
Investigating the Mitoprotective Effects of S1P Receptor Modulators Ex Vivo Using a Novel Semi-Automated Live Imaging Set-Up
 , , and
, , and
Abstract
:1. Introduction
2. Results
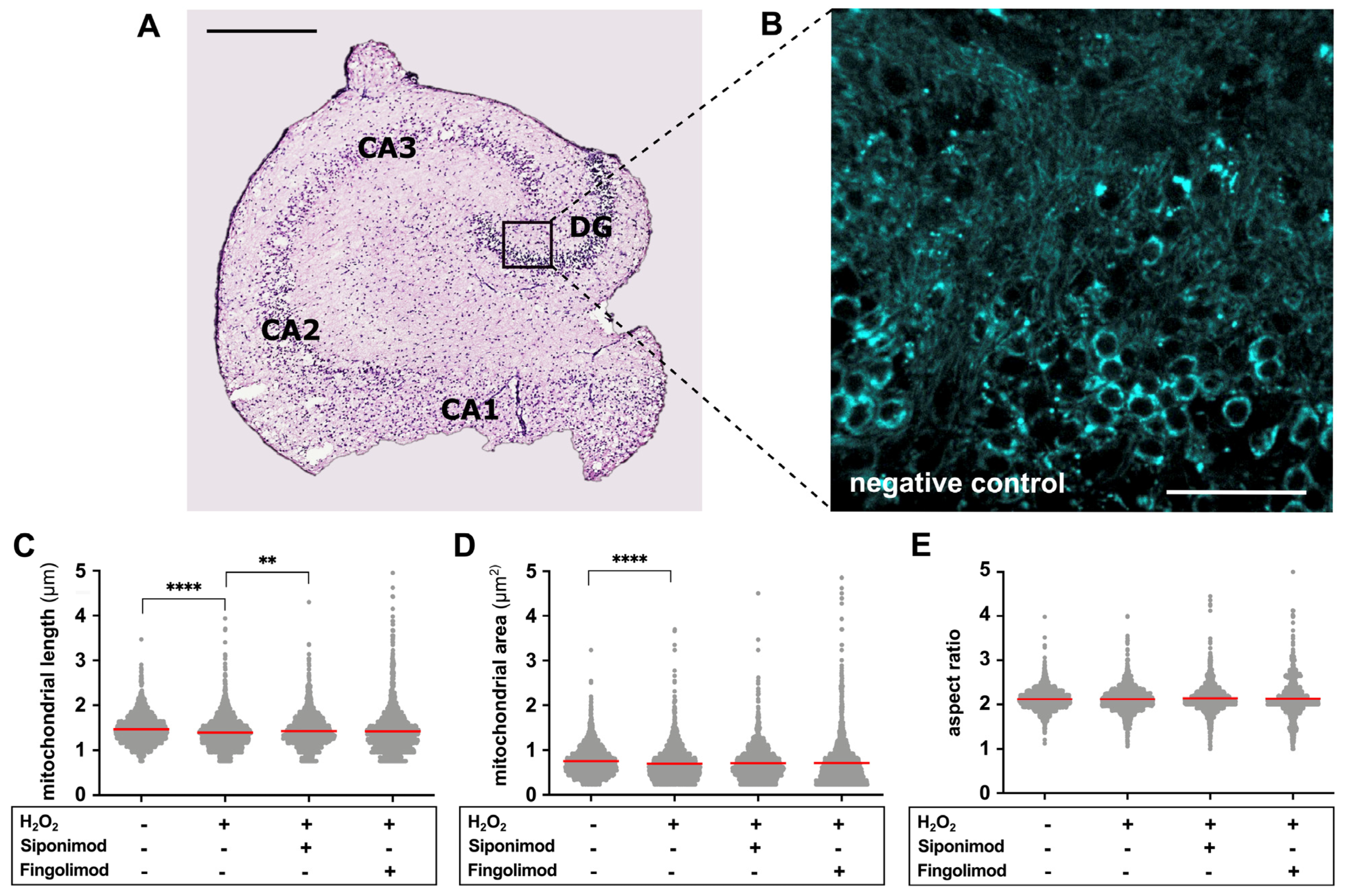
2.1. Establishment of a Semi-Automated Analysis of Mitochondrial Motility and Morphology
2.2. Siponimod Prevents Oxidative Stress-Induced Alterations in Mitochondrial Morphology
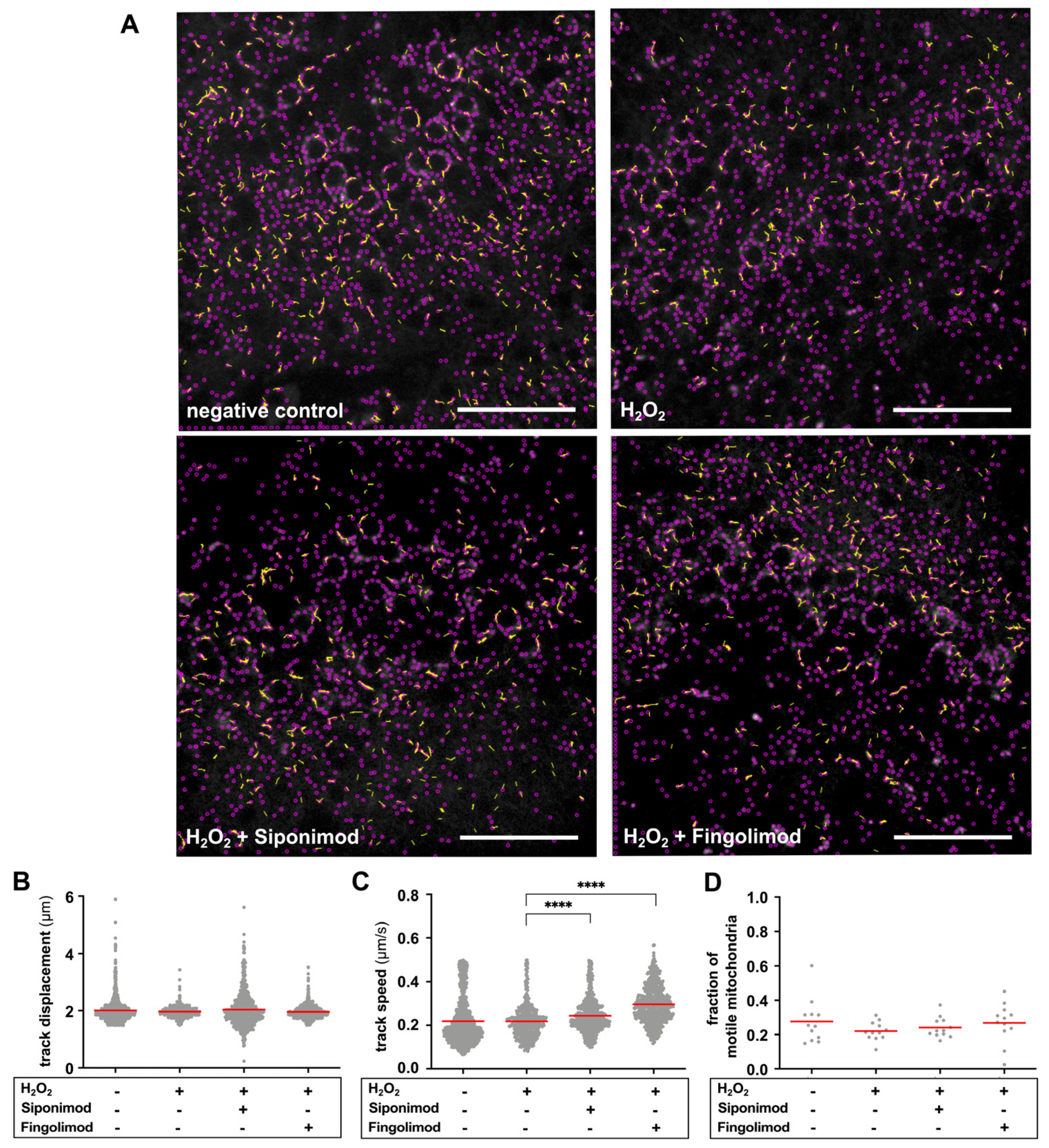
2.3. Siponimod Reduces Oxidative Stress-Induced Changes in Mitochondrial Motility
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Animals
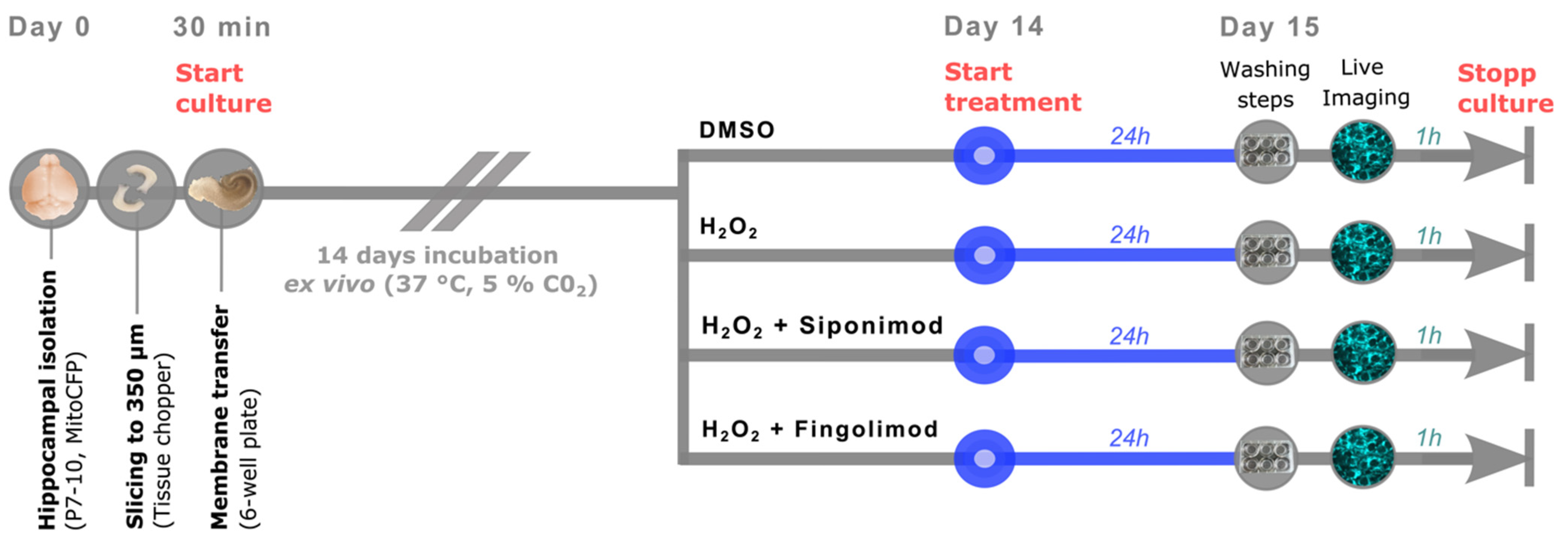
4.2. Generation of Organotypic Hippocampal Slice Cultures
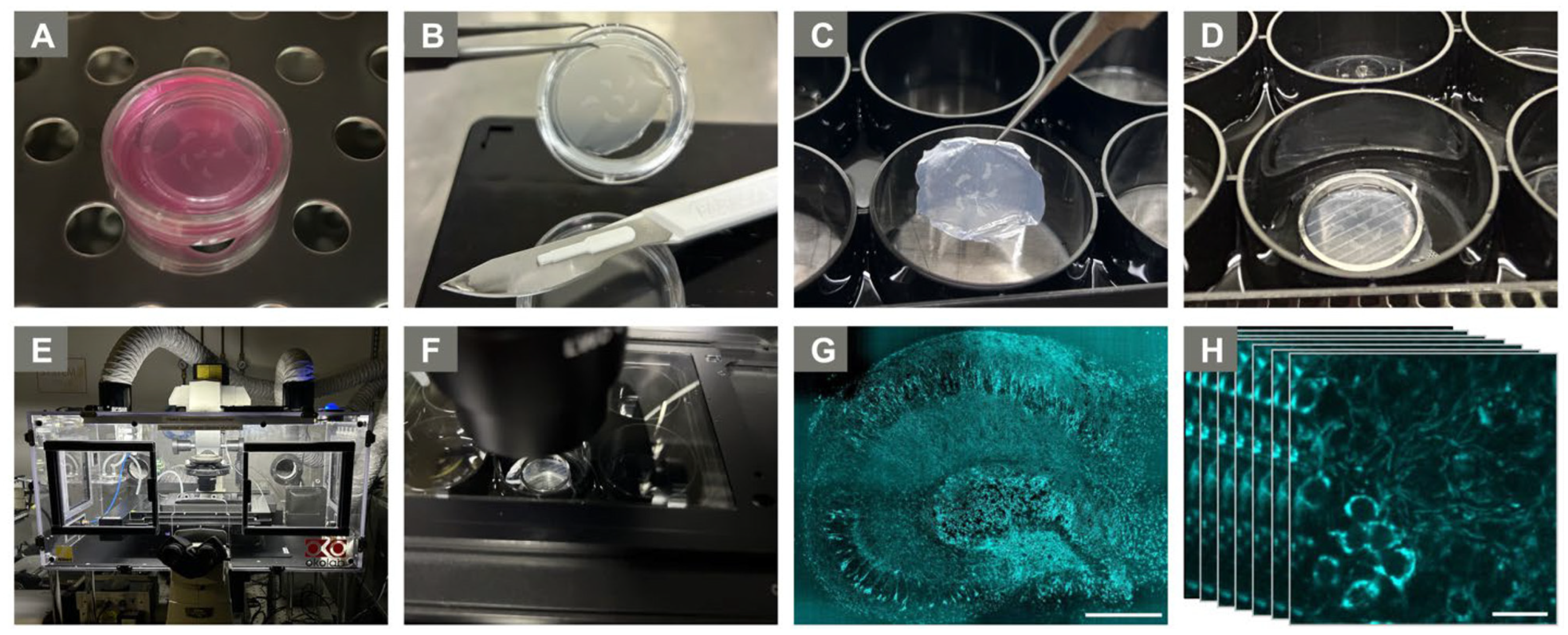
4.3. Experimental Ex Vivo Setup
4.4. Live Imaging
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, G.; Thompson, A.; Berg, J.; Gannedahl, M.; Eriksson, J.; MSCOI Study Group; European Multiple Sclerosis Platform. New insights into the burden and costs of multiple sclerosis in Europe. Mult. Scler. 2017, 23, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Klineova, S.; Lublin, F.D. Clinical Course of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028928. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Weiner, H.L. A shift from adaptive to innate immunity: A potential mechanism of disease progression in multiple sclerosis. J. Neurol. 2008, 255 (Suppl. S1), 3–11. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Salapa, H.E.; Lee, S.; Shin, Y.; Levin, M.C. Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sci. 2017, 7, 69. [Google Scholar] [CrossRef]
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar] [CrossRef]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef] [PubMed]
- De Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2015, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Kozin, M.S.; Kulakova, O.G.; Favorova, O.O. Involvement of Mitochondria in Neurodegeneration in Multiple Sclerosis. Biochemistry 2018, 83, 813–830. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Comi, G.; Kappos, L.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.-P.; Montalban, X.; Havrdová, E.K.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): A multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019, 18, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Bayas, A.; Christ, M.; Faissner, S.; Klehmet, J.; Pul, R.; Skripuletz, T.; Meuth, S.G. Disease-modifying therapies for relapsing/active secondary progressive multiple sclerosis—A review of population-specific evidence from randomized clinical trials. Ther. Adv. Neurol. Disord. 2023, 16, 17562864221146836. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, A.B.; Grebenciucova, E.; Shoemaker, T.; Graham, E.L. Current Updates on the Diagnosis and Management of Multiple Sclerosis for the General Neurologist. J. Clin. Neurol. 2023, 19, 217–229. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium; Harroud, A.; Stridh, P.; McCauley, J.L.; Saarela, J.; Bosch, A.M.R.v.D.; Engelenburg, H.J.; Beecham, A.H.; Alfredsson, L.; Alikhani, K.; et al. Locus for severity implicates CNS resilience in progression of multiple sclerosis. Nature 2023, 619, 323–331. [Google Scholar] [CrossRef]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The Immune Modulator FTY720 Targets Sphingosine 1-Phosphate Receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y. FTY720, a Novel Immunosuppressant, Induces Sequestration of Circulating Mature Lymphocytes by Acceleration of Lymphocyte Homing in Rats. I. FTY720 Selectively Decreases the Number of Circulating Mature Lymphocytes by Acceleration of Lymphocyte Homing. J. Immunol. 1998, 160, 5037–5044. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Martín-Montañez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.-Y.; Fernández, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells 2020, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M. Does Siponimod Exert Direct Effects in the Central Nervous System? Cells 2020, 9, 1771. [Google Scholar] [CrossRef] [PubMed]
- Cohan, S.L.; Benedict, R.H.B.; Cree, B.A.C.; DeLuca, J.; Hua, L.H.; Chun, J. The Two Sides of Siponimod: Evidence for Brain and Immune Mechanisms in Multiple Sclerosis. CNS Drugs 2022, 36, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Siponimod: A Review in Secondary Progressive Multiple Sclerosis. CNS Drugs 2020, 34, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Gajofatto, A. Spotlight on siponimod and its potential in the treatment of secondary progressive multiple sclerosis: The evidence to date. Drug Des. Dev. Ther. 2017, 11, 3153–3157. [Google Scholar] [CrossRef]
- Basavarajappa, D.; Gupta, V.; Chitranshi, N.; Wall, R.; Rajput, R.; Pushpitha, K.; Sharma, S.; Mirzaei, M.; Klistorner, A.; Graham, S. Siponimod exerts neuroprotective effects on the retina and higher visual pathway through neuronal S1PR1 in experimental glaucoma. Neural Regen. Res. 2022, 18, 840–848. [Google Scholar] [CrossRef]
- Basavarajappa, D.; Gupta, V.; Chitranshi, N.; Viswanathan, D.; Gupta, V.; Wall, R.V.; Palanivel, V.; Mirzaei, M.; You, Y.; Klistorner, A.; et al. Anti-inflammatory Effects of Siponimod in a Mouse Model of Excitotoxicity-Induced Retinal Injury. Mol. Neurobiol. 2023, 60, 7222–7237. [Google Scholar] [CrossRef]
- Vinnenberg, L.; Rychlik, N.; Oniani, T.; Williams, B.; White, J.A.; Kovac, S.; Meuth, S.G.; Budde, T.; Hundehege, P. Assessing neuroprotective effects of diroximel fumarate and siponimod via modulation of pacemaker channels in an experimental model of remyelination. Exp. Neurol. 2024, 371, 114572. [Google Scholar] [CrossRef]
- Bigaud, M.; Rudolph, B.; Briard, E.; Beerli, C.; Hofmann, A.; Hermes, E.; Muellershausen, F.; Schubart, A.; Gardin, A. Siponimod (BAF312) penetrates, distributes, and acts in the central nervous system: Preclinical insights. Mult. Scler. J.-Exp. Transl. Clin. 2021, 7, 20552173211049168. [Google Scholar] [CrossRef] [PubMed]
- Vališ, M.; Achiron, A.; Hartung, H.P.; Mareš, J.; Tichá, V.; Štourač, P.; Halusková, S.; Angelucci, F.; Pavelek, Z. The Benefits and Risks of Switching from Fingolimod to Siponimod for the Treatment of Relapsing–Remitting and Secondary Progressive Multiple Sclerosis. Drugs R D 2023, 23, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Hundehege, P.; Cerina, M.; Eichler, S.; Thomas, C.; Herrmann, A.M.; Göbel, K.; Müntefering, T.; Fernandez-Orth, J.; Bock, S.; Narayanan, V.; et al. The next-generation sphingosine-1 receptor modulator BAF312 (siponimod) improves cortical network functionality in focal autoimmune encephalomyelitis. Neural Regen. Res. 2019, 14, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Thevenaz, P.; Ruttimann, U.; Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998, 7, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Malla, B.; Niesner, R.; Hauser, A.; Infante-Duarte, C. Imaging and analysis of neuronal mitochondria in murine acute brain slices. J. Neurosci. Methods 2022, 372, 109558. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.-Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Malla, B.; Cotten, S.; Ulshoefer, R.; Paul, F.; Hauser, A.E.; Niesner, R.; Bros, H.; Infante-Duarte, C. Teriflunomide preserves peripheral nerve mitochondria from oxidative stress-mediated alterations. Ther. Adv. Chronic Dis. 2020, 11, 2040622320944773. [Google Scholar] [CrossRef]
- Malla, B.; Liotta, A.; Bros, H.; Ulshöfer, R.; Paul, F.; Hauser, A.E.; Niesner, R.; Infante-Duarte, C. Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1538. [Google Scholar] [CrossRef]
- Ulshöfer, R.; Bros, H.; Hauser, A.E.; Niesner, R.A.; Paul, F.; Malla, B.; Infante-Duarte, C. Preventing Axonal Sodium Overload or Mitochondrial Calcium Uptake Protects Axonal Mitochondria from Oxidative Stress-Induced Alterations. Oxidative Med. Cell. Longev. 2022, 2022, 6125711. [Google Scholar] [CrossRef]
- Bros, H.; Millward, J.M.; Paul, F.; Niesner, R.; Infante-Duarte, C. Oxidative damage to mitochondria at the nodes of Ranvier precedes axon degeneration in ex vivo transected axons. Exp. Neurol. 2014, 261, 127–135. [Google Scholar] [CrossRef]
- Butler, R.; Bradford, D.; Rodgers, K.E. Analysis of shared underlying mechanism in neurodegenerative disease. Front. Aging Neurosci. 2022, 14, 1006089. [Google Scholar] [CrossRef] [PubMed]
- Anderhalten, L.; Silva, R.V.; Morr, A.; Wang, S.; Smorodchenko, A.; Saatz, J.; Traub, H.; Mueller, S.M.; Boehm-Sturm, P.; Rodriguez-Sillke, Y.; et al. Different Impact of Gadopentetate and Gadobutrol on Inflammation-Promoted Retention and Toxicity of Gadolinium within the Mouse Brain. Investig. Radiol. 2022, 57, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C. Organotypic Brain Slice Cultures. Curr. Protoc. Immunol. 2018, 123, e59. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, J.; Suuronen, T.; Miettinen, R.; van Groen, T.; Salminen, A. A refined in vitro model to study inflammatory responses in organotypic membrane culture of postnatal rat hippocampal slices. J. Neuroinflamm. 2005, 2, 25. [Google Scholar] [CrossRef]
- Trumbeckaite, S.; Gizatullina, Z.; Arandarcikaite, O.; Röhnert, P.; Vielhaber, S.; Malesevic, M.; Fischer, G.; Seppet, E.; Striggow, F.; Gellerich, F.N. Oxygen glucose deprivation causes mitochondrial dysfunction in cultivated rat hippocampal slices: Protective effects of CsA, its immunosuppressive congener [D-Ser]8CsA, the novel non-immunosuppressive cyclosporin derivative Cs9, and the NMDA receptor antagonist MK 801. Mitochondrion 2013, 13, 539–547. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.-M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 175. [Google Scholar] [CrossRef]
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria–Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214. [Google Scholar] [CrossRef]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial Dynamics: A Key Role in Neurodegeneration and a Potential Target for Neurodegenerative Disease. Front. Neurosci. 2021, 15, 654785. [Google Scholar] [CrossRef]
- van der Bliek, A.M. Fussy mitochondria fuse in response to stress. EMBO J. 2009, 28, 1533–1534. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2010, 108, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.-C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Stratton, J.A.; Kuhlmann, T.; Antel, J. The role of glial cells in multiple sclerosis disease progression. Nat. Rev. Neurol. 2022, 18, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; Schlegel, C.; Gudi, V.; Prajeeth, C.K.; Skripuletz, T.; Trebst, C.; Stangel, M. Effect of FTY720-phosphate on the expression of inflammation-associated molecules in astrocytes in vitro. Mol. Med. Rep. 2015, 12, 6171–6177. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Huo, Y.; Kwang, W.; Pushparaj, P.; Kumar, S.; Ling, E.-A.; Dheen, S. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 2010, 166, 132–144. [Google Scholar] [CrossRef]
- Hoffmann, F.S.; Hofereiter, J.; Rübsamen, H.; Melms, J.; Schwarz, S.; Faber, H.; Weber, P.; Pütz, B.; Loleit, V.; Weber, F.; et al. Fingolimod induces neuroprotective factors in human astrocytes. J. Neuroinflamm. 2015, 12, 184. [Google Scholar] [CrossRef]
- Agudo-López, A.; Miguel, B.G.; Fernández, I.; Martínez, A.M. Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 2010, 470, 130–133. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2010, 25, 600–612. [Google Scholar] [CrossRef]
- Villanueva, J.; Gimenez-Molina, Y.; Davletov, B.; Gutiérrez, L.M. Vesicle Fusion as a Target Process for the Action of Sphingosine and Its Derived Drugs. Int. J. Mol. Sci. 2022, 23, 1086. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflamm. 2011, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.L.; Fang, J.; Almazan, G.; Antel, J. Sphingosine 1-phosphate receptor agonists promote axonal ensheathment by human fetal oligodendrocyte progenitors. Mult. Scler. J. 2011, 17, S366. [Google Scholar]
- O’sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflamm. 2016, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.; Hinterding, K.; Cooke, N.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Futur. Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Persson, A.-K.; Kim, I.; Zhao, P.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium Channels Contribute to Degeneration of Dorsal Root Ganglion Neurites Induced by Mitochondrial Dysfunction in an In Vitro Model of Axonal Injury. J. Neurosci. 2013, 33, 19250–19261. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Zhang, Q.; Chen, L.; Huang, X.; Zhang, Y.; Liu, X.; Liu, W.; Li, W. Mechanisms Underlying H2O2-Evoked Carbonyl Modification of Cytoskeletal Protein and Axon Injury in PC-12 Cells. Cell. Physiol. Biochem. 2018, 48, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959. [Google Scholar]
- Gorzalczany, S.B.; Basso, A.G.R. Strategies to apply 3Rs in preclinical testing. Pharmacol. Res. Perspect. 2021, 9, e00863. [Google Scholar] [CrossRef] [PubMed]
- Gülden, M.; Jess, A.; Kammann, J.; Maser, E.; Seibert, H. Cytotoxic potency of H2O2 in cell cultures: Impact of cell concentration and exposure time. Free Radic. Biol. Med. 2010, 49, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Kerschensteiner, M.; Bareyre, F.M.; Burgess, R.W.; Lichtman, J.W. Imaging axonal transport of mitochondria in vivo. Nat. Methods 2007, 4, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Andreasson, K. The Organotypic Hippocampal Slice Culture Model for Examining Neuronal Injury. J. Vis. Exp. 2010, 44, e2106. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative stress inhibits axonal transport: Implications for neurodegenerative diseases. Mol. Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef]
- Noraberg, J.; Poulsen, F.R.; Blaabjerg, M.; Kristensen, B.W.; Bonde, C.; Montero, M.; Meyer, M.; Gramsbergen, J.B.; Zimmer, J. Organotypic Hippocampal Slice Cultures for Studies of Brain Damage, Neuroprotection and Neurorepair. Curr. Drug Target-CNS Neurol. Disord. 2005, 4, 435–452. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Length (µm) | Mitochondrial Area (µm2) | Aspect Ratio (1–∞). | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | KW | Mean ± SD | Median | KW | Mean ± SD | Median | KW | ||||
| Negative control | 1.469 ± 0.317 | 1.443 | 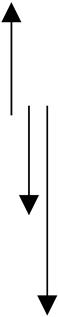 | **** | 0.755 ± 0.324 | 0.712 | | **** | 2.118 ± 0.228 | 2.108 | | >0.1 |
| H2O2 | 1.400 ± 0.353 | 1.370 | 0.698 ± 0.371 | 0.630 | 2.119 ± 0.280 | 2.110 | ||||||
| H2O2 + Siponimod | 1.433 ± 0.362 | 1.398 | ** | 0.711 ± 0.358 | 0.665 | >0.1 | 2.140 ± 0.306 | 2.106 | >0.1 | |||
| H2O2 + Fingolimod | 1.426 ± 0.493 | 1.368 | >0.1 | 0.713 ± 0.509 | 0.607 | >0.1 | 2.130 ± 0.410 | 2.076 | >0.1 | |||
| Track Displacement (µm) | Track Speed (µm/s) | Fraction of Motile Mitoch. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | KW | Mean ± SD | Median | KW | Mean ± SD | Median | KW | ||||
| Negative control | 2.009 ± 0.416 | 1.935 | | >0.1 | 0.219 ± 0.107 | 0.186 | | >0.1 | 0.277 ± 0.128 | 0.252 | | >0.1 |
| H2O2 | 1.968 ± 0.197 | 1.956 | 0.219 ± 0.081 | 0.209 | 0.220 ± 0.054 | 0.212 | ||||||
| H2O2 + Siponimod | 2.037 ± 0.628 | 1.938 | >0.1 | 0.244 ± 0.081 | 0.231 | **** | 0.241 ± 0.059 | 0.223 | >0.1 | |||
| H2O2 + Fingolimod | 1.962 ± 0.209 | 1.931 | >0.1 | 0.296 ± 0.084 | 0.289 | **** | 0.269 ± 0.087 | 0.284 | <0.1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, R.; Malla, B.; Höhrhan, M.; Infante-Duarte, C.; Anderhalten, L. Investigating the Mitoprotective Effects of S1P Receptor Modulators Ex Vivo Using a Novel Semi-Automated Live Imaging Set-Up. Int. J. Mol. Sci. 2024, 25, 261. https://doi.org/10.3390/ijms25010261
Ludwig R, Malla B, Höhrhan M, Infante-Duarte C, Anderhalten L. Investigating the Mitoprotective Effects of S1P Receptor Modulators Ex Vivo Using a Novel Semi-Automated Live Imaging Set-Up. International Journal of Molecular Sciences. 2024; 25(1):261. https://doi.org/10.3390/ijms25010261
Chicago/Turabian StyleLudwig, Rebecca, Bimala Malla, Maria Höhrhan, Carmen Infante-Duarte, and Lina Anderhalten. 2024. "Investigating the Mitoprotective Effects of S1P Receptor Modulators Ex Vivo Using a Novel Semi-Automated Live Imaging Set-Up" International Journal of Molecular Sciences 25, no. 1: 261. https://doi.org/10.3390/ijms25010261
APA StyleLudwig, R., Malla, B., Höhrhan, M., Infante-Duarte, C., & Anderhalten, L. (2024). Investigating the Mitoprotective Effects of S1P Receptor Modulators Ex Vivo Using a Novel Semi-Automated Live Imaging Set-Up. International Journal of Molecular Sciences, 25(1), 261. https://doi.org/10.3390/ijms25010261