
Beta Blockade Prevents Cardiac Morphological and Molecular Remodelling in Experimental Uremia
Abstract
:1. Introduction
2. Results
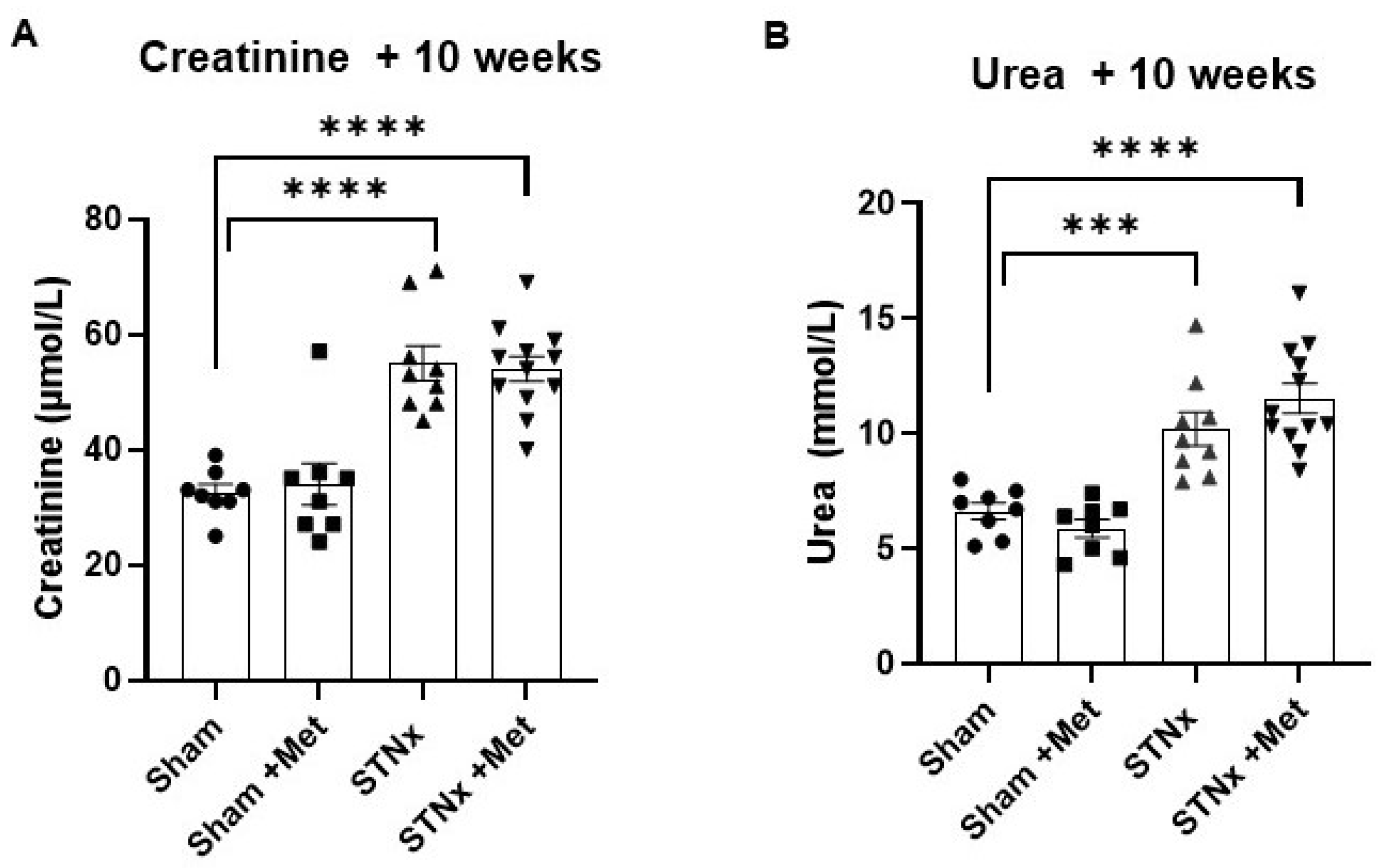
2.1. Biochemical Effects of Subtotal Nephrectomy
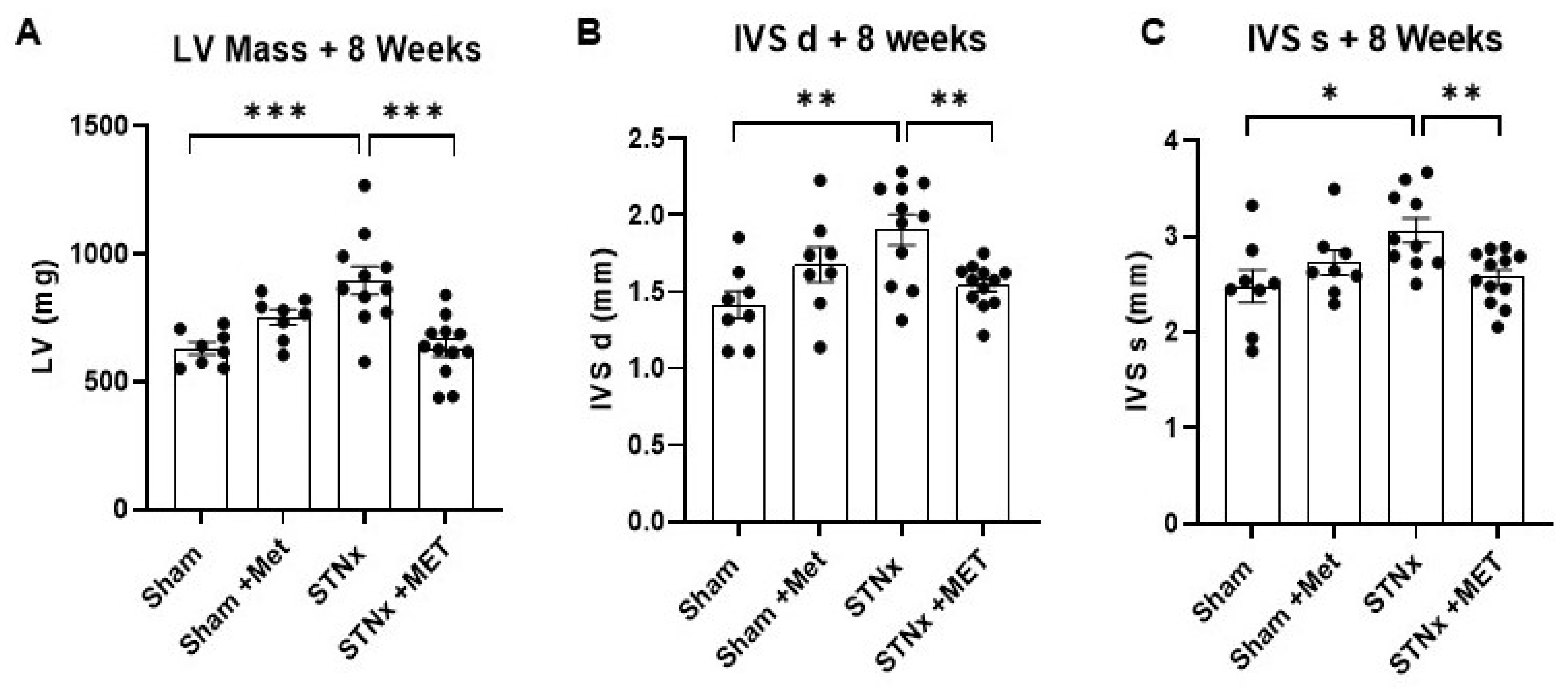
2.2. Assessment of Cardiac Structure
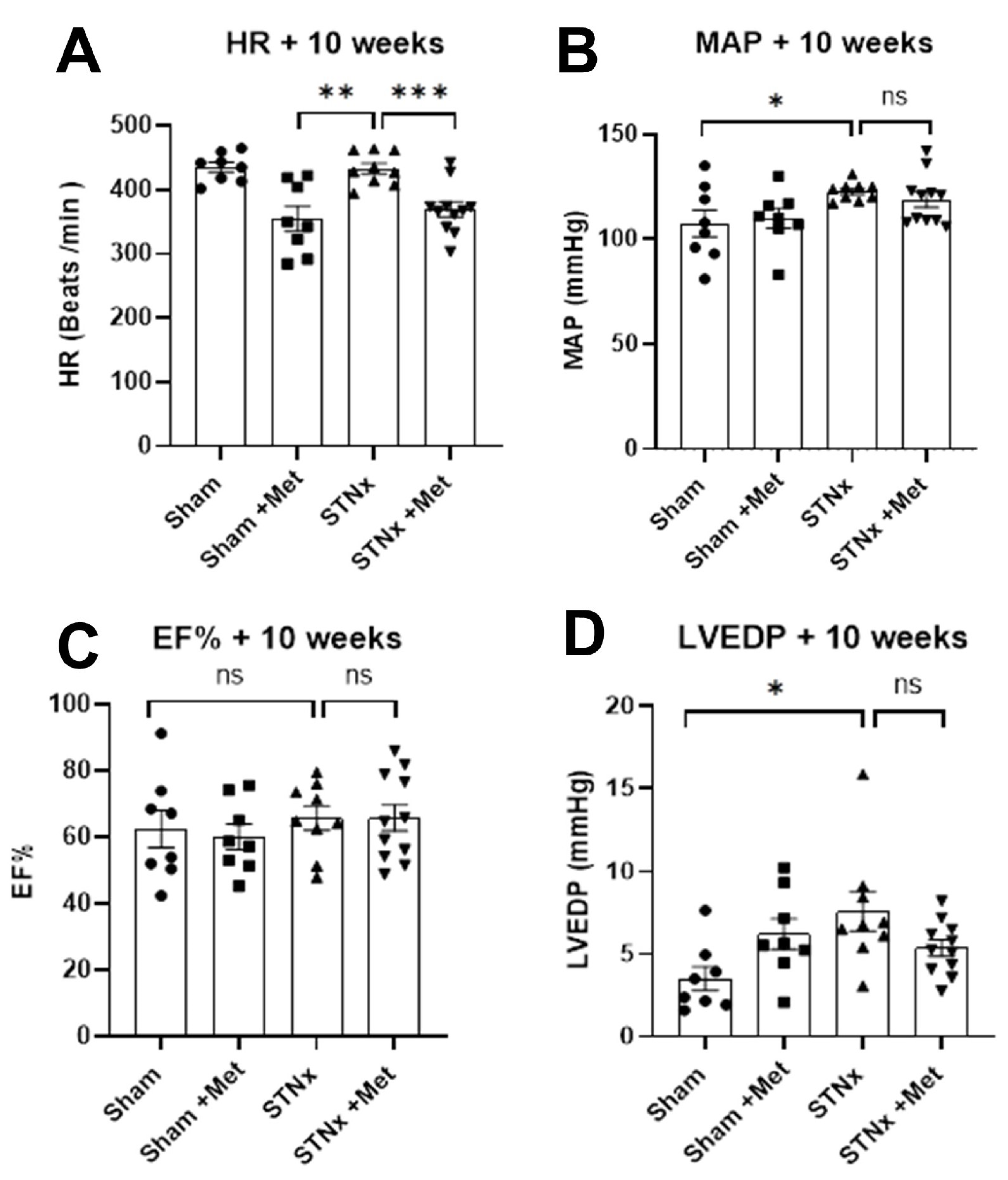
2.3. Assessment of Cardiac Function
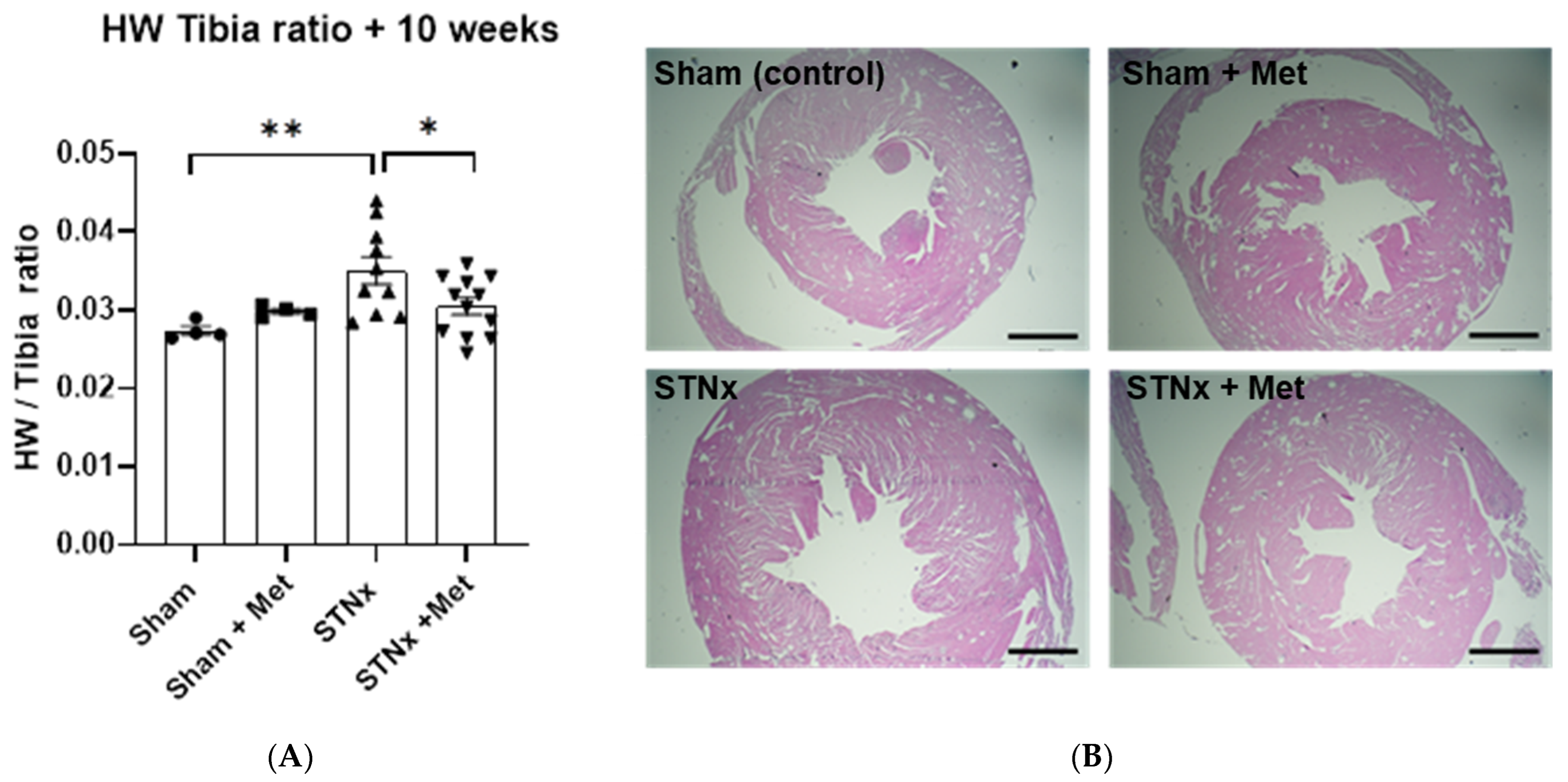
2.4. Quantifying Cardiac Hypertrophy
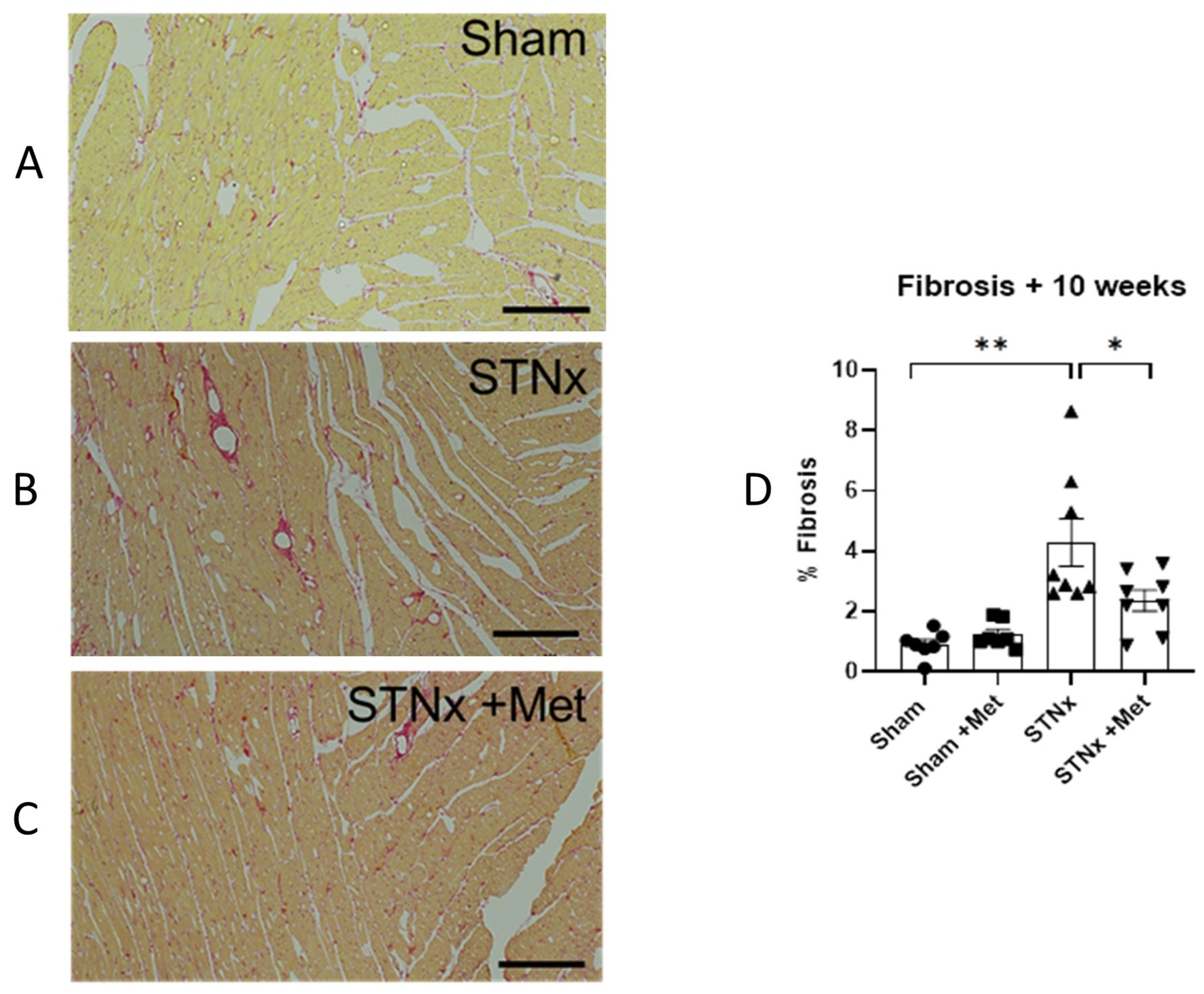
2.5. Cardiac Fibrosis
2.6. Mechanism of Action of Beta Blockade
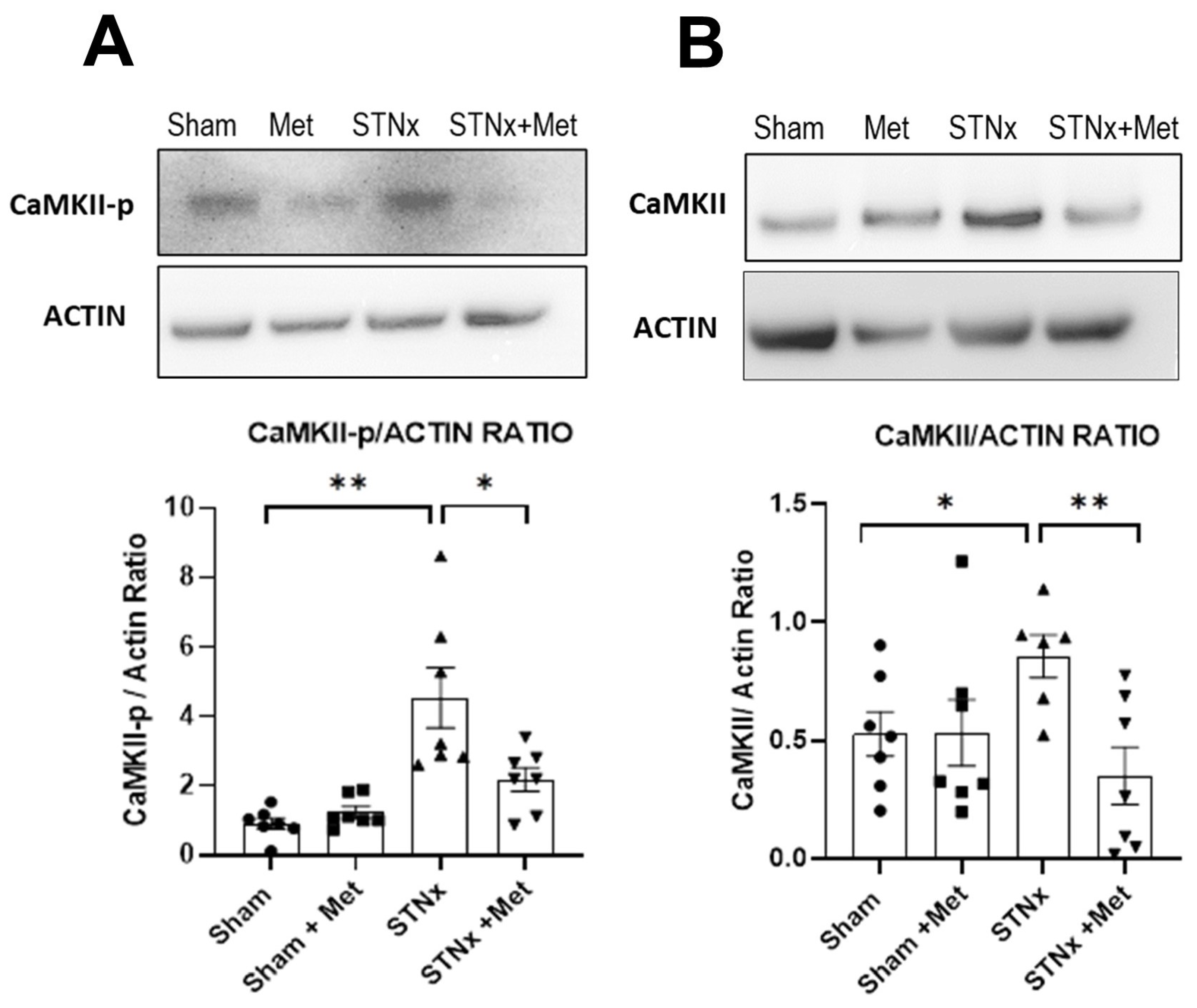
2.6.1. Inhibition of Calcium Calmodulin Kinase II (CaMKII) Pathway
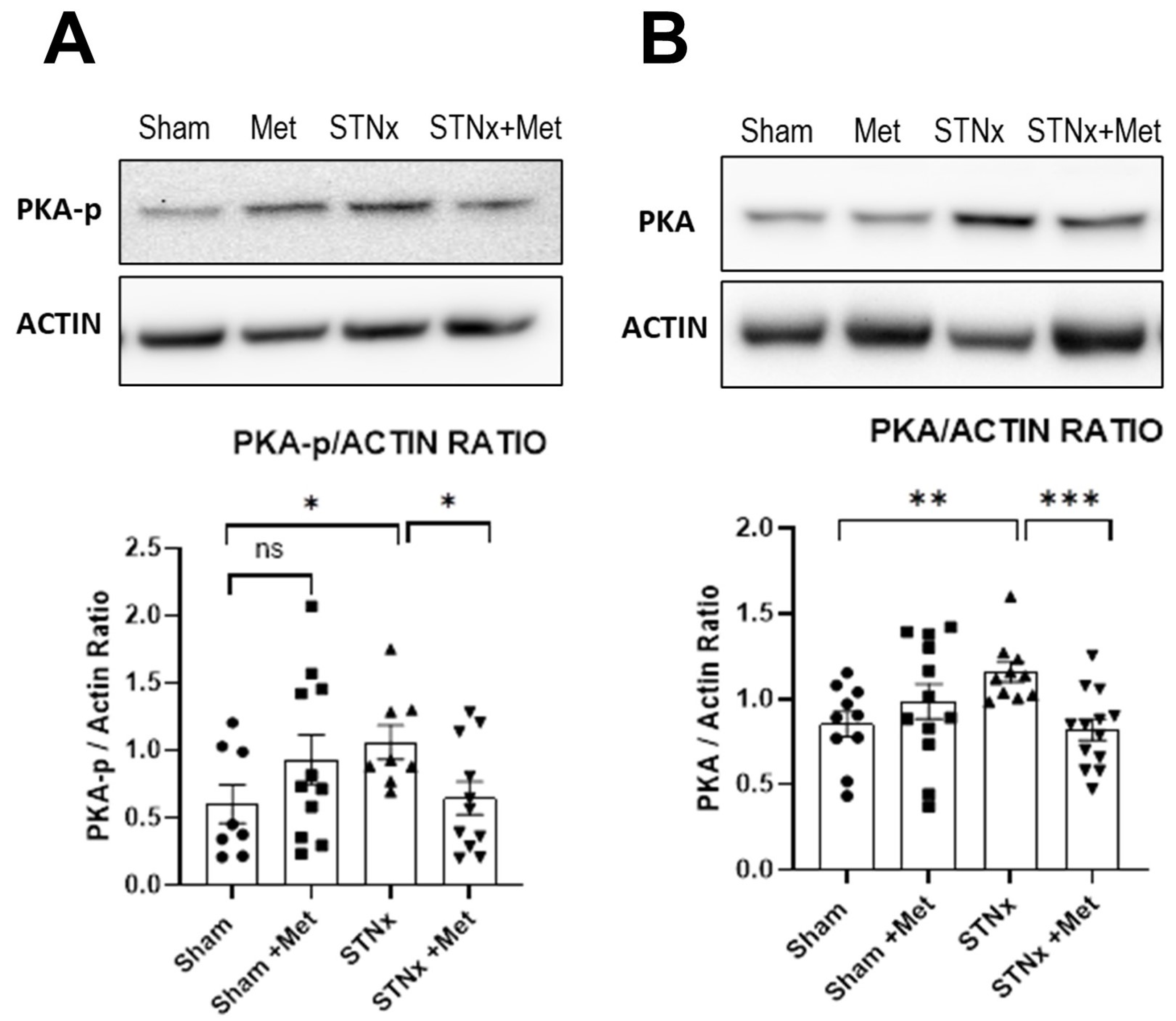
2.6.2. Inhibition of Protein Kinase A Pathway
3. Discussion
4. Materials and Methods
5. Conclusions
Disclosures
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, M.; Hsu, C.Y.; Li, Y.; Mishra, R.K.; Keane, M.; Rosas, S.E.; Dries, D.; Xie, D.; Chen, J.; He, J.; et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J. Am. Soc. Nephrol. 2012, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.F.; Deo, R.; Bansal, N.; Anderson, A.H.; Yang, P.; Go, A.S.; Keane, M.; Townsend, R.; Porter, A.; Budoff, M.; et al. Associations of Conventional Echocardiographic Measures with Incident Heart Failure and Mortality: The Chronic Renal Insufficiency Cohort. Clin. J. Am. Soc. Nephrol. 2017, 12, 60–68. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- System USRD. 2020 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2020. [Google Scholar]
- Braunwald, E.; Bristow, M.R. Congestive heart failure: Fifty years of progress. Circulation 2000, 102, IV14–IV23. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Socratous, F.; Hennebry, S.; Eikelis, N.; Lambert, E.A.; Straznicky, N.; Esler, M.D.; Lambert, G.W. Sympathetic activation in chronic renal failure. J. Am. Soc. Nephrol. 2009, 20, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Iwao, H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar] [PubMed]
- Massy, Z.A.; Stenvinkel, P.; Drueke, T.B. The role of oxidative stress in chronic kidney disease. Semin. Dial. 2009, 22, 405–408. [Google Scholar] [CrossRef]
- Middleton, R.J.; Parfrey, P.S.; Foley, R.N. Left ventricular hypertrophy in the renal patient. J. Am. Soc. Nephrol. 2001, 12, 1079–1084. [Google Scholar] [CrossRef]
- Gu, H.; Sinha, M.D.; Li, Y.; Simpson, J.; Chowienczyk, P.J. Elevated ejection-phase myocardial wall stress in children with chronic kidney disease. Hypertension 2015, 66, 823–829. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Lee, T.T.; Massie, B.M. Effect of beta-blockade on mortality in patients with heart failure: A meta-analysis of randomized clinical trials. J. Am. Coll. Cardiol. 1997, 30, 27–34. [Google Scholar] [CrossRef]
- Foody, J.M.; Farrell, M.H.; Krumholz, H.M. beta-Blocker therapy in heart failure: Scientific review. JAMA 2002, 287, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Colucci, W.S.; Kolias, T.J.; Adams, K.F.; Armstrong, W.F.; Ghali, J.K.; Gottlieb, S.S.; Greenberg, B.; Klibaner, M.I.; Kukin, M.L.; Sugg, J.E. Metoprolol reverses left ventricular remodeling in patients with asymptomatic systolic dysfunction: The REversal of VEntricular Remodeling with Toprol-XL (REVERT) trial. Circulation 2007, 116, 49–56. [Google Scholar] [PubMed]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Investig. 2000, 105, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Babler, A.; Floege, J.; Kramann, R. Cardiac Remodeling in Chronic Kidney Disease. Toxins 2020, 12, 161. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolic mechanisms in physiological and pathological cardiac hypertrophy: New paradigms and challenges. Nat. Rev. Cardiol. 2023, 20, 812–829. [Google Scholar] [CrossRef]
- Houser, S.R.; Piacentino, V., III; Weisser, J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J. Mol. Cell Cardiol. 2000, 32, 1595–1607. [Google Scholar] [CrossRef]
- Olson, E.N. A decade of discoveries in cardiac biology. Nat. Med. 2004, 10, 467–474. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef]
- Bristow, M.R. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: A convergence of receptor pharmacology and clinical cardiology. Circ. Res. 2011, 109, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Investig. 2013, 123, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-dependent protein kinase II: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Khoo, M.S.; Wu, Y.; Yang, Y.; Grueter, C.E.; Ni, G.; Price, E.E., Jr.; Thiel, W.; Guatimosim, S.; Song, L.S.; et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 2005, 11, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, D.; Gill, S.K.; Flather, M.D.; Holmes, J.; Packer, M.; Rosano, G.; Böhm, M.; McMurray, J.J.; Wikstrand, J.; Anker, S.D.; et al. Impact of Renal Impairment on Beta-Blocker Efficacy in Patients with Heart Failure. J. Am. Coll. Cardiol. 2019, 74, 2893–2904. [Google Scholar] [CrossRef]
- Cice, G.; Ferrara, L.; Di Benedetto, A.; Russo, P.E.; Marinelli, G.; Pavese, F.; Iacono, A. Dilated cardiomyopathy in dialysis patients--beneficial effects of carvedilol: A double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2001, 37, 407–411. [Google Scholar] [CrossRef]
- Rambausek, M.; Ritz, E.; Mall, G.; Mehls, O.; Katus, H. Myocardial hypertrophy in rats with renal insufficiency. Kidney Int. 1985, 28, 775–782. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Finnie, S.L.; Teenan, O.; Cairns, C.; Boyd, A.; Bailey, M.A.; Thomson, A.; Hughes, J.; Bénézech, C.; Conway, B.R.; et al. Refining the Mouse Subtotal Nephrectomy in Male 129S2/SV Mice for Consistent Modeling of Progressive Kidney Disease with Renal Inflammation and Cardiac Dysfunction. Front. Physiol. 2019, 10, 1365. [Google Scholar] [CrossRef]
- Fowler, E.D.; Drinkhill, M.J.; Norman, R.; Pervolarakia, E. Beta1-adrenoceptor antagonist, metoprolol attenuates cardiac myocyte Ca(2+) handling dysfunction in rats with pulmonary artery hypertension. J. Mol. Cell Cardiol. 2018, 120, 74–83. [Google Scholar] [CrossRef]
- Temple, I.P.; Logantha, S.J.; Absi, M.; Zhang, Y.; Pervolaraki, E.; Yanni, J.; Atkinson, A.; Petkova, M.; Quigley, G.M.; Castro, S.; et al. Atrioventricular Node Dysfunction and Ion Channel Transcriptome in Pulmonary Hypertension. Circ. Arrhythm Electrophysiol. 2016, 9, e003432. [Google Scholar] [CrossRef] [PubMed]
- Frentzou, G.A.; Drinkhill, M.J.; Turner, N.A.; Ball, S.G.; Ainscough, J.F. A state of reversible compensated ventricular dysfunction precedes pathological remodelling in response to cardiomyocyte-specific activity of angiotensin II type-1 receptor in mice. Dis. Model Mech. 2015, 8, 783–794. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | Sham + Metoprolol | STNx | STNx + Metoprolol | |
|---|---|---|---|---|
| Creatinine (µmol/L) | 32.05 ± 1.44 | 30.71 ± 1.81 | 55.00 ± 3.06 | 54.00 ± 2.20 |
| Urea (mmol/L) | 6.63 ± 0.36 | 5.88 ± 0.39 | 10.20 ± 0.72 | 11.53 ± 0.65 |
| Sham | Sham + Metoprolol | STNx | STNx + Metoprolol | |
|---|---|---|---|---|
| HR (min−1) | 434.74 ± 7.84 | 354.64 ± 19.45 | 432.72 ± 8.54 | 369.49 ± 11.80 |
| MAP (mmHg) | 107.50 ± 6.35 | 109.88 ± 4.72 | 122.78 ± 1.47 | 118.82 ± 3.60 |
| Echo and PV loop parameters at 10 weeks | ||||
| EF (%) | 62.54 ± 5.58 | 60.13 ± 3.81 | 65.74 ± 3.60 | 65.84 ± 3.95 |
| LVEDP (mmHg) | 3.51 ± 0.72 | 6.21 ± 0.93 | 7.57 ± 1.18 | 5.37 ± 0.49 |
| Left ventricular mass at 8 weeks | ||||
| LV mass (mg) | 629.81 ± 24.27 | 750.90 ± 29.45 | 896.37 ± 54.59 | 632.17 ± 34.04 |
| Heart weight to tibial length ratio at 10 weeks | ||||
| HW:TL | 0.0274 ± 0.0006 | 0.0299 ± 0.0004 | 0.0350 ± 0.0018 | 0.0304 ± 0.0011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinnappa, S.; Maqbool, A.; Viswambharan, H.; Mooney, A.; Denby, L.; Drinkhill, M. Beta Blockade Prevents Cardiac Morphological and Molecular Remodelling in Experimental Uremia. Int. J. Mol. Sci. 2024, 25, 373. https://doi.org/10.3390/ijms25010373
Chinnappa S, Maqbool A, Viswambharan H, Mooney A, Denby L, Drinkhill M. Beta Blockade Prevents Cardiac Morphological and Molecular Remodelling in Experimental Uremia. International Journal of Molecular Sciences. 2024; 25(1):373. https://doi.org/10.3390/ijms25010373
Chicago/Turabian StyleChinnappa, Shanmugakumar, Azhar Maqbool, Hema Viswambharan, Andrew Mooney, Laura Denby, and Mark Drinkhill. 2024. "Beta Blockade Prevents Cardiac Morphological and Molecular Remodelling in Experimental Uremia" International Journal of Molecular Sciences 25, no. 1: 373. https://doi.org/10.3390/ijms25010373