


Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease

 , , , and
, , , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Structure and Role of αSyn
2.1. αSynuclein Structure
2.2. αSyn Aggregation and Variety of αSyn Species
2.3. Mechanisms Implicated in αSyn Modifications
2.3.1. Mutations
2.3.2. Ubiquitination
2.3.3. SUMOylation
2.3.4. Phosphorylation
2.3.5. Nitration
2.3.6. Truncation of the C-Terminal
2.4. The Significance of Synaptic αSyn Interactions in Health and Disease
3. αSyn-Induced Toxicity in Distinct Organelles
3.1. αSyn in the Mitochondria
3.2. αSyn-Induced Disturbances in the Ubiquitin-Proteasome System (UPS) and Autophagy-Lysosomal Pathway (ALP)
3.3. Endoplasmic Reticulum (ER)/Golgi Damage and Unfolded Protein Response (UPR) Signaling Pathway
3.4. Nuclear Dysfunction
4. αSyn Impact on the Extraneuronal Space
4.1. Seeding and Propagation of αSynuclein
4.2. Glial Interplay Contributes to Neuroinflammatory Response and Aggravation of αSyn-Induced Damage
4.2.1. Reactive Microglia and Adaptive Immune Response
4.2.2. Astrocytes
4.2.3. Oligodendrocytes
4.2.4. Excitotoxicity
5. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Hustad, E.; Aasly, J.O. Clinical and Imaging Markers of Prodromal Parkinson’s Disease. Front. Neurol. 2020, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and Molecular Diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular Milieu Imparts Distinct Pathological α-Synuclein Strains in α-Synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-Synuclein Strains in Parkinson’s Disease and Multiple System Atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Borghammer, P.; Just, M.K.; Horsager, J.; Skjærbæk, C.; Raunio, A.; Kok, E.H.; Savola, S.; Murayama, S.; Saito, Y.; Myllykangas, L.; et al. A Postmortem Study Suggests a Revision of the Dual-Hit Hypothesis of Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 166. [Google Scholar] [CrossRef]
- Seidel, K.; Mahlke, J.; Siswanto, S.; Krüger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.W.; et al. The Brainstem Pathologies of Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2015, 25, 121–135. [Google Scholar] [CrossRef]
- Schulz, J.; Pagano, G.; Fernández Bonfante, J.A.; Wilson, H.; Politis, M. Nucleus Basalis of Meynert Degeneration Precedes and Predicts Cognitive Impairment in Parkinson’s Disease. Brain 2018, 141, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Swallow, D.; Grosset, K.A.; Anichtchik, O.; Spillantini, M.; Grosset, D.G. Alpha-Synuclein in Peripheral Tissues and Body Fluids as a Biomarker for Parkinson’s Disease—A Systematic Review. Acta Neurol. Scand. 2014, 130, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Maass, F.; Rikker, S.; Dambeck, V.; Warth, C.; Tatenhorst, L.; Csoti, I.; Schmitz, M.; Zerr, I.; Leha, A.; Bähr, M.; et al. Increased Alpha-Synuclein Tear Fluid Levels in Patients with Parkinson’s Disease. Sci. Rep. 2020, 10, 8507. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.F.; Ekmark-Léwen, S.; Perdiki, M.; Klingstedt, T.; Hoffmann, A.; Wiechec, E.; Nilsson, P.; Nilsson, K.P.R.; Alafuzoff, I.; Ingelsson, M.; et al. Accumulation of Alpha-Synuclein within the Liver, Potential Role in the Clearance of Brain Pathology Associated with Parkinson’s Disease. Acta Neuropathol. Commun. 2021, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef] [PubMed]
- Courte, J.; Bousset, L.; Boxberg, Y.V.; Villard, C.; Melki, R.; Peyrin, J.M. The Expression Level of Alpha-Synuclein in Different Neuronal Populations Is the Primary Determinant of Its Prion-like Seeding. Sci. Rep. 2020, 10, 4895. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from α-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease Gene SNCA: Evolutionary and Structural Insights with Pathological Implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic Modifiers of Risk and Age at Onset in GBA Associated Parkinson’s Disease and Lewy Body Dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.; Mor, D.E.; Lightfoot, R.; Malkus, K.; Giasson, B.; Ischiropoulos, H. Evidence of Native α-Synuclein Conformers in the Human Brain. J. Biol. Chem. 2014, 289, 7929–7934. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-Synuclein. Nature 2013, E4–E6. [Google Scholar] [CrossRef]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in Central Nervous System and from Erythrocytes, Mammalian Cells, and Escherichia Coli Exists Predominantly as Disordered Monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef]
- Lucas, H.; Fernández, R. Navigating the Dynamic Landscape of Alpha-Synuclein Morphology: A Review of the Physiologically Relevant Tetrameric Conformation. Neural Regen. Res. 2020, 15, 407. [Google Scholar] [CrossRef]
- Sandal, M.; Valle, F.; Tessari, I.; Mammi, S.; Bergantino, E.; Musiani, F.; Brucale, M.; Bubacco, L.; Samorì, B. Conformational Equilibria in Monomeric α-Synuclein at the Single-Molecule Level. PLoS Biol. 2008, 6, e6. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Mondal, J. Conformational Plasticity in α-Synuclein and How Crowded Environment Modulates It. J. Phys. Chem. B 2023, 127, 4032–4049. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-Terminus of the Intrinsically Disordered Protein α-Synuclein Triggers Membrane Binding and Helix Folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Man, W.K.; Tahirbegi, B.; Vrettas, M.D.; Preet, S.; Ying, L.; Vendruscolo, M.; De Simone, A.; Fusco, G. The Docking of Synaptic Vesicles on the Presynaptic Membrane Induced by α-Synuclein Is Modulated by Lipid Composition. Nat. Commun. 2021, 12, 927. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; Hirpa, D.; Zheng, K.H.; Banerjee, R.; Gunawardena, S. The Non-Amyloidal Component Region of α-Synuclein Is Important for α-Synuclein Transport Within Axons. Front. Cell Neurosci. 2020, 13, 540. [Google Scholar] [CrossRef] [PubMed]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and Propagation of α-Synuclein Aggregation in the Nervous System. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A Short Motif in the N-Terminal Region of α-Synuclein Is Critical for Both Aggregation and Function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- Stephens, A.D.; Zacharopoulou, M.; Moons, R.; Fusco, G.; Seetaloo, N.; Chiki, A.; Woodhams, P.J.; Mela, I.; Lashuel, H.A.; Phillips, J.J.; et al. Extent of N-Terminus Exposure of Monomeric Alpha-Synuclein Determines Its Aggregation Propensity. Nat. Commun. 2020, 11, 2820. [Google Scholar] [CrossRef] [PubMed]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-Terminal Calcium Binding of α-Synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, X.; Xu, D.; Zheng, W.; Liu, M.; Li, C. Calcium Accelerates SNARE-Mediated Lipid Mixing through Modulating α-Synuclein Membrane Interaction. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Paik, S.R.; Yang, C.H. Structural and Functional Implications of C-Terminal Regions of α-Synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef] [PubMed]
- Izawa, Y.; Tateno, H.; Kameda, H.; Hirakawa, K.; Hato, K.; Yagi, H.; Hongo, K.; Mizobata, T.; Kawata, Y. Role of C-Terminal Negative Charges and Tyrosine Residues in Fibril Formation of α-Synuclein. Brain Behav. 2012, 2, 595–605. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping Long-Range Interactions in Alpha-Synuclein Using Spin-Label NMR and Ensemble Molecular Dynamics Simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Ullman, O.; Fisher, C.K.; Stultz, C.M. Explaining the Structural Plasticity of α-Synuclein. J. Am. Chem. Soc. 2011, 133, 19536–19546. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of Long-Range Tertiary Interactions Potentiates Aggregation of Natively Unstructured α-Synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef]
- Farzadfard, A.; Pedersen, J.N.; Meisl, G.; Somavarapu, A.K.; Alam, P.; Goksøyr, L.; Nielsen, M.A.; Sander, A.F.; Knowles, T.P.J.; Pedersen, J.S.; et al. The C-Terminal Tail of α-Synuclein Protects against Aggregate Replication but Is Critical for Oligomerization. Commun. Biol. 2022, 5, 123. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein Assembles into Higher-Order Multimers upon Membrane Binding to Promote SNARE Complex Formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein Occurs Physiologically as a Helically Folded Tetramer That Resists Aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A Soluble α-Synuclein Construct Forms a Dynamic Tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Von Saucken, V.E.; Bartels, T.; Selkoe, D. KTKEGV Repeat Motifs Are Key Mediators of Normal α-Synuclein Tetramerization: Their Mutation Causes Excess Monomers and Neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; Von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-Causing α-Synuclein Missense Mutations Shift Native Tetramers to Monomers as a Mechanism for Disease Initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 Deficiency Negatively Affects Physiological α-Synuclein Tetramers and Related Multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Ornelas, L.; Stricker, J.M.S.; Soriano-Cruz, S.; Weykopf, B.; Dettmer, U.; Muratore, C.R.; Scherzer, C.R.; Selkoe, D.J. Parkinson-Causing Mutations in LRRK2 Impair the Physiological Tetramerization of Endogenous α-Synuclein in Human Neurons. NPJ Park. Dis. 2022, 8, 118. [Google Scholar] [CrossRef]
- Glajch, K.E.; Moors, T.E.; Chen, Y.; Bechade, P.A.; Nam, A.Y.; Rajsombath, M.M.; McCaffery, T.D.; Dettmer, U.; Weihofen, A.; Hirst, W.D.; et al. Wild-Type GBA1 Increases the α-Synuclein Tetramer-Monomer Ratio, Reduces Lipid-Rich Aggregates, and Attenuates Motor and Cognitive Deficits in Mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2103425118. [Google Scholar] [CrossRef]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014.e8. [Google Scholar] [CrossRef]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.H.; Fanning, S.; Stirtz, G.; Kim, T.E.; Levy, O.A.; Young-Pearse, T.L.; et al. Cell Models of Lipid-Rich α-Synuclein Aggregation Validate Known Modifiers of α-Synuclein Biology and Identify Stearoyl-CoA Desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; Van Rossum, M.; Goldfarb, D.; et al. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Bell, R.; Castellana-Cruz, M.; Nene, A.; Thrush, R.J.; Xu, C.K.; Kumita, J.R.; Vendruscolo, M. Effects of N-Terminal Acetylation on the Aggregation of Disease-Related α-Synuclein Variants. J. Mol. Biol. 2022, 167825. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.; Thrush, R.J.; Castellana-Cruz, M.; Oeller, M.; Staats, R.; Nene, A.; Flagmeier, P.; Xu, C.K.; Satapathy, S.; Galvagnion, C.; et al. N-Terminal Acetylation of α-Synuclein Slows down Its Aggregation Process and Alters the Morphology of the Resulting Aggregates. Biochemistry 2022, 61, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and Spread of Pathogenic Protein Assemblies in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution Conditions Determine the Relative Importance of Nucleation and Growth Processes in α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.T.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Lipid Vesicles Trigger α-Synuclein Aggregation by Stimulating Primary Nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early Stages of Aggregation of Engineered α-Synuclein Monomers and Oligomers in Solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.; Meisl, G.; Buell, A.K.; Young, L.; Kaminski, C.F.; Knowles, T.P.J.; Sparr, E.; Linse, S. Secondary Nucleation of Monomers on Fibril Surface Dominates α-Synuclein Aggregation and Provides Autocatalytic Amyloid Amplification. Q. Rev. Biophys. 2017, 50, e6. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de Novo Compound Targeting α-Synuclein Improves Deficits in Models of Parkinson’s Disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef]
- Roostaee, A.; Beaudoin, S.; Staskevicius, A.; Roucou, X. Aggregation and Neurotoxicity of Recombinant α-Synuclein Aggregates Initiated by Dimerization. Mol. Neurodegener. 2013, 8, 5. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure Based Aggregation Studies Reveal the Presence of Helix-Rich Intermediate during α-Synuclein Aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [PubMed]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.M.; Raussens, V. Toxic Prefibrillar α-Synuclein Amyloid Oligomers Adopt a Distinctive Antiparallel β-Sheet Structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small Soluble α-Synuclein Aggregates Are the Toxic Species in Parkinson’s Disease. Nat. Commun. 2022, 13, 5512. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Fagerqvist, T.; Näsström, T.; Ihse, E.; Lindström, V.; Sahlin, C.; Fangmark Tucker, S.M.; Kasaryan, A.; Karlsson, M.; Nikolajeff, F.; Schell, H.; et al. Off-Pathway α-Synuclein Oligomers Seem to Alter α-Synuclein Turnover in a Cell Model but Lack Seeding Capability in Vivo. Amyloid 2013, 20, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed]
- Miti, T.; Mulaj, M.; Schmit, J.D.; Muschol, M. Stable, Metastable, and Kinetically Trapped Amyloid Aggregate Phases. Biomacromolecules 2015, 16, 326–335. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural Basis of Membrane Disruption and Cellular Toxicity by α-Synuclein Oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-Resolution Structure of a Vesicle Disrupting α-Synuclein Oligomer That Accumulates during Fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, S.; Kim, T.H.; Lee, J.H.; Yang, J.E.; Joo, K.; Lee, J.; Char, K.; Paik, S.R. Morphological Evaluation of Meta-Stable Oligomers of α-Synuclein with Small-Angle Neutron Scattering. Sci. Rep. 2018, 8, 14295. [Google Scholar] [CrossRef]
- Ono, K. The Oligomer Hypothesis in α-Synucleinopathy. Neurochem. Res. 2017, 42, 3362–3371. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM Structure of Alpha-Synuclein Fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of Full-Length α-Synuclein Reveals Fibril Polymorphs with a Common Structural Kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid Fibril Structure of α-Synuclein Determined by Cryo-Electron Microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van Den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Assessment of Heterogeneity among Participants in the Parkinson’s Progression Markers Initiative Cohort Using α-Synuclein Seed Amplification: A Cross-Sectional Study. Lancet Neurol. 2023, 22, 407–417. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Pritzkow, S.; Shahnawaz, M.; Farris, C.M.; Soto, C. Seed Amplification Assay for the Detection of Pathologic Alpha-Synuclein Aggregates in Cerebrospinal Fluid. Nat. Protoc. 2023, 18, 1179–1196. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Y.; Liu, Z.; Long, H.; Zhao, C.; Luo, F.; Sun, Y.; Tao, Y.; Su, X.; Li, D.; et al. Parkinson’s Disease Associated Mutation E46K of α-Synuclein Triggers the Formation of a Distinct Fibril Structure. Nat. Commun. 2020, 11, 2643. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Thüne, K.; Cramm, M.; Rabano, A.; Zafar, S.; Stoops, E.; Vanderstichele, H.; Villar-Pique, A.; Llorens, F.; et al. Effect of the Micro-Environment on α-Synuclein Conversion and Implication in Seeded Conversion Assays. Transl. Neurodegener. 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-Synuclein Post-Translational Modifications as Potential Biomarkers for Parkinson Disease and Other Synucleinopathies. Mol. Cell Proteom. 2013, 12, 3543–3558. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated α-Synuclein Is Ubiquitinated in α-Synucleinopathy Lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.Y. Oxidative Damage Linked to Neurodegeneration by Selective α-Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jäkälä, P.; Hartmann, T.; Price, D.L.; et al. Aggregation Promoting C-Terminal Truncation of α-Synuclein Is a Normal Cellular Process and Is Enhanced by the Familial Parkinson’s Disease-Linked Mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Elghani, F.A.; Bandopadhyay, R.; Engelender, S. SUMOylation and Ubiquitination Reciprocally Regulate α-Synuclein Degradation and Pathological Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc Modification Blocks the Aggregation and Toxicity of the Protein α-Synuclein Associated with Parkinson’s Disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic Comparison of the Effects of Alpha-Synuclein Mutations on Its Oligomerization and Aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Giasson, B.I. The A53E α-Synuclein Pathological Mutation Demonstrates Reduced Aggregation Propensity in Vitro and in Cell Culture. Neurosci. Lett. 2015, 597, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of Fibrils Formed by α-Synuclein Hereditary Disease Mutant H50Q Reveal New Polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, Site-Specific Ubiquitin Modification of α-Synuclein Reveals Differential Effects on Aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of α-Synuclein by Siah-1 Promotes α-Synuclein Aggregation and Apoptotic Cell Death. Hum. Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Manzanza, N.d.O.; Sedlackova, L.; Kalaria, R.N. Alpha-Synuclein Post-Translational Modifications: Implications for Pathogenesis of Lewy Body Disorders. Front. Aging Neurosci. 2021, 13, 690293. [Google Scholar] [CrossRef]
- Abeywardana, T.; Pratt, M.R. Extent of Inhibition of α-Synuclein Aggregation in Vitro by SUMOylation Is Conjugation Site- and SUMO Isoform-Selective. Biochemistry 2015, 54, 959–961. [Google Scholar] [CrossRef]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and Serine Phosphorylation of α-Synuclein Have Opposing Effects on Neurotoxicity and Soluble Oligomer Formation. J. Clin. Investig. 2009, 119. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits α-Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Iwasaki, Y.; Yamashita, Y.; Irie, K.; Hosokawa, M.; Satoh, K.; Mishima, K. Tyrosine 136 Phosphorylation of α-Synuclein Aggregates in the Lewy Body Dementia Brain: Involvement of Serine 129 Phosphorylation by Casein Kinase 2. Acta Neuropathol. Commun. 2021, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.Y.; Ischiropoulos, H. Functional Consequences of α-Synuclein Tyrosine Nitration: Diminished Binding to Lipid Vesicles and Increased Fibril Formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamashita, H.; Nakamura, T.; Nagano, Y.; Nakamura, S. Tyrosine 125 of α-Synuclein Plays a Critical Role for Dimerization Following Nitrative Stress. Brain Res. 2002, 938, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-Terminal Truncation Exacerbates the Aggregation and Cytotoxicity of α-Synuclein: A Vicious Cycle in Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wateren, I.M.; Knowles, T.P.J.; Buell, A.K.; Dobson, C.M.; Galvagnion, C. C-Terminal Truncation of α-Synuclein Promotes Amyloid Fibril Amplification at Physiological PH. Chem. Sci. 2018, 9, 5506–5516. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.R.; Rhoades, E. Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. α-Synuclein Shares Physical and Functional Homology with 14-3-3 Proteins. J. Neurosci. 1999, 19, 5782–5791. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-Synuclein Cooperates with CSPα in Preventing Neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. α-Synuclein Negatively Regulates Protein Kinase Cδ Expression to Suppress Apoptosis in Dopaminergic Neurons by Reducing P300 Histone Acetyltransferase Activity. J. Neurosci. 2011, 31, 2035–2051. [Google Scholar] [CrossRef]
- Plotegher, N.; Kumar, D.; Tessari, I.; Brucale, M.; Munari, F.; Tosatto, L.; Belluzzi, E.; Greggio, E.; Bisaglia, M.; Capaldi, S.; et al. The Chaperone-like Protein 14-3-3η Interacts with Human α-Synuclein Aggregation Intermediates Rerouting the Amyloidogenic Pathway and Reducing α-Synuclein Cellular Toxicity. Hum. Mol. Genet. 2014, 23, 5615–5629. [Google Scholar] [CrossRef] [PubMed]
- Micheli, L.; Creanza, T.M.; Ceccarelli, M.; D’Andrea, G.; Giacovazzo, G.; Ancona, N.; Coccurello, R.; Scardigli, R.; Tirone, F. Transcriptome Analysis in a Mouse Model of Premature Aging of Dentate Gyrus: Rescue of Alpha-Synuclein Deficit by Virus-Driven Expression or by Running Restores the Defective Neurogenesis. Front. Cell Dev. Biol. 2021, 9, 696684. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Garcia Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice Lacking α-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef]
- Peng, X.; Peng, X.M.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. Alpha-Synuclein Activation of Protein Phosphatase 2A Reduces Tyrosine Hydroxylase Phosphorylation in Dopaminergic Cells. J. Cell Sci. 2005, 118, 3523–3530. [Google Scholar] [CrossRef]
- Tehranian, R.; Montoya, S.E.; Van Laar, A.D.; Hastings, T.G.; Perez, R.G. Alpha-Synuclein Inhibits Aromatic Amino Acid Decarboxylase Activity in Dopaminergic Cells. J. Neurochem. 2006, 99, 1188–1196. [Google Scholar] [CrossRef]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by α-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Björklund, A. Impaired Neurotransmission Caused by Overexpression of α-Synuclein in Nigral Dopamine Neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef]
- Gaugler, M.N.; Genc, O.; Bobela, W.; Mohanna, S.; Ardah, M.T.; El-Agnaf, O.M.; Cantoni, M.; Bensadoun, J.C.; Schneggenburger, R.; Knott, G.W.; et al. Nigrostriatal Overabundance of α-Synuclein Leads to Decreased Vesicle Density and Deficits in Dopamine Release That Correlate with Reduced Motor Activity. Acta Neuropathol. 2012, 123, 653–669. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Dovonou, A.; Bolduc, C.; Soto Linan, V.; Gora, C.; Peralta III, M.R.; Lévesque, M. Animal Models of Parkinson’s Disease: Bridging the Gap between Disease Hallmarks and Research Questions. Transl. Neurodegener. 2023, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Salvá, M.; Van Der Perren, A.; Casadei, N.; Stroobants, S.; Nuber, S.; D’Hooge, R.; Van Den Haute, C.; Baekelandt, V. RAAV2/7 Vector-Mediated Overexpression of Alpha-Synuclein in Mouse Substantia Nigra Induces Protein Aggregation and Progressive Dose-Dependent Neurodegeneration. Mol. Neurodegener. 2013, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Expression of α-Synuclein Is Regulated in a Neuronal Cell Type-Dependent Manner. Anat. Sci. Int. 2019, 94, 11–22. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 5999. [Google Scholar] [CrossRef] [PubMed]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. α-Synuclein and Synapsin III Cooperatively Regulate Synaptic Function in Dopamine Neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of Vesicular Monoamine Transporter-2 Activity in Alpha-Synuclein Stably Transfected SH-SY5Y Cells. Cell Mol. Neurobiol. 2008, 28, 35–47. [Google Scholar] [CrossRef]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of Oligomer-Prone α-Synuclein Exacerbates Synaptic and Neuronal Degeneration in Vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein Oligomers Induce Early Axonal Dysfunction in Human IPSC-Based Models of Synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Zhu, M.; Qin, Z.-J.; Hu, D.; Munishkina, L.A.; Fink, A.L. Alpha-Synuclein Can Function as an Antioxidant Preventing Oxidation of Unsaturated Lipid in Vesicles. Biochemistry 2006, 45, 8135–8142. [Google Scholar] [CrossRef]
- Menges, S.; Minakaki, G.; Schaefer, P.M.; Meixner, H.; Prots, I.; Schlötzer-Schrehardt, U.; Friedland, K.; Winner, B.; Outeiro, T.F.; Winklhofer, K.F.; et al. Alpha-Synuclein Prevents the Formation of Spherical Mitochondria and Apoptosis under Oxidative Stress. Sci. Rep. 2017, 7, 42942. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Marchesan, E.; Zonta, L.; Bono, F.; Bottani, E.; Longhena, F.; Ziviani, E.; Valerio, A.; Bellucci, A. Alpha-Synuclein Preserves Mitochondrial Fusion and Function in Neuronal Cells. Oxid. Med. Cell Longev. 2019, 2019, 4246350. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region Specific Mitochondrial Impairment in Mice with Widespread Overexpression of Alpha-Synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- Floor, E.; Wetzel, M.G. Increased Protein Oxidation in Human Substantia Nigra Pars Compacta in Comparison with Basal Ganglia and Prefrontal Cortex Measured with an Improved Dinitrophenylhydrazine Assay. J. Neurochem. 1998, 70, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative Stress Involvement in Alpha-Synuclein Oligomerization in Parkinson’s Disease Cybrids. Antioxid. Redox Signal 2009, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High Levels of Mitochondrial DNA Deletions in Substantia Nigra Neurons in Aging and Parkinson Disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA Deletions Are Abundant and Cause Functional Impairment in Aged Human Substantia Nigra Neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective Mitochondrial DNA Homeostasis in the Substantia Nigra in Parkinson Disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein Binds to TOM20 and Inhibits Mitochondrial Protein Import in Parkinson’s Disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage-Dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.H.; Fuentes, F.; Vanasco, V.; Alvarez, S.; Alaimo, A.; Cassina, A.; Coluccio Leskow, F.; Velazquez, F. Alpha-Synuclein Mitochondrial Interaction Leads to Irreversible Translocation and Complex I Impairment. Arch. Biochem. Biophys. 2018, 651, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal 2016, 24, 376–391. [Google Scholar] [CrossRef]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP Induces Alpha-Synuclein Aggregation in the Substantia Nigra of Baboons. Neuroreport 2000, 11, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kame, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Env. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Zigoneanu, I.G.; Yang, Y.J.; Krois, A.S.; Haque, M.E.; Pielak, G.J. Interaction of α-Synuclein with Vesicles That Mimic Mitochondrial Membranes. Biochim. Biophys. Acta Biomembr. 2012, 1818, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.L.; Chappard, A.; Singh, B.P.; Maclachlan, C.; Rodrigues, M.; Fedotova, E.I.; Berezhnov, A.V.; De, S.; Peddie, C.J.; Athauda, D.; et al. Pathological Structural Conversion of α-Synuclein at the Mitochondria Induces Neuronal Toxicity. Nat. Neurosci. 2022, 25, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, Structure, and Function of the Mitochondrial Permeability Transition Pore: Controversies, Consensus, Recent Advances, and Future Directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Beccano-Kelly, D.A.; Cherubini, M.; Mousba, Y.; Cramb, K.M.L.; Giussani, S.; Caiazza, M.C.; Rai, P.; Vingill, S.; Bengoa-Vergniory, N.; Ng, B.; et al. Calcium Dysregulation Combined with Mitochondrial Failure and Electrophysiological Maturity Converge in Parkinson’s IPSC-Dopamine Neurons. iScience 2023, 26, 107044. [Google Scholar] [CrossRef]
- Wilson, E.L.; Metzakopian, E. ER-Mitochondria Contact Sites in Neurodegeneration: Genetic Screening Approaches to Investigate Novel Disease Mechanisms. Cell Death Differ. 2021, 28, 1804–1821. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein Aggregates Activate Calcium Pump SERCA Leading to Calcium Dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef] [PubMed]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 Relocalize at Mitochondria-Associated Membranes during Mitophagy and Promote ER-Mitochondria Tethering and Autophagosome Formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial Alpha-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy in Vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Liu, K.; Liu, Z.F.; Cong, L.; Lei, M.Y.; Ma, Z.; Li, J.; Deng, Y.; Liu, W.; Xu, B. Manganese-Induced Alpha-Synuclein Overexpression Aggravates Mitochondrial Damage by Repressing PINK1/Parkin-Mediated Mitophagy. Food Chem. Toxicol. 2021, 152, 112213. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Lenkiewicz, A.M.; Babiec, L.; Murawska, E.; Jęśko, H.M.; Cieślik, M.; Culmsee, C.; Adamczyk, A. Exogenous Alpha-Synuclein Evoked Parkin Downregulation Promotes Mitochondrial Dysfunction in Neuronal Cells. Implications for Parkinson’s Disease Pathology. Front. Aging Neurosci. 2021, 13, 591475. [Google Scholar] [CrossRef]
- Stykel, M.G.; Humphries, K.M.; Kamski-Hennekam, E.; Buchner-Duby, B.; Porte-Trachsel, N.; Ryan, T.; Coackley, C.L.; Bamm, V.V.; Harauz, G.; Ryan, S.D. α-Synuclein Mutation Impairs Processing of Endomembrane Compartments and Promotes Exocytosis and Seeding of α-Synuclein Pathology. Cell Rep. 2021, 35, 109099. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin Exposure on the Outer Mitochondrial Membrane Modulates α-Synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of Mitochondrial Fusion by α-Synuclein Is Rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-Associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef]
- Eschbach, J.; Von Einem, B.; Müller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual Exacerbation of Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Deregulation and α-Synuclein Oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef]
- Griffey, C.J.; Yamamoto, A. Macroautophagy in CNS Health and Disease. Nat. Rev. Neurosci. 2022, 23, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant Alpha-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct Roles in Vivo for the Ubiquitin-Proteasome System and the Autophagy-Lysosomal Pathway in the Degradation of α-Synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Wills, J.; Jones, J.; Haggerty, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Elevated Tauopathy and Alpha-Synuclein Pathology in Postmortem Parkinson’s Disease Brains with and without Dementia. Exp. Neurol. 2010, 225, 210–218. [Google Scholar] [CrossRef]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a New Form of α-Synuclein by Parkin from Human Brain: Implications for Parkinson’s Disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Granek, Z.; Barczuk, J.; Siwecka, N.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. Int. J. Mol. Sci. 2023, 24, 2044. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Ugolino, J.; Fang, S.; Kubisch, C.; Monteiro, M.J. Mutant Atp13a2 Proteins Involved in Parkinsonism Are Degraded by ER-Associated Degradation and Sensitize Cells to ER-Stress Induced Cell Death. Hum. Mol. Genet. 2011, 20, 3565–3577. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to- Golgi Retrieval of Lamp2a, a Receptor of Chaperone- Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-Type ATPase ATP13A2/PARK9 Function Induces General Lysosomal Deficiency and Leads to Parkinson Disease Neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Van den Haute, C.; Lobbestael, E.; Martin, S.; van Veen, S.; Vangheluwe, P.; Baekelandt, V. ATP13A2 Regulates Cellular α-Synuclein Multimerization, Membrane Association, and Externalization. Int. J. Mol. Sci. 2021, 22, 2689. [Google Scholar] [CrossRef]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible Expression of Mutant α-Synuclein Decreases Proteasome Activity and Increases Sensitivity to Mitochondria-Dependent Apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome Impairment by α-Synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and Monomeric α-Synuclein Bind to the S6′ Proteasomal Protein and Inhibit Proteasomal Function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef]
- Popova, B.; Galka, D.; Häffner, N.; Wang, D.; Schmitt, K.; Valerius, O.; Knop, M.; Braus, G.H. α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast. Cells 2021, 10, 2229. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-Modified α-Synuclein Blocks Chaperone-Mediated Autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein Impairs Macroautophagy: Implications for Parkinson’s Disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase Deficiency in Substantia Nigra of Parkinson Disease Brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, J.; Xie, W.; Huang, L.; Zhong, C.; Chen, Z. Beclin1 and HMGB1 Ameliorate the Aα-Synuclein-Mediated Autophagy Inhibition in PC12 Cells. Diagn. Pathol. 2016, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective Molecular Alterations in the Autophagy Pathway in Patients with Lewy Body Disease and in Models of α-Synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-Induced Lysosomal Dysfunction Occurs through Disruptions in Protein Trafficking in Human Midbrain Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Gao, P.; Arzberger, T.; Höllerhage, M.; Herms, J.; Höglinger, G.; Koeglsperger, T. Alpha-Synuclein Defects Autophagy by Impairing SNAP29-Mediated Autophagosome-Lysosome Fusion. Cell Death Dis. 2021, 12, 854. [Google Scholar] [CrossRef]
- Ma, Z.; Liang, H.; Hu, B.; Cai, S.; Yan, D. Autophagy-regulating MiRNAs: Novel Therapeutic Targets for Parkinson’s Disease (Review). Int. J. Mol. Med. 2023, 51, 50. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the Unfolded Protein Response by α-Synuclein in Experimental Models of Parkinson’s Disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of α-Synucleinopathy in Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of Dopaminergic Neuron Survival by the Unfolded Protein Response Transcription Factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-Synuclein Induces the Unfolded Protein Response in Parkinson’s Disease SNCA Triplication IPSC-Derived Neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An Unfolded Putative Transmembrane Polypeptide, Which Can Lead to Endoplasmic Reticulum Stress, Is a Substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s IPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the Unfolded Protein Response in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Makioka, K.; Yamazaki, T.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Involvement of Endoplasmic Reticulum Stress Defined by Activated Unfolded Protein Response in Multiple System Atrophy. J. Neurol. Sci. 2010, 297, 60–65. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the Unfolded Protein Response Is an Early Event in Alzheimer’s and Parkinson’s Disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.J.; Ron, D.; Przedborski, S.; et al. CHOP/GADD153 Is a Mediator of Apoptotic Death in Substantia Nigra Dopamine Neurons in an in Vivo Neurotoxin Model of Parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and Rescue of α-Synuclein Toxicity in Parkinson Patient-Derived Neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Shin, K.S.; Kwon, Y.K. A-Synuclein Induces Unfolded Protein Response via Distinct Signaling Pathway Independent of Er-membrane Kinases. Integr. Biosci. 2006, 10, 115–120. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Zhang, Y.; Guerrero-Muñoz, M.J.; Kayed, R.; Rincon-Limas, D.E.; Fernandez-Funez, P. Differential Activation of the ER Stress Factor XBP1 by Oligomeric Assemblies. Neurochem. Res. 2012, 37, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-Mediated Inhibition of ATF6 Processing into COPII Vesicles Disrupts UPR Signaling in Parkinson’s Disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Schekman, R. In Vitro Reconstitution of ER-Stress Induced ATF6 Transport in COPII Vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s Disease Protein Alpha-Synuclein Is a Modulator of Processing Bodies and MRNA Stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Jiang, K.; Rocha, S.; Westling, A.; Kesarimangalam, S.; Dorfman, K.D.; Wittung-Stafshede, P.; Westerlund, F. Alpha-Synuclein Modulates the Physical Properties of DNA. Chem.—A Eur. J. 2018, 24, 15685–15690. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-Synuclein Is a DNA Binding Protein That Modulates DNA Repair with Implications for Lewy Body Disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef]
- Mukherjee, S.K.; Knop, J.M.; Oliva, R.; Möbitz, S.; Winter, R. Untangling the Interaction of α-Synuclein with DNA i-Motifs and Hairpins by Volume-Sensitive Single-Molecule FRET Spectroscopy. RSC Chem. Biol. 2021, 2, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear Localization of Alpha-Synuclein and Its Interaction with Histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-Synuclein Acts in the Nucleus to Inhibit Histone Acetylation and Promote Neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Jos, S.; Gogoi, H.; Prasad, T.K.; Hurakadli, M.A.; Kamariah, N.; Padmanabhan, B.; Padavattan, S. Molecular Insights into α-Synuclein Interaction with Individual Human Core Histones, Linker Histone, and DsDNA. Protein Sci. 2021, 30, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Nishie, M.; Mori, F.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. A Quantitative Investigation of Neuronal Cytoplasmic and Intranuclear Inclusions in the Pontine and Inferior Olivary Nuclei in Multiple System Atrophy. Neuropathol. Appl. Neurobiol. 2004, 30, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Pinho, R.; Paiva, I.; Jerčić, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear Localization and Phosphorylation Modulate Pathological Effects of Alpha-Synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef]
- Koss, D.J.; Erskine, D.; Porter, A.; Palmoski, P.; Menon, H.; Todd, O.G.J.; Leite, M.; Attems, J.; Outeiro, T.F. Nuclear Alpha-Synuclein Is Present in the Human Brain and Is Modified in Dementia with Lewy Bodies. Acta Neuropathol. Commun. 2022, 10, 98. [Google Scholar] [CrossRef]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and Neuritic Distribution of Serine-129 Phosphorylated α-Synuclein in Transgenic Mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef]
- Weston, L.J.; Bowman, A.M.; Osterberg, V.R.; Meshul, C.K.; Woltjer, R.L.; Unni, V.K. Aggregated Alpha-Synuclein Inclusions within the Nucleus Predict Impending Neuronal Cell Death in a Mouse Model of Parkinsonism. Int. J. Mol. Sci. 2022, 23, 15294. [Google Scholar] [CrossRef]
- Beck, M.; Hurt, E. The Nuclear Pore Complex: Understanding Its Function through Structural Insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Rousseaux, M.W.C.; de Haro, M.; Lasagna-Reeves, C.A.; de Maio, A.; Park, J.; Jafar-Nejad, P.; Al-Ramahi, I.; Sharma, A.; See, L.; Lu, N.; et al. TRIM28 Regulates the Nuclear Accumulation and Toxicity of Both Alpha-Synuclein and Tau. Elife 2016, 5, e19809. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The Mechanistic Role of Alpha-Synuclein in the Nucleus: Impaired Nuclear Function Caused by Familial Parkinson’s Disease SNCA Mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef]
- Jiang, K.; Rocha, S.; Kumar, R.; Westerlund, F.; Wittung-Stafshede, P. C-Terminal Truncation of α-Synuclein Alters DNA Structure from Extension to Compaction. Biochem. Biophys. Res. Commun. 2021, 568, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovičić, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The Novel Parkinson’s Disease Linked Mutation G51D Attenuates in Vitro Aggregation and Membrane Binding of α-Synuclein, and Enhances Its Secretion and Nuclear Localization in Cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, H.; Jo, A.; Khang, R.; Park, C.H.; Park, S.J.; Kwag, E.; Shin, J.H. α-Synuclein A53T Binds to Transcriptional Adapter 2-Alpha and Blocks Histone H3 Acetylation. Int. J. Mol. Sci. 2021, 22, 5392. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of Cellular Processes by Histone and Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, X.; Zhang, L.; Zhang, Y.; Feng, L. Nuclear Accumulation of Histone Deacetylase 4 (HDAC4) Exerts Neurotoxicity in Models of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6970–6983. [Google Scholar] [CrossRef]
- de Oliveira, R.M.; Vicente Miranda, H.; Francelle, L.; Pinho, R.; Szegö, É.M.; Martinho, R.; Munari, F.; Lázaro, D.F.; Moniot, S.; Guerreiro, P.; et al. The Mechanism of Sirtuin 2–Mediated Exacerbation of Alpha-Synuclein Toxicity in Models of Parkinson Disease. PLoS Biol. 2017, 15, e2000374. [Google Scholar] [CrossRef]
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schütz, A.L.; Rajput, A.; Szego, É.M.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium Butyrate Rescues Dopaminergic Cells from Alpha-Synuclein-Induced Transcriptional Deregulation and DNA Damage. Hum. Mol. Genet. 2017, 26, 2231–2246. [Google Scholar] [CrossRef]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. α-Synuclein Enhances Histone H3 Lysine-9 Dimethylation and H3K9me2-Dependent Transcriptional Responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive Patterns of DNA Methylation Associated with Parkinson Disease: Identification of Concordant Epigenetic Changes in Brain and Peripheral Blood Leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein Sequesters Dnmt1 from the Nucleus: A Novel Mechanism for Epigenetic Alterations in Lewy Body Diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG Demethylation Enhances Alpha-Synuclein Expression and Affects the Pathogenesis of Parkinson’s Disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy Bodies in Grafted Neurons in Subjects with Parkinson’s Disease Suggest Host-to-Graft Disease Propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.Y. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, V.R.; Spinelli, K.J.; Weston, L.J.; Luk, K.C.; Woltjer, R.L.; Unni, V.K. Progressive Aggregation of Alpha-Synuclein and Selective Degeneration of Lewy Inclusion-Bearing Neurons in a Mouse Model of Parkinsonism. Cell Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Kubo, M.; Shimozawa, A.; Akiyama, H.; Hasegawa, M. Pathological Alpha-Synuclein Propagates through Neural Networks. Acta Neuropathol. Commun. 2014, 2, 88. [Google Scholar] [CrossRef]
- Tremblay, C.; Rahayel, S.; Vo, A.; Morys, F.; Shafiei, G.; Abbasi, N.; Markello, R.D.; Gan-Or, Z.; Misic, B.; Dagher, A. Brain Atrophy Progression in Parkinson’s Disease Is Shaped by Connectivity and Local Vulnerability. Brain Commun. 2021, 3, fcab269. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of Oligomeric Forms of A-synuclein Protein in Human Plasma as a Potential Biomarker for Parkinson’s Disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Singer, W.; Schmeichel, A.M.; Shahnawaz, M.; Schmelzer, J.D.; Boeve, B.F.; Sletten, D.M.; Gehrking, T.L.; Gehrking, J.A.; Olson, A.D.; Savica, R.; et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain in Spinal Fluid Differentiate Multiple System Atrophy from Lewy Body Synucleinopathies. Ann. Neurol. 2020, 88, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Steiner, J.A.; Angot, E.; Brundin, P. A Deadly Spread: Cellular Mechanisms of α-Synuclein Transfer. Cell Death Differ. 2011, 18, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.-M.; Gravel, C.; Lévesque, M. Treating Parkinson’s Disease with Antibodies: Previous Studies and Future Directions. J. Park. Dis. 2021, 11, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Chedid, J.; Labrador-Garrido, A.; Zhong, S.; Gao, J.; Zhao, Y.; Perera, G.; Kim, W.S.; Halliday, G.M.; Dzamko, N. A Small Molecule Toll-like Receptor Antagonist Rescues α-Synuclein Fibril Pathology. J. Biol. Chem. 2022, 298, 102260. [Google Scholar] [CrossRef] [PubMed]
- Neupane, S.; De Cecco, E.; Aguzzi, A. The Hidden Cell-to-Cell Trail of α-Synuclein Aggregates. J. Mol. Biol. 2023, 435, 167930. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial Exosomes Facilitate α-Synuclein Transmission in Parkinson’s Disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling Nanotubes Spread Fibrillar A-synuclein by Intercellular Trafficking of Lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Ito, N.; Tsuji, M.; Adachi, N.; Nakamura, S.; Sarkar, A.K.; Ikenaka, K.; Aguirre, C.; Kimura, A.M.; Kiuchi, Y.; Mochizuki, H.; et al. Extracellular High Molecular Weight α-Synuclein Oligomers Induce Cell Death by Disrupting the Plasma Membrane. NPJ Park. Dis. 2023, 9, 139. [Google Scholar] [CrossRef]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M.Y. α-Synuclein (Asyn) Preformed Fibrils Induce Endogenous Asyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, Y.; Xia, D.; Meng, L.; He, M.; Liu, C.; Zhang, Z. Silica Nanoparticles Promote α-Synuclein Aggregation and Parkinson’s Disease Pathology. Front. Neurosci. 2022, 15, 807988. [Google Scholar] [CrossRef]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The Release of Toxic Oligomers from α-Synuclein Fibrils Induces Dysfunction in Neuronal Cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of α-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding Induced by α-Synuclein Oligomers Provides Evidence for Spreading of α-Synuclein Pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Nawa, M.; Konno, H.; Kodera, N.; Ando, T.; Teplow, D.B.; Ono, K. Self- And Cross-Seeding on α-Synuclein Fibril Growth Kinetics and Structure Observed by High-Speed Atomic Force Microscopy. ACS Nano 2020, 14, 9979–9989. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 2020, 105, 260–275.e6. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Ono, K. Interactions of Amyloid Coaggregates with Biomolecules and Its Relevance to Neurodegeneration. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Y.; Lindqvist, D.; Chen, H.; Thomas, P.M.; Brundin, P.; et al. The Vermiform Appendix Impacts the Risk of Developing Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.L.; Tasselli, M.; Aubert, P.; Kruse, N.; et al. Bidirectional Gut-to-Brain and Brain-to-Gut Propagation of Synucleinopathy in Non-Human Primates. Brain 2020, 143, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-Analysis of the Parkinson’s Disease Gut Microbiome Suggests Alterations Linked to Intestinal Inflammation. NPJ Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the: Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-Analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Gao, H.M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M.Y. Neuroinflammation and Oxidation/Nitration of α-Synuclein Linked to Dopaminergic Neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated α-Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Rodríguez, P.V.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early Microglial Activation and Peripheral Inflammation in Dementia with Lewy Bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.P.; Marmion, D.J.; Schonhoff, A.M.; Jurkuvenaite, A.; Won, W.J.; Standaert, D.G.; Kordower, J.H.; Harms, A.S. T Cell Infiltration in Both Human Multiple System Atrophy and a Novel Mouse Model of the Disease. Acta Neuropathol. 2020, 139, 855–874. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-Released Oligomeric α-Synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-Dependent TLR1/2 Signaling by Misfolded α-Synuclein, a Protein Linked to Neurodegenerative Disorders. Sci. Signal 2015, 8, ra45. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like Receptor 4 Is Required for α-Synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Cao, S.; Standaert, D.G.; Harms, A.S. The Gamma Chain Subunit of Fc Receptors Is Required for Alpha-Synuclein-Induced pro-Inflammatory Signaling in Microglia. J. Neuroinflammation 2012, 9, 259. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α-Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Wang, W.; Nguyen, L.T.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 Causes Truncation and Aggregation of the Parkinson’s Disease-Associated Protein α-Synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 Mitigates LRRK2- and α-Synuclein-Induced Neurodegeneration by Modulating Proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia Affect α-Synuclein Cell-to-Cell Transfer in a Mouse Model of Parkinson’s Disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Harm, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII Is Required for α-Synuclein-Induced Activation of Microglia, CD4 T Cell Proliferation, and Dopaminergic Neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T Cells from Patients with Parkinson’s Disease Recognize α-Synuclein Peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I Expression Renders Catecholaminergic Neurons Susceptible to T-Cell-Mediated Degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef] [PubMed]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bové, J. CD8 T Cell Nigral Infiltration Precedes Synucleinopathy in Early Stages of Parkinson’s Disease. Brain 2020, 143, 3717–3733. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-Specific T Cell Reactivity Is Associated with Preclinical and Early Parkinson’s Disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s Disease Patients Have a Complex Phenotypic and Functional Th1 Bias: Cross-Sectional Studies of CD4+ Th1/Th2/T17 and Treg in Drug-Naïve and Drug-Treated Patients. J. Neuroinflammation 2018, 15, 205. [Google Scholar] [CrossRef]
- Liu, S.Y.; Qiao, H.W.; Song, T.B.; Liu, X.L.; Yao, Y.X.; Zhao, C.S.; Barret, O.; Xu, S.L.; Cai, Y.N.; Tamagnan, G.D.; et al. Brain Microglia Activation and Peripheral Adaptive Immunity in Parkinson’s Disease: A Multimodal PET Study. J. Neuroinflammation 2022, 19, 209. [Google Scholar] [CrossRef]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Kim, C.; Park, S.; Joo, J.; Choi, B.; Yang, D.; Jun, K.; Eom, J.; Lee, S.J.; Chung, S.J.; et al. Characterization of Altered Molecular Mechanisms in Parkinson’s Disease through Cell Type–Resolved Multiomics Analyses. Sci. Adv. 2023, 9, eabo2467. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Wang, X.; Liu, C.; Zhang, H.-L. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front. Cell Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of Region-Specific Astrocyte Subtypes at Single Cell Resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G.; Yan, Y.-Q.; Ma, C.-G. Astrocytes: A Double-Edged Sword in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 1702. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-Synuclein-Positive Filamentous Inclusions in Astrocytes and Oligodendrocytes of Parkinson’s Disease Brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Rockenstein, E.; Spencer, B.; Kim, H.K.; Adame, A.; Trejo, M.; Stafa, K.; Lee, H.J.; Lee, S.J.; Masliah, E. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015, 13, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.J.; et al. Immunotherapy Targeting Toll-like Receptor 2 Alleviates Neurodegeneration in Models of Synucleinopathy by Modulating α-Synuclein Transmission and Neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Kim, C.; Kwon, S.; Iba, M.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Shin, S.J.; Fields, J.A.; Rissman, R.A.; et al. Effects of Innate Immune Receptor Stimulation on Extracellular α-Synuclein Uptake and Degradation by Brain Resident Cells. Exp. Mol. Med. 2021, 53, 281–290. [Google Scholar] [CrossRef]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botía, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar Concentrations of Oligomeric Alpha-Synuclein Sensitizes TLR4 to Play an Initiating Role in Parkinson’s Disease Pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive Uptake of α-Synuclein Oligomers in Astrocytes Results in Sustained Intracellular Deposits and Mitochondrial Damage. Mol. Cell Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar α-Synuclein Induces Neurotoxic Astrocyte Activation via RIP Kinase Signaling and NF-ΚB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.M.; Chakrabarty, P.; Saha, K.; Khoshbouei, H.; Golde, T.E.; Giasson, B.I. Proteolysis of α-Synuclein Fibrils in the Lysosomal Pathway Limits Induction of Inclusion Pathology. J. Neurochem. 2017, 140, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of α-Synuclein in an Astrocyte Cell Line Promotes Autophagy Inhibition and Apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein Transfer between Neurons and Astrocytes Indicates That Astrocytes Play a Role in Degradation Rather than in Spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between Astrocytes and Microglia Results in Increased Degradation of α-Synuclein and Amyloid-β Aggregates. J. Neuroinflammation 2021, 18, 124. [Google Scholar] [CrossRef]
- Wang, R.; Ren, H.; Kaznacheyeva, E.; Lu, X.; Wang, G. Association of Glial Activation and α-Synuclein Pathology in Parkinson’s Disease. Neurosci. Bull. 2023, 39, 479–490. [Google Scholar] [CrossRef]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: An in Vitro and in Vivo Study Using Rodent and Human Models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-Synuclein Transfers from Neurons to Oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef]
- Pukaß, K.; Goldbaum, O.; Richter-Landsberg, C. Mitochondrial Impairment and Oxidative Stress Compromise Autophagosomal Degradation of α-Synuclein in Oligodendroglial Cells. J. Neurochem. 2015, 135, 194–205. [Google Scholar] [CrossRef]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and Alpha-Synuclein in Neurodegeneration: A Complex Interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef]
- Bryois, J.; Skene, N.G.; Hansen, T.F.; Kogelman, L.J.A.; Watson, H.J.; Liu, Z.; Adan, R.; Alfredsson, L.; Ando, T.; Andreassen, O.; et al. Genetic Identification of Cell Types Underlying Brain Complex Traits Yields Insights into the Etiology of Parkinson’s Disease. Nat. Genet. 2020, 52, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A Single-Cell Atlas of the Human Substantia Nigra Reveals Cell-Specific Pathways Associated with Neurological Disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Pérez-Acuña, D.; Rhee, K.H.; Lee, S.J. Changes in Oligodendroglial Subpopulations in Parkinson’s Disease. Mol. Brain 2023, 16, 65. [Google Scholar] [CrossRef]
- Azevedo, C.; Teku, G.; Pomeshchik, Y.; Reyes, J.F.; Chumarina, M.; Russ, K.; Savchenko, E.; Hammarberg, A.; Lamas, N.J.; Collin, A.; et al. Parkinson’s Disease and Multiple System Atrophy Patient IPSC-Derived Oligodendrocytes Exhibit Alpha-Synuclein–Induced Changes in Maturation and Immune Reactive Properties. Proc. Natl. Acad. Sci. USA 2022, 119. [Google Scholar] [CrossRef]
- Boshkovski, T.; Cohen-Adad, J.; Misic, B.; Arnulf, I.; Corvol, J.C.; Vidailhet, M.; Lehéricy, S.; Stikov, N.; Mancini, M. The Myelin-Weighted Connectome in Parkinson’s Disease. Mov. Disord. 2022, 37, 724–733. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Kanazawa, T.; Itoh, Y.; Wakabayashi, K.; Kakita, A.; Takahashi, H. Unmyelinated Axons Are More Vulnerable to Degeneration than Myelinated Axons of the Cardiac Nerve in Parkinson’s Disease. Neuropathol. Appl. Neurobiol. 2011, 37, 791–802. [Google Scholar] [CrossRef]
- Del Campo, N.; Phillips, O.; Ory-Magne, F.; Brefel-Courbon, C.; Galitzky, M.; Thalamas, C.; Narr, K.L.; Joshi, S.; Singh, M.K.; Péran, P.; et al. Broad White Matter Impairment in Multiple System Atrophy. Hum. Brain Mapp. 2021, 42, 357–366. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- de Ceglia, R.; Ledonne, A.; Litvin, D.G.; Lind, B.L.; Carriero, G.; Latagliata, E.C.; Bindocci, E.; Di Castro, M.A.; Savtchouk, I.; Vitali, I.; et al. Specialized Astrocytes Mediate Glutamatergic Gliotransmission in the CNS. Nature 2023. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic Expression of Parkinson’s Disease-Related A53T Alpha-Synuclein Causes Neurodegeneration in Mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Littlejohn, K.; Yuan, S.; Fernandez, C.; Cilluffo, M.; Koo, B.K.; Whitelegge, J.P.; Watson, J.B. Stimulation of Synaptoneurosome Glutamate Release by Monomeric and Fibrillated α-Synuclein. J. Neurosci. Res. 2017, 95, 1871–1887. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Kuś, J.; Saramowicz, K.; Czerniawska, M.; Wiese, W.; Siwecka, N.; Rozpędek-Kamińska, W.; Kucharska-Lusina, A.; Strzelecki, D.; Majsterek, I. Molecular Mechanisms Underlying NMDARs Dysfunction and Their Role in ADHD Pathogenesis. Int. J. Mol. Sci. 2023, 24, 12983. [Google Scholar] [CrossRef] [PubMed]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Vicente Miranda, H.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.A.; Castanho, M.A.R.B.; et al. Extracellular Alpha-Synuclein Oligomers Modulate Synaptic Transmission and Impair LTP via NMDA-Receptor Activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef]
- Hüls, S.; Högen, T.; Vassallo, N.; Danzer, K.M.; Hengerer, B.; Giese, A.; Herms, J. AMPA-Receptor-Mediated Excitatory Synaptic Transmission Is Enhanced by Iron-Induced α-Synuclein Oligomers. J. Neurochem. 2011, 117, 868–878. [Google Scholar] [CrossRef]
- Yang, J.; Hertz, E.; Zhang, X.; Leinartaité, L.; Lundius, E.G.; Li, J.; Svenningsson, P. Overexpression of α-Synuclein Simultaneously Increases Glutamate NMDA Receptor Phosphorylation and Reduces Glucocerebrosidase Activity. Neurosci. Lett. 2016, 611, 51–58. [Google Scholar] [CrossRef]
- Vos, T.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; Aboyans, V.; et al. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 328 Diseases and Injuries for 195 Countries, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune Attack: The Role of Inflammation in Alzheimer Disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.J.; Orru, C.D.; Concha-Marambio, L.; Giaisi, S.; Groveman, B.R.; Farris, C.M.; Holguin, B.; Hughson, A.G.; LaFontant, D.E.; Caspell-Garcia, C.; et al. High Diagnostic Performance of Independent Alpha-Synuclein Seed Amplification Assays for Detection of Early Parkinson’s Disease. Acta Neuropathol. Commun. 2021, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Siwecka, N.; Saramowicz, K.; Galita, G.; Rozpędek-Kamińska, W.; Majsterek, I. Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy. Pharmaceutics 2023, 15, 2051. [Google Scholar] [CrossRef]
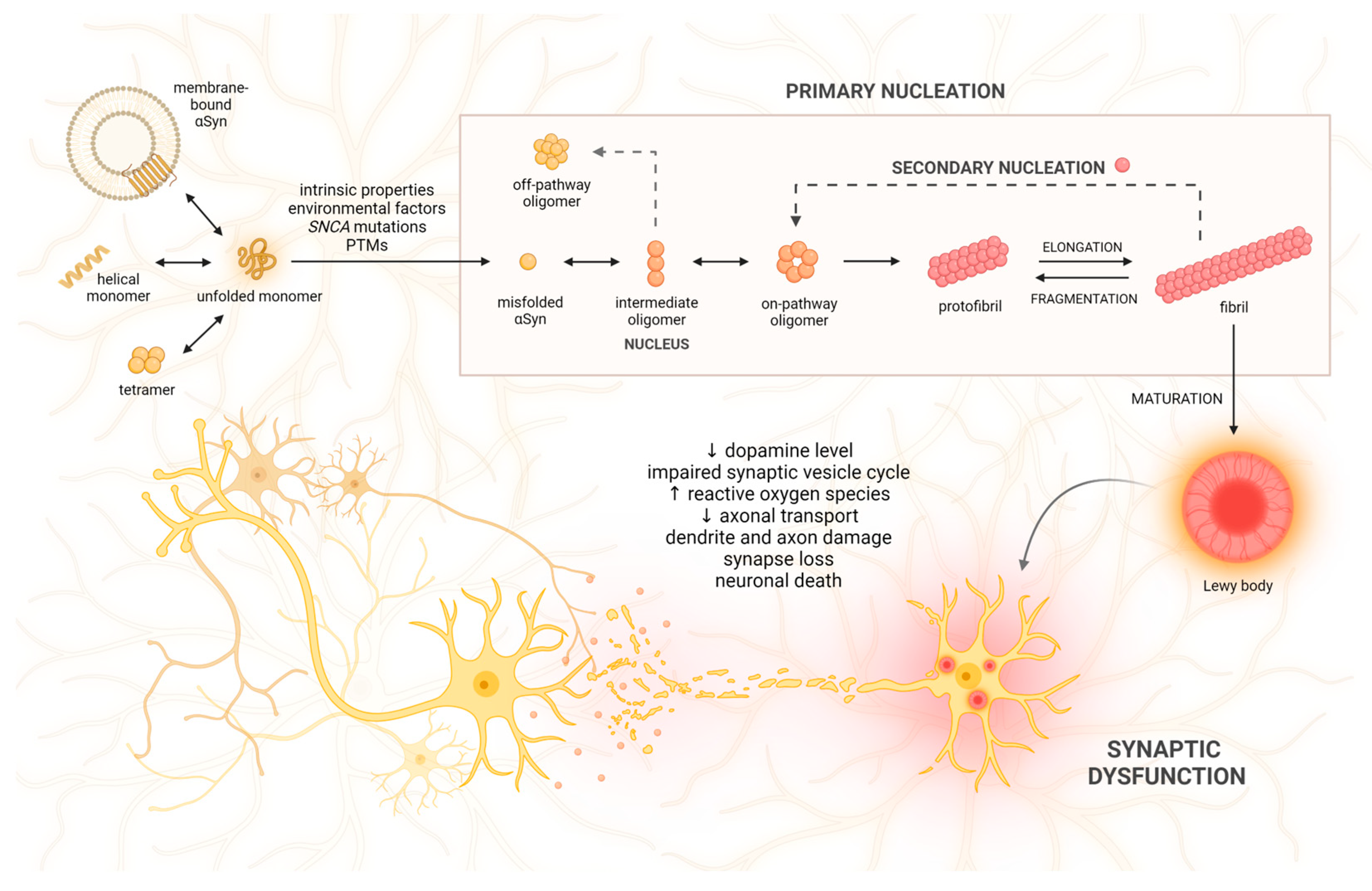

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saramowicz, K.; Siwecka, N.; Galita, G.; Kucharska-Lusina, A.; Rozpędek-Kamińska, W.; Majsterek, I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 360. https://doi.org/10.3390/ijms25010360
Saramowicz K, Siwecka N, Galita G, Kucharska-Lusina A, Rozpędek-Kamińska W, Majsterek I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(1):360. https://doi.org/10.3390/ijms25010360
Chicago/Turabian StyleSaramowicz, Kamil, Natalia Siwecka, Grzegorz Galita, Aleksandra Kucharska-Lusina, Wioletta Rozpędek-Kamińska, and Ireneusz Majsterek. 2024. "Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 1: 360. https://doi.org/10.3390/ijms25010360
APA StyleSaramowicz, K., Siwecka, N., Galita, G., Kucharska-Lusina, A., Rozpędek-Kamińska, W., & Majsterek, I. (2024). Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. International Journal of Molecular Sciences, 25(1), 360. https://doi.org/10.3390/ijms25010360