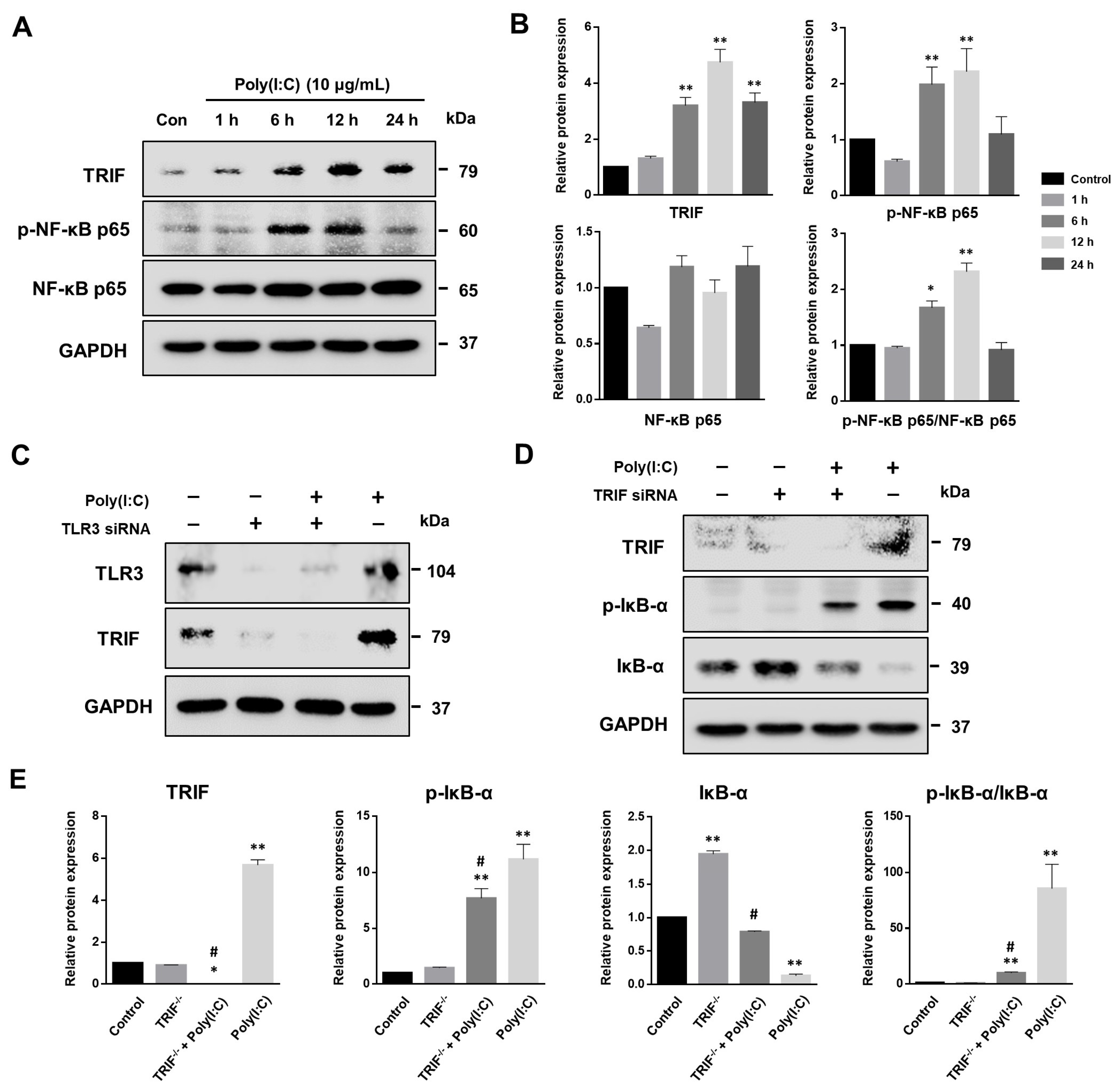
2.5. NF-κB Signaling Pathway Induced by Poly(I:C)-Stimulated TLR3-TRIF Signaling in Bronchial Epithelial Cells
2.6. MAVS Degradation Affected by Poly(I:C)-Induced mtROS and TLR3-TRIF Signaling in Bronchial Epithelial Cells
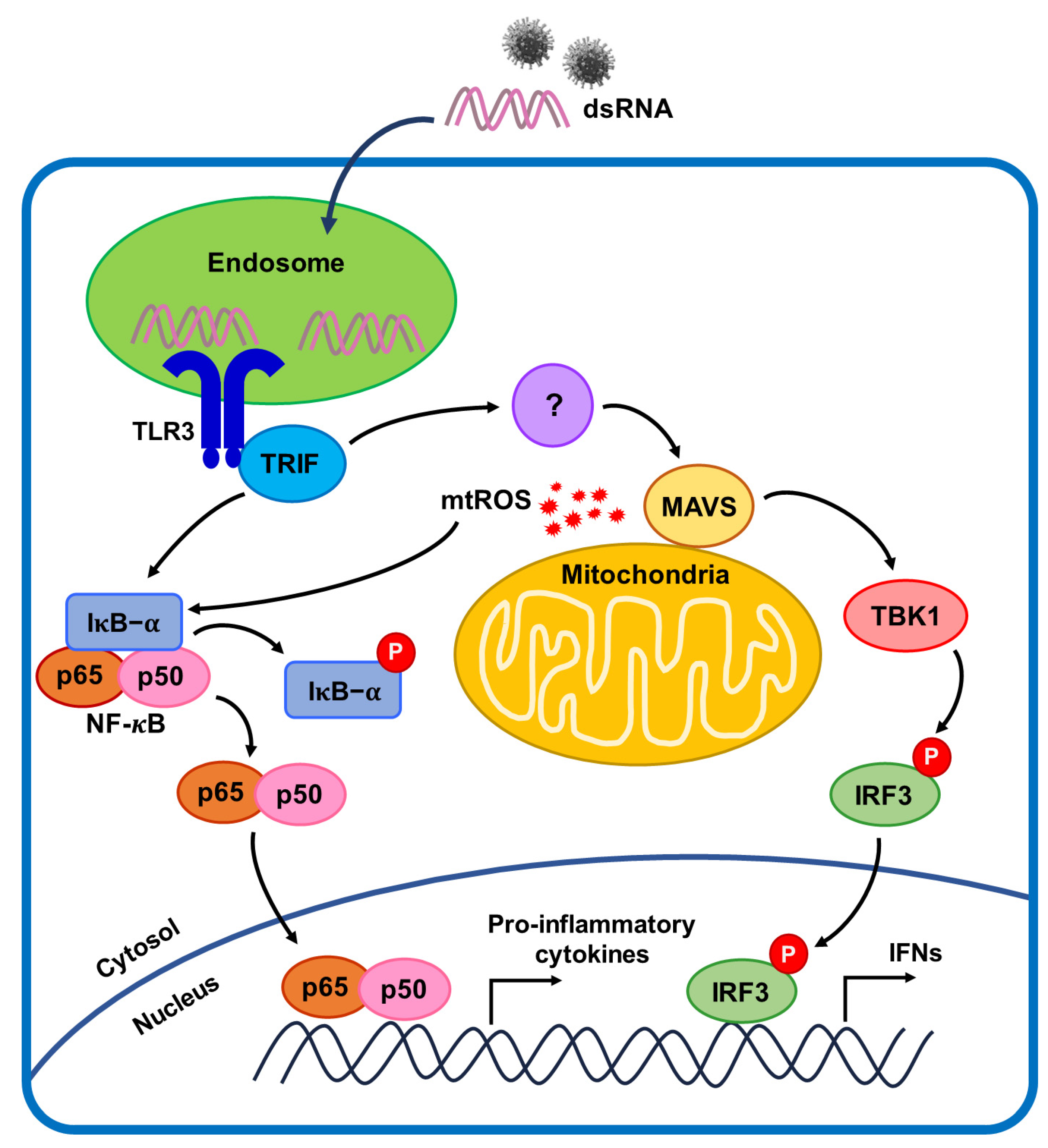
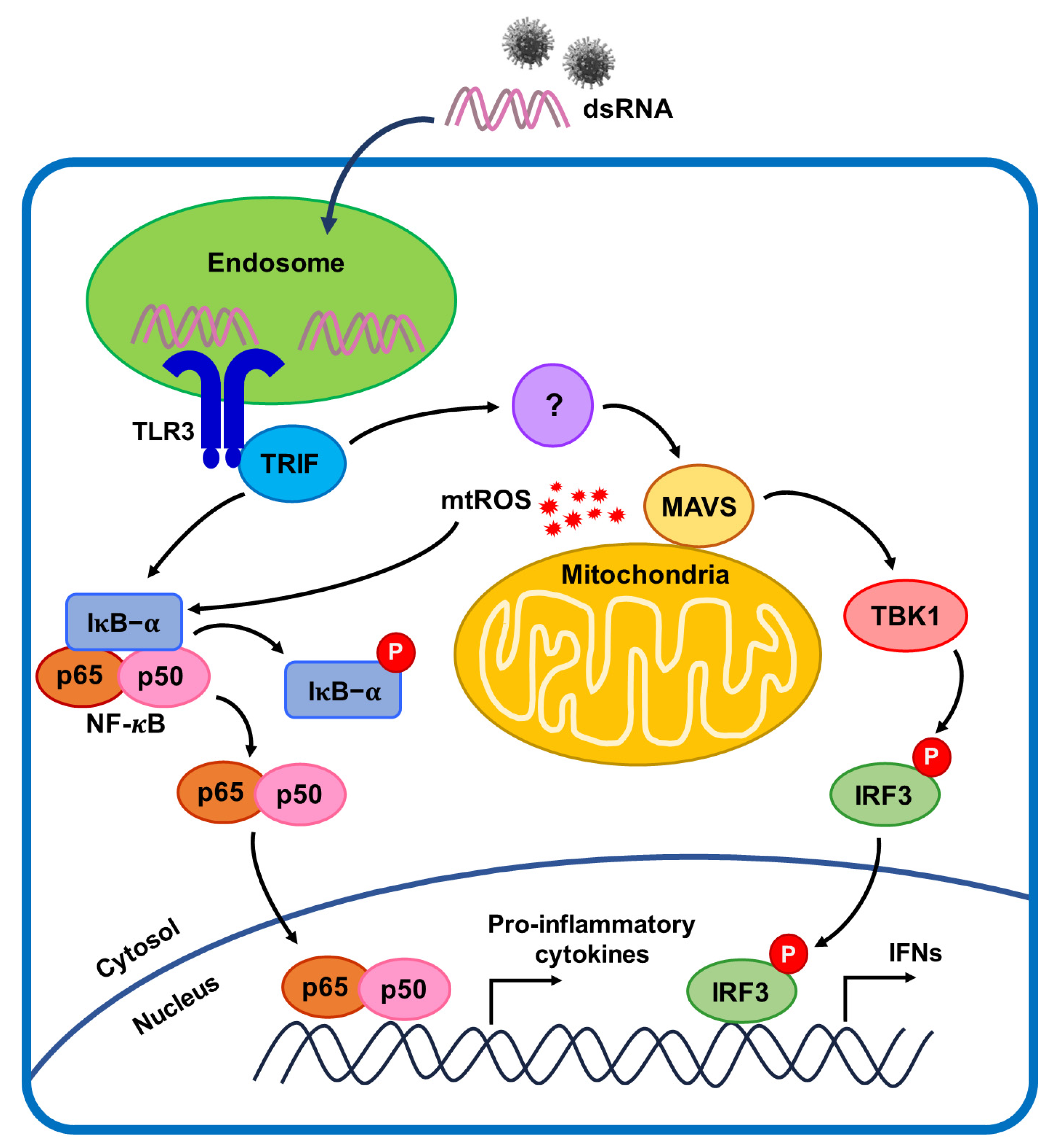
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Poly(I:C) Treatment
4.2. Transient siRNA Transfections
4.3. Western Blot Analysis
4.4. Flow Cytometry
4.5. Confocal Fluorescence Microscopy
4.6. Nano Live Imaging
4.7. Real-Time Quantitative PCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Albiger, B.; Dahlberg, S.; Henriques-Normark, B.; Normark, S. Role of the innate immune system in host defence against bacterial infections: Focus on the Toll-like receptors. J. Intern. Med. 2007, 261, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kawai, T.; Akira, S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016246. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 2011, 3, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Sen, G.C. dsRNA-activation of TLR3 and RLR signaling: Gene induction-dependent and independent effects. J. Interferon Cytokine Res. 2014, 34, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like Receptors in the Vascular System: Sensing the Dangers within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, J.; Zhao, Y.; Ma, X.; Yi, H. Toll-like receptor 3 (TLR3) regulation mechanisms and roles in antiviral innate immune responses. J. Zhejiang Univ. Sci. B. 2021, 22, 609–632. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, M.; Wang, X.; Li, J.; Wang, Y.; Ye, L.; Dai, M.; Zhou, L.; Persidsky, Y.; Ho, W. TLR3 activation efficiency by high or low molecular mass poly I:C. Innate Immun. 2013, 19, 184–192. [Google Scholar] [CrossRef]
- Komal, A.; Noreen, M.; El-Kott, A.F. TLR3 agonists: RGC100, ARNAX, and poly-IC: A comparative review. Immunol. Res. 2021, 69, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: Possible interactions with cellular processes. Can. J. Psychiatry. 2016, 61, 457–669. [Google Scholar] [CrossRef]
- Koopman, W.J.; Distelmaier, F.; Smeitink, J.A.; Willems, P.H. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological signaling functions of reactive oxygen species in stem cells: From flies to man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.M.; Basile, J.I.; Corra Feo, L.; López, B.; Ritacco, V.; Alemán, M. Reactive oxygen species production by human dendritic cells involves TLR2 and dectin-1 and is essential for efficient immune response against Mycobacteria. Cell. Microbiol. 2016, 18, 875–886. [Google Scholar] [CrossRef]
- Shekhova, E. Mitochondrial reactive oxygen species as major effectors of antimicrobial immunity. PLoS Pathog. 2020, 16, e1008470. [Google Scholar] [CrossRef]
- Myszor, I.T.; Gudmundsson, G.H. Modulation of innate immunity in airway epithelium for host-directed therapy. Front. Immunol. 2023, 14, 1197908. [Google Scholar] [CrossRef]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, E.; Staal, J.; Beyaert, R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 2008, 21, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Kim, J.J.; Lee, S.J.; Hwang, J.H.; Lee, C.H.; Lee, M.S.; Jo, E.K. TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. J. Immunol. 2013, 190, 6368–6377. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Roy, D.; Jaworski, A.; Edwards, A.; Abizaid, A.; Kumar, A.; Golshani, A.; Cassol, E. Differential remodeling of the electron transport chain is required to support TLR3 and TLR4 signaling and cytokine production in macrophages. Sci. Rep. 2019, 9, 18801. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Horner, S.M. MAVS coordination of antiviral innate immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, Q.; Xu, P. TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim. Biophys. Sinica. 2020, 52, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Seya, T.; Matsumoto, M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem. J. 2014, 458, 195–201. [Google Scholar] [CrossRef]
- Faksh, A.; Britt, R.D., Jr.; Vogel, E.R.; Thompson, M.A.; Pandya, H.C.; Martin, R.J.; Pabelick, C.M.; Prakash, Y.S. TLR3 activation increases chemokine expression in human fetal airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L202–L211. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.K. Mitochondrial reactive oxygen species: Double-edged weapon in host defense and pathological inflammation during infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef]
- Lever, A.R.; Park, H.; Mulhern, T.J.; Jackson, G.R.; Comolli, J.C.; Borenstein, J.T.; Hayden, P.J.; Prantil-Baun, R. Comprehensive evaluation of poly(I:C) induced inflammatory response in an airway epithelial model. Physiol. Rep. 2015, 3, e12334. [Google Scholar] [CrossRef]
- Ramezanpour, M.; Bolt, H.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Primary human nasal epithelial cells: A source of poly (I:C) LMW-induced IL-6 production. Sci. Rep. 2018, 8, 11325. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Jiang, T.; Lu, H.; Han, H.; Luo, R. Combination of Poly I:C and arsenic trioxide triggers apoptosis synergistically via activation of TLR3 and mitochondrial pathways in hepatocellular carcinoma cells. Cell Biol. Int. 2011, 35, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. USA 2004, 101, 3533–3538. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Matsumiya, T.; Onomoto, K.; Hayakari, R.; Imaizumi, T.; Yoshida, H.; Yoneyama, M.; Fujita, T.; Satoh, K. Foreign RNA induces the degradation of mitochondrial antiviral signaling protein (MAVS): The role of intracellular antiviral factors. PLoS ONE 2012, 7, e45136. [Google Scholar] [CrossRef] [PubMed]
- Lakhdari, O.; McAllister, C.S.; Wang, M.; Minev, I.; Prince, L.S.; Eckmann, L.; Kagnoff, M.F. TLR3 signaling is downregulated by a MAVS isoform in epithelial cells. Cell. Immunol. 2016, 310, 205–210. [Google Scholar] [CrossRef]
- Weiss, R.; Laengle, J.; Sachet, M.; Shurygina, A.P.; Kiselev, O.; Egorov, A.; Bergmann, M. Interleukin-24 inhibits influenza A virus replication in vitro through induction of toll-like receptor 3 dependent apoptosis. Antivir. Res. 2015, 123, 93–104. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Teijaro, J.R. Type I interferons in viral control and immune regulation. Curr. Opin. Virol. 2016, 16, 31–40. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Sander, W.J.; Fourie, C.; Sabiu, S.; O’Neill, F.H.; Pohl, C.H.; O’Neill, H.G. Reactive oxygen species as potential antiviral targets. Rev. Med. Virol. 2022, 32, e2240. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.U.; Lim, D.; Kim, Y.S.; Ku, B.; Kim, D.J. Influenza viral matrix 1 protein aggravates viral pathogenicity by inducing TLR4-mediated reactive oxygen species production and apoptotic cell death. Cell Death Dis. 2023, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T. Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta 2013, 1833, 225–232. [Google Scholar] [CrossRef]
- Park, C.; Lee, S.; Cho, I.H.; Lee, H.K.; Kim, D.; Choi, S.Y.; Oh, S.B.; Park, K.; Kim, J.S.; Lee, S.J. TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: Differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia 2006, 53, 248–256. [Google Scholar] [CrossRef]
- Su, J.; Yu, X. Editorial: Ligands, Adaptors and pathways of TLRs in non-mammals. Front. Immunol. 2019, 17, 2439. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Nobre, L.; Wise, D.; Ron, D.; Volmer, R. Modulation of Innate Immune Signalling by Lipid-Mediated MAVS Transmembrane Domain Oligomerization. PLoS ONE 2015, 10, e0136883. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| IL-6 | forward primer: 5′-GGT ACT CCT CCA CGG CAT CT-3′ reverse primer: 5′-GTG CCT CTT TGC TGC TTT CAC-3′ |
| TNF-α | forward primer: 5′-GCC CAT GTT GTA GCA AAC CC-3′ reverse primer: 5′-TAT CTC TCA GCT CCA CGC CA-3′ |
| 18S | forward primer: 5′-ACC GCA GCT AGG AAT AAT GGA-3′ reverse primer: 5′-CGG TCA GTT CCG AAA ACC A-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, G.E.; Park, J.Y.; Park, C.H.; Cho, W.G. Mitochondrial Reactive Oxygen Species in TRIF-Dependent Toll-like Receptor 3 Signaling in Bronchial Epithelial Cells against Viral Infection. Int. J. Mol. Sci. 2024, 25, 226. https://doi.org/10.3390/ijms25010226
Chu GE, Park JY, Park CH, Cho WG. Mitochondrial Reactive Oxygen Species in TRIF-Dependent Toll-like Receptor 3 Signaling in Bronchial Epithelial Cells against Viral Infection. International Journal of Molecular Sciences. 2024; 25(1):226. https://doi.org/10.3390/ijms25010226
Chicago/Turabian StyleChu, Ga Eul, Jun Young Park, Chan Ho Park, and Won Gil Cho. 2024. "Mitochondrial Reactive Oxygen Species in TRIF-Dependent Toll-like Receptor 3 Signaling in Bronchial Epithelial Cells against Viral Infection" International Journal of Molecular Sciences 25, no. 1: 226. https://doi.org/10.3390/ijms25010226
APA StyleChu, G. E., Park, J. Y., Park, C. H., & Cho, W. G. (2024). Mitochondrial Reactive Oxygen Species in TRIF-Dependent Toll-like Receptor 3 Signaling in Bronchial Epithelial Cells against Viral Infection. International Journal of Molecular Sciences, 25(1), 226. https://doi.org/10.3390/ijms25010226