
Long COVID: Molecular Mechanisms and Detection Techniques


 , and
, and 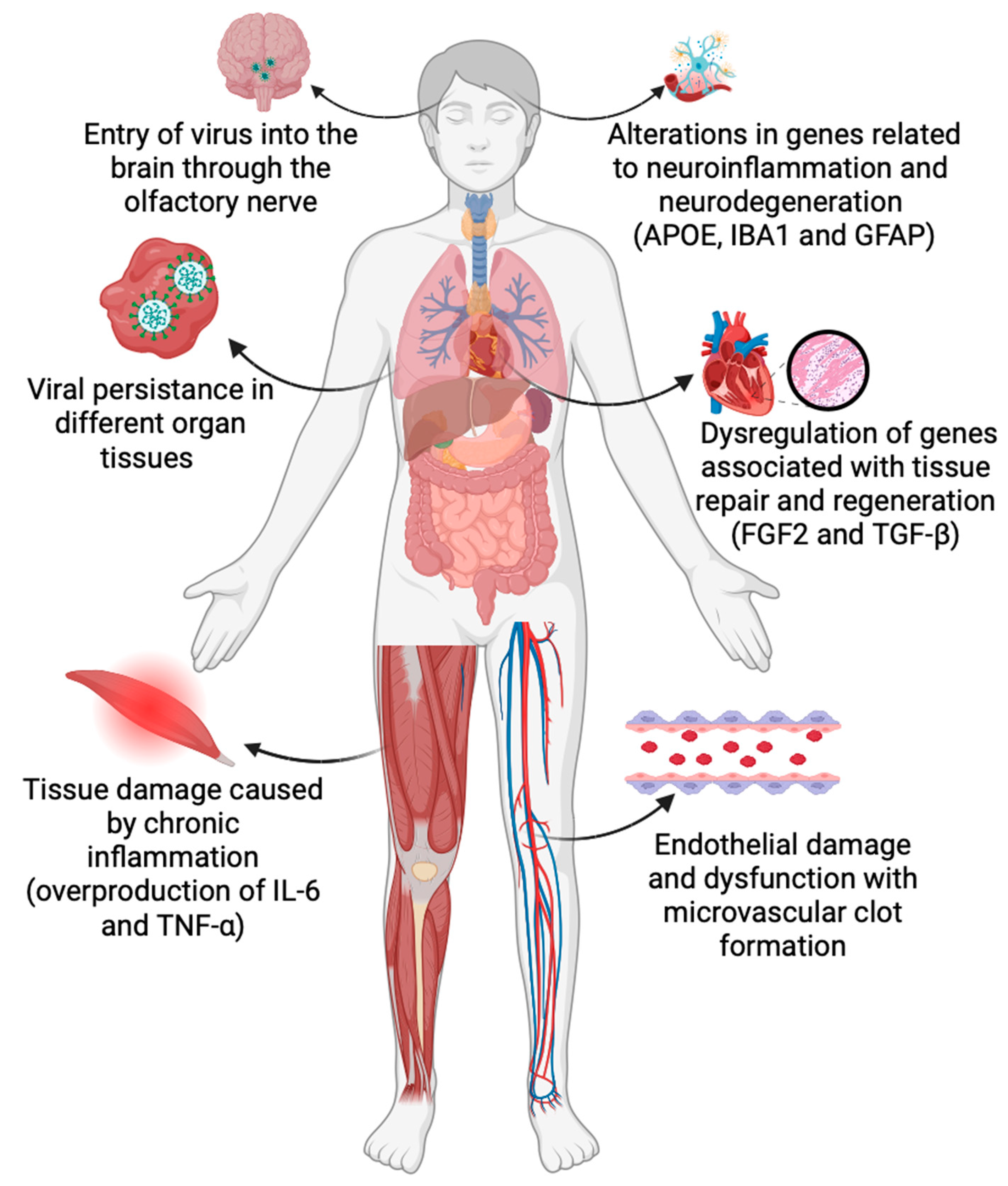{kind=link}
{kind=link}
Abstract
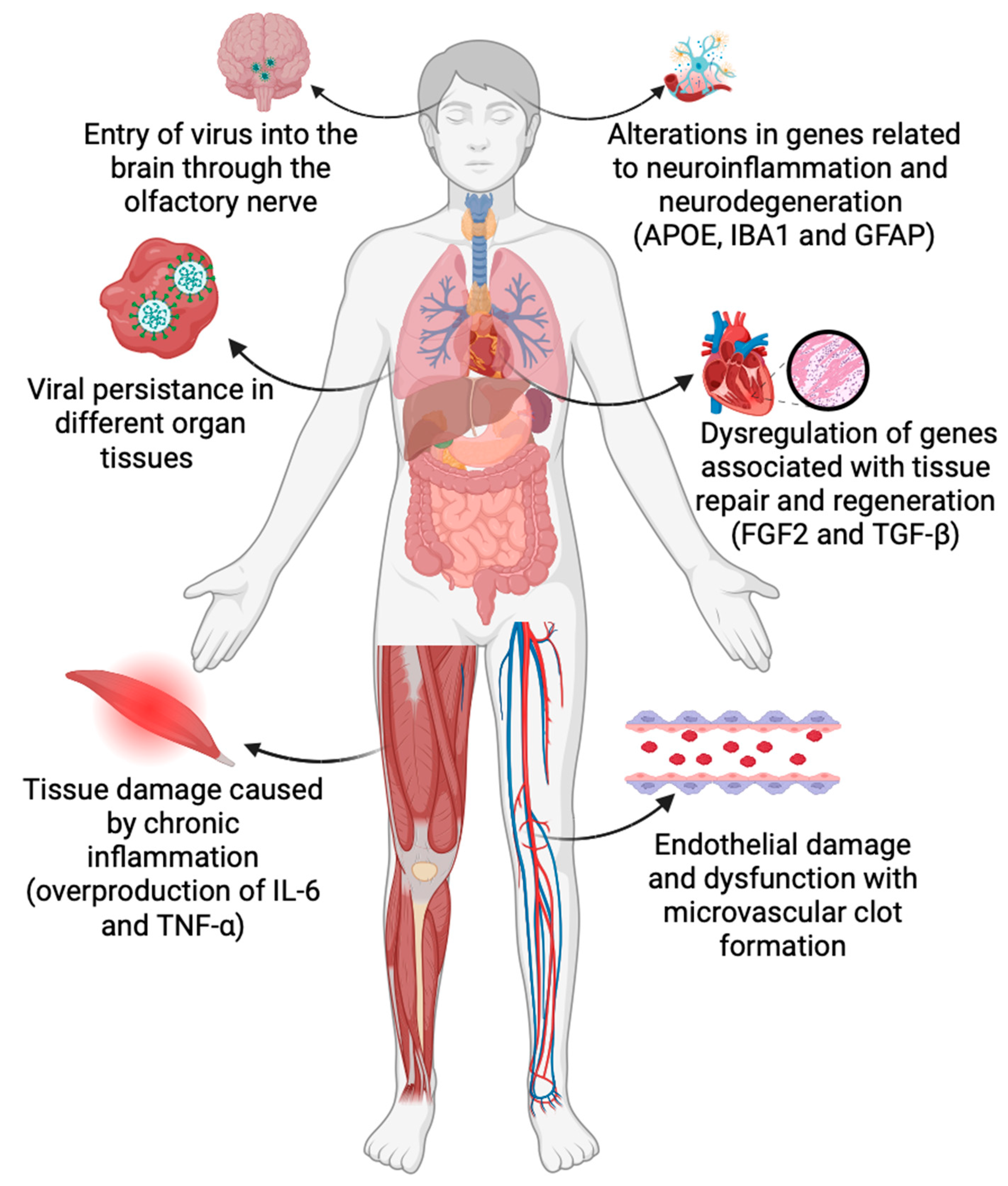
:1. Introduction
2. Altered Gene Expression Profiles in Long COVID
2.1. Long COVID and Altered Immune Response
2.2. Long COVID and Impaired Tissue Repair
2.3. Long COVID and Neurological Impairments
3. Mechanisms Leading to Altered Gene Expression in Long COVID
3.1. Dysregulated miRNA Profiles
3.2. Transcriptional Factors
3.3. Long Noncoding RNAs
4. Implications of Altered Molecular Processes in Long COVID Symptoms
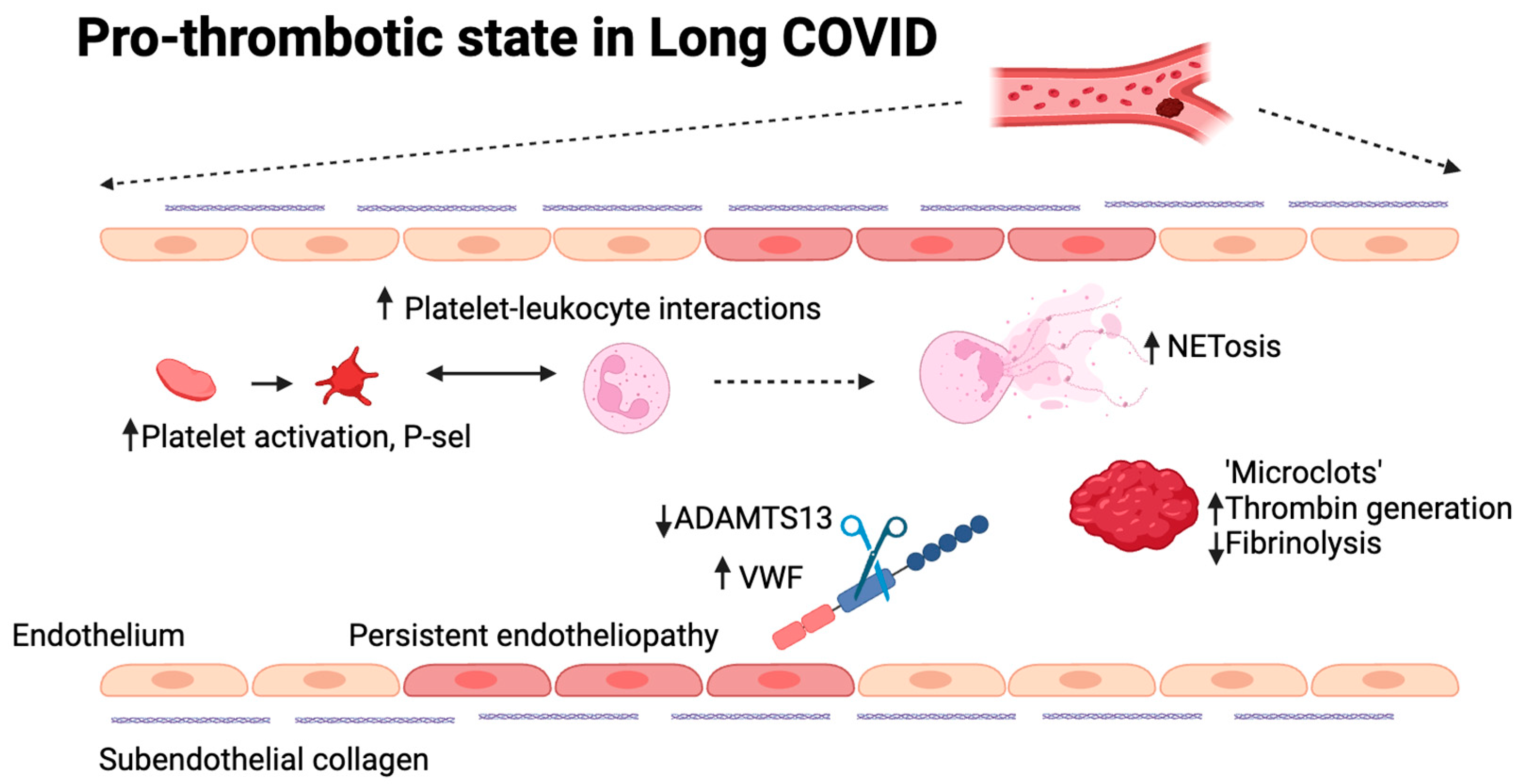
4.1. Long COVID and Vascular Dysfunctions
4.2. Long COVID and Inflammation
5. Molecular Methods of Detection and Quantification with Applications in Long COVID
5.1. Digital PCR
5.2. Microarray
5.3. Sequencing Techniques
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Talla, A.; Vasaikar, S.V.; Szeto, G.L.; Lemos, M.P.; Czartoski, J.L.; MacMillan, H.; Moodie, Z.; Cohen, K.W.; Fleming, L.B.; Thomson, Z.; et al. Persistent serum protein signatures define an inflammatory subcategory of long COVID. Nat. Commun. 2023, 14, 3417. [Google Scholar] [CrossRef] [PubMed]
- Espín, E.; Yang, C.; Shannon, C.P.; Assadian, S.; He, D.; Tebbutt, S.J. Cellular and molecular biomarkers of long COVID: A scoping review. eBioMedicine 2023, 91, 104552. [Google Scholar] [CrossRef] [PubMed]
- Kopplin, N.; Garcia, A.; Reczek, A.; Wilkinson, K.; Yekkaluri, S.; Murphy, C.C.; Tiro, J.; Muthukumar, A.R.; Masica, A.; Singal, A.G. Post-acute sequelae of COVID-19 and longitudinal antibody levels in a community-based cohort. PLoS ONE 2023, 18, e0291259. [Google Scholar] [CrossRef]
- Mitroi, R.; Padureanu, V.; Mitrea, A.; Protasiewicz Timofticiuc, D.C.; Rosu, M.M.; Clenciu, D.; Enescu, A.; Padureanu, R.; Tenea Cojan, T.S.; Vladu, I.M. Prothrombotic status in COVID-19 with diabetes mellitus (Review). Biomed. Rep. 2023, 19, 65. [Google Scholar] [CrossRef]
- Reiss, A.B.; Greene, C.; Dayaramani, C.; Rauchman, S.H.; Stecker, M.M.; De Leon, J.; Pinkhasov, A. Long COVID, the Brain, Nerves, and Cognitive Function. Neurol. Int. 2023, 15, 821–841. [Google Scholar] [CrossRef]
- Monje, M.; Iwasaki, A. The neurobiology of long COVID. Neuron 2022, 110, 3484–3496. [Google Scholar] [CrossRef]
- Flamier, A.; Bisht, P.; Richards, A.; Tomasello, D.L.; Jaenisch, R. Human iPS cell-derived sensory neurons can be infected by SARS-CoV-2. iScience 2023, 26, 107690. [Google Scholar] [CrossRef]
- Leng, A.; Shah, M.; Ahmad, S.A.; Premraj, L.; Wildi, K.; Li Bassi, G.; Pardo, C.A.; Choi, A.; Cho, S.-M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells 2023, 12, 816. [Google Scholar] [CrossRef]
- Seibert, F.S.; Stervbo, U.; Wiemers, L.; Skrzypczyk, S.; Hogeweg, M.; Bertram, S.; Kurek, J.; Anft, M.; Westhoff, T.H.; Babel, N. Severity of neurological Long-COVID symptoms correlates with increased level of autoantibodies targeting vasoregulatory and autonomic nervous system receptors. Autoimmun. Rev. 2023, 22, 103445. [Google Scholar] [CrossRef]
- Merad, M.; Blish, C.A.; Sallusto, F.; Iwasaki, A. The immunology and immunopathology of COVID-19. Science 2022, 375, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Langton, D.J.; Bourke, S.C.; Lie, B.A.; Reiff, G.; Natu, S.; Darlay, R.; Burn, J.; Echevarria, C. The influence of HLA genotype on the severity of COVID-19 infection. HLA 2021, 98, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrera, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2022, 12, 746021. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.-S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Garg, S.; Kim, L.; Whitaker, M.; O’Halloran, A.; Cummings, C.; Holstein, R.; Prill, M.; Chai, S.J.; Kirley, P.D.; Alden, N.B.; et al. Hospitalization Rates and Characteristics of Patients Hospitalized with Laboratory-Confirmed Coronavirus Disease 2019–COVID-NET, 14 States, March 1–30, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 458–464. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Whalley, J.P.; Knight, J.C.; Wicker, L.S.; Todd, J.A.; Ferreira, R.C. SARS-CoV-2 infection induces a long-lived pro-inflammatory transcriptional profile. Genome Med. 2023, 15, 69. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.-S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef]
- Bohnacker, S.; Hartung, F.; Henkel, F.; Quaranta, A.; Kolmert, J.; Priller, A.; Ud-Dean, M.; Giglberger, J.; Kugler, L.M.; Pechtold, L.; et al. Mild COVID-19 imprints a long-term inflammatory eicosanoid- and chemokine memory in monocyte-derived macrophages. Mucosal Immunol. 2022, 15, 515–524. [Google Scholar] [CrossRef]
- Yin, K.; Peluso, M.J.; Luo, X.; Thomas, R.; Shin, M.-G.; Neidleman, J.; Andrew, A.; Young, K.; Ma, T.; Hoh, R.; et al. Long COVID manifests with T cell dysregulation, inflammation, and an uncoordinated adaptive immune response to SARS-CoV-2. bioRxiv, 2023; preprint. [Google Scholar]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Santopaolo, M.; Gregorova, M.; Hamilton, F.; Arnold, D.; Long, A.; Lacey, A.; Oliver, E.; Halliday, A.; Baum, H.; Hamilton, K.; et al. Prolonged T-cell activation and long COVID symptoms independently associate with severe COVID-19 at 3 months. eLife 2023, 12, e85009. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Khan, A.W.; Kim, M.S.; Choi, S. The Role of Fibroblast Growth Factor (FGF) Signaling in Tissue Repair and Regeneration. Cells 2021, 10, 3242. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Luo, S.; Qin, R.; Yang, M.; Wang, X.; Yang, Q.; Zhang, Y.; Wang, Q.; Zhu, R.; Fan, H.; et al. Long-term infection of SARS-CoV-2 changed the body’s immune status. Clin. Immunol. 2020, 218, 108524. [Google Scholar] [CrossRef] [PubMed]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.-R.; Mottaghi, S.S.; Mehrabi, M.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. 2021, 90, 104773. [Google Scholar] [CrossRef]
- Möhlendick, B.; Schönfelder, K.; Breuckmann, K.; Elsner, C.; Babel, N.; Balfanz, P.; Dahl, E.; Dreher, M.; Fistera, D.; Herbstreit, F.; et al. ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Pharmacogenet. Genom. 2021, 31, 165–171. [Google Scholar] [CrossRef]
- Paruchuri, S.S.H.; Farwa, U.E.; Jabeen, S.; Pamecha, S.; Shan, Z.; Parekh, R.; Lakkimsetti, M.; Alamin, E.; Sharma, V.; Haider, S.; et al. Myocarditis and Myocardial Injury in Long COVID Syndrome: A Comprehensive Review of the Literature. Cureus 2023, 15, e42444. [Google Scholar] [CrossRef]
- Qi, P.; Huang, M.; Zhu, H. Exploring potential biomarkers and therapeutic targets of long COVID-associated inflammatory cardiomyopathy. Front. Med. 2023, 10, 1191354. [Google Scholar] [CrossRef]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.-H.; Wood, J.; O’Dea, M.R.; Dutton, S.; Shamardani, K.; Nwangwu, K.; et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 2022, 185, 2452–2468.e16. [Google Scholar] [CrossRef]
- Taquet, M.; Skorniewska, Z.; Hampshire, A.; Chalmers, J.D.; Ho, L.-P.; Horsley, A.; Marks, M.; Poinasamy, K.; Raman, B.; Leavy, O.C.; et al. Acute blood biomarker profiles predict cognitive deficits 6 and 12 months after COVID-19 hospitalization. Nat. Med. 2023, 29, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-J.; Liu, S.-H.; Manachevakul, S.; Lee, T.-A.; Kuo, C.-T.; Bello, D. Biomarkers in long COVID-19: A systematic review. Front. Med. 2023, 10, 1085988. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Sans, H.M.; Forman, C.A.; Nylander, A.N.; Ho, H.; Lu, S.; Goldberg, S.A.; Hoh, R.; Tai, V.; Munter, S.E.; et al. Plasma Markers of Neurologic Injury and Inflammation in People With Self-Reported Neurologic Postacute Sequelae of SARS-CoV-2 Infection. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e200003. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Mayilsamy, K.; McGill, A.R.; Martinez, T.E.; Chandran, B.; Blair, L.J.; Bickford, P.C.; Mohapatra, S.S.; Mohapatra, S. SARS-CoV-2 infection increases the gene expression profile for Alzheimer’s disease risk. Mol. Ther.-Methods Clin. Dev. 2022, 27, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Long, S.; Cortés-Altamirano, J.L.; Bandala, C.; Avendaño-Ortiz, K.; Bonilla-Jaime, H.; Bueno-Nava, A.; Ávila-Luna, A.; Sánchez-Aparicio, P.; Clavijo-Cornejo, D.; Dotor-Llerena, A.L. Role of the MicroRNAs in the Pathogenic Mechanism of Painful Symptoms in Long COVID. Syst. Rev. Int. J. Mol. Sci. 2023, 24, 3574. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Wu, D.; Yang, X.O. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 2020, 53, 368–370. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Widiasta, A.; Sribudiani, Y.; Nugrahapraja, H.; Hilmanto, D.; Sekarwana, N.; Rachmadi, D. Potential role of ACE2-related microRNAs in COVID-19-associated nephropathy. Non-Coding RNA Res. 2020, 5, 153–166. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef] [PubMed]
- Trogstad, L.; Laake, I.; Robertson, A.H.; Mjaaland, S.; Caspersen, I.H.; Juvet, L.K.; Magnus, P.; Blix, K.; Feiring, B. Heavy bleeding and other menstrual disturbances in young women after COVID-19 vaccination. Vaccine 2023, 41, 5271–5282. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, G.; Hou, H.; Liao, Q.; Chen, J.; Bai, H.; Lee, S.; Wang, C.; Li, H.; Cheng, L.; et al. Analysis of sex hormones and menstruation in COVID-19 women of child-bearing age. Reprod. BioMed. Online 2021, 42, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Schoenbaum, E.E.; Hartel, D.; Lo, Y.; Howard, A.A.; Floris-Moore, M.; Arnsten, J.H.; Santoro, N. HIV Infection, Drug Use, and Onset of Natural Menopause. Clin. Infect. Dis. 2005, 41, 1517–1524. [Google Scholar] [CrossRef]
- Hajjo, R.; Momani, E.; Sabbah, D.A.; Baker, N.; Tropsha, A. Identifying a causal link between prolactin signaling pathways and COVID-19 vaccine-induced menstrual changes. npj Vaccines 2023, 8, 129. [Google Scholar] [CrossRef]
- Udomsinprasert, W.; Nontawong, N.; Saengsiwaritt, W.; Panthan, B.; Jiaranai, P.; Thongchompoo, N.; Santon, S.; Runcharoen, C.; Sensorn, I.; Jittikoon, J.; et al. Host genetic polymorphisms involved in long-term symptoms of COVID-19. Emerg. Microbes Infect. 2023, 12, 2239952. [Google Scholar] [CrossRef]
- Downes, D.J.; Cross, A.R.; Hua, P.; Roberts, N.; Schwessinger, R.; Cutler, A.J.; Munis, A.M.; Brown, J.; Mielczarek, O.; De Andrea, C.E.; et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat. Genet. 2021, 53, 1606–1615. [Google Scholar] [CrossRef]
- Santoni, D.; Ghosh, N.; Derelitto, C.; Saha, I. Transcription Factor Driven Gene Regulation in COVID-19 Patients. Viruses 2023, 15, 1188. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Antoniadou, C.; Natsi, A.-M.; Gavriilidis, E.; Papadopoulos, V.; Xingi, E.; Didaskalou, S.; Mikroulis, D.; Tsironidou, V.; Kambas, K.; et al. Down-regulation of KLF2 in lung fibroblasts is linked with COVID-19 immunofibrosis and restored by combined inhibition of NETs, JAK-1/2 and IL-6 signaling. Clin. Immunol. 2023, 247, 109240. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, Y.; Ding, Y.; Luo, S.; Zheng, X.; Wu, X.; Liu, Z.; Ilyas, I.; Chen, S.; Han, S.; et al. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct. Target. Ther. 2021, 6, 266. [Google Scholar] [CrossRef] [PubMed]
- Shahjaman, M.; Rezanur Rahman, M.; Rabiul Auwul, M. A network-based systems biology approach for identification of shared Gene signatures between male and female in COVID-19 datasets. Inform. Med. Unlocked 2021, 25, 100702. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wei, Y.; Zou, J.; Jaffery, R.; Liang, S.; Zheng, C.; Chen, K.; Shi, P.-Y.; Chen, Y.; Xie, X.; et al. Integrated multi-omics analyses identify key anti-viral host factors and pathways controlling SARS-CoV-2 infection. Res. Sq. 2022; preprint. [Google Scholar]
- Shaw, R.J.; Bradbury, C.; Abrams, S.T.; Wang, G.; Toh, C. COVID-19 and immunothrombosis: Emerging understanding and clinical management. Br. J. Haematol. 2021, 194, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Chen, L.; Yang, W.; Li, K.; Zhao, J.; Yan, C.; You, C.; Jiang, M.; Zhou, M.; Shen, X. Transcriptional landscape of circulating platelets from patients with COVID-19 reveals key subnetworks and regulators underlying SARS-CoV-2 infection: Implications for immunothrombosis. Cell Biosci. 2022, 12, 15. [Google Scholar] [CrossRef]
- Wolny, M.; Rozanova, S.; Knabbe, C.; Pfeiffer, K.; Barkovits, K.; Marcus, K.; Birschmann, I. Changes in the Proteome of Platelets from Patients with Critical Progression of COVID-19. Cells 2023, 12, 2191. [Google Scholar] [CrossRef]
- Barrett, T.J.; Cornwell, M.; Myndzar, K.; Rolling, C.C.; Xia, Y.; Drenkova, K.; Biebuyck, A.; Fields, A.T.; Tawil, M.; Luttrell-Williams, E.; et al. Platelets amplify endotheliopathy in COVID-19. Sci. Adv. 2021, 7, eabh2434. [Google Scholar] [CrossRef]
- Lammi, V.; Nakanishi, T.; Jones, S.E.; Andrews, S.J.; Karjalainen, J.; Cortés, B.; O’Brien, H.E.; Fulton-Howard, B.E.; Haapaniemi, H.H.; Schmidt, A.; et al. Genome-wide Association Study of Long COVID. medRxiv, 2023; preprint. [Google Scholar]
- Ferreira, L.C.; Gomes, C.E.M.; Rodrigues-Neto, J.F.; Jeronimo, S.M.B. Genome-wide association studies of COVID-19: Connecting the dots. Infect. Genet. Evol. 2022, 106, 105379. [Google Scholar] [CrossRef] [PubMed]
- Wiehagen, K.R.; Corbo-Rodgers, E.; Li, S.; Staub, E.S.; Hunter, C.A.; Morrisey, E.E.; Maltzman, J.S. Foxp4 Is Dispensable for T Cell Development, but Required for Robust Recall Responses. PLoS ONE 2012, 7, e42273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Giralt, N.; Du, J.; Marin-Corral, J.; Bódalo-Torruella, M.; Blasco-Hernando, F.; Muñoz-Bermúdez, R.; Clarós, M.; Nonell, L.; Perera-Bel, J.; Fernandez-González, M.; et al. Circulating microRNA profiling is altered in the acute respiratory distress syndrome related to SARS-CoV-2 infection. Sci. Rep. 2022, 12, 6929. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, Q.; Zhang, B.; Zhao, W.; Shen, C. The regulation of lncRNAs and miRNAs in SARS-CoV-2 infection. Front. Cell Dev. Biol. 2023, 11, 1229393. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Aggarwal, D.; Spector, D.L. MALAT1 Long Non-Coding RNA: Functional Implications. ncRNA 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Ashwin, H.; Milross, L.; Hunter, B.; Majo, J.; Filby, A.J.; Fisher, A.J.; Kaye, P.M.; Lagos, D. Downregulation of MALAT1 is a hallmark of tissue and peripheral proliferative T cells in COVID-19. Clin. Exp. Immunol. 2023, 212, 262–275. [Google Scholar] [CrossRef]
- Huang, K.; Wang, C.; Vagts, C.; Raguveer, V.; Finn, P.W.; Perkins, D.L. Long non-coding RNAs (lncRNAs) NEAT1 and MALAT1 are differentially expressed in severe COVID-19 patients: An integrated single-cell analysis. PLoS ONE 2022, 17, e0261242. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, G.; Cheng, Z.; Wang, X.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect. Tissue Res. 2018, 59, 581–592. [Google Scholar] [CrossRef]
- Knutsen, E.; Harris, A.L.; Perander, M. Expression and functions of long non-coding RNA NEAT1 and isoforms in breast cancer. Br. J. Cancer 2022, 126, 551–561. [Google Scholar] [CrossRef]
- Tayel, S.I.; El-Masry, E.A.; Abdelaal, G.A.; Shehab-Eldeen, S.; Essa, A.; Muharram, N.M. Interplay of LncRNAs NEAT1 and TUG1 in Incidence of Cytokine Storm in Appraisal of COVID-19 Infection. Int. J. Biol. Sci. 2022, 18, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.; Adamoski, D.; Genelhould, G.; Zhen, F.; Yamaguto, G.E.; Araujo-Souza, P.S.; Nogueira, M.B.; Raboni, S.M.; Bonatto, A.C.; Gradia, D.F. NEAT1 and MALAT1 are highly expressed in saliva and nasopharyngeal swab samples of COVID-19 patients. Mol. Oral. Microbiol. 2021, 36, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Abbasi-Kolli, M.; Nahand, J.S.; Kiani, S.J.; Khanaliha, K.; Khatami, A.; Taghizadieh, M.; Torkamani, A.R.; Babakhaniyan, K.; Bokharaei-Salim, F. The expression patterns of MALAT-1, NEAT-1, THRIL, and miR-155-5p in the acute to the post-acute phase of COVID-19 disease. Braz. J. Infect. Dis. 2022, 26, 102354. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhou, X.; Feng, W.; Jia, M.; Zhang, X.; An, T.; Luan, M.; Pan, Y.; Zhang, S.; Zhou, Z. Risk stratification by long non-coding RNAs profiling in COVID-19 patients. J. Cell. Mol. Med. 2021, 25, 4753–4764. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895. [Google Scholar] [CrossRef]
- Littlefield, K.M.; Watson, R.O.; Schneider, J.M.; Neff, C.P.; Yamada, E.; Zhang, M.; Campbell, T.B.; Falta, M.T.; Jolley, S.E.; Fontenot, A.P.; et al. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PLoS Pathog. 2022, 18, e1010359. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deeks, S.G.; Mustapic, M.; Kapogiannis, D.; Henrich, T.J.; Lu, S.; Goldberg, S.A.; Hoh, R.; Chen, J.Y.; Martinez, E.O.; et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 2022, 91, 772–781. [Google Scholar] [CrossRef]
- Salari, N.; Khodayari, Y.; Hosseinian-Far, A.; Zarei, H.; Rasoulpoor, S.; Akbari, H.; Mohammadi, M. Global prevalence of chronic fatigue syndrome among long COVID-19 patients: A systematic review and meta-analysis. BioPsychoSoc. Med. 2022, 16, 21. [Google Scholar] [CrossRef]
- Evans, R.A.; Leavy, O.C.; Richardson, M.; Elneima, O.; McAuley, H.J.C.; Shikotra, A.; Singapuri, A.; Sereno, M.; Saunders, R.M.; Harris, V.C.; et al. Clinical characteristics with inflammation profiling of long COVID and association with 1-year recovery following hospitalisation in the UK: A prospective observational study. Lancet Respir. Med. 2022, 10, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Shao, Q.; Yu, H.; Liu, J.; Li, Y.; Wang, B.; Sang, H.; Li, D.; Bing, A.; Hou, Y.; et al. Tight Junctions, the Key Factor in Virus-Related Disease. Pathogens 2022, 11, 1200. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Aenlle, K.K.; Cohen, J.; Mathew, A.; Isler, D.; Pangeni, R.P.; Nathanson, L.; Theoharides, T.C.; Klimas, N.G. COVID-19 and Long COVID: Disruption of the Neurovascular Unit, Blood-Brain Barrier, and Tight Junctions. Neuroscientist 2023, 10738584231194927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, W.; Roy, S.; Liu, H.; Roberts, R.M.; Wang, L.; Shi, L.; Ma, W. Tight junction protein occludin is an internalization factor for SARS-CoV-2 infection and mediates virus cell-to-cell transmission. Proc. Natl. Acad. Sci. USA 2023, 120, e2218623120. [Google Scholar] [CrossRef] [PubMed]
- Yende, S.; Parikh, C.R. Long COVID and kidney disease. Nat. Rev. Nephrol. 2021, 17, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, K.; Reinshagen, L.; Puccini, M.; Friebel, J.; Wilde, A.-C.B.; Alsheik, A.; Rroku, A.; Landmesser, U.; Haghikia, A.; Kränkel, N.; et al. Disease Severity in Moderate-to-Severe COVID-19 Is Associated With Platelet Hyperreactivity and Innate Immune Activation. Front. Immunol. 2022, 13, 844701. [Google Scholar] [CrossRef]
- Martins-Gonçalves, R.; Campos, M.M.; Palhinha, L.; Azevedo-Quintanilha, I.G.; Abud Mendes, M.; Ramos Temerozo, J.; Toledo-Mendes, J.; Rosado-de-Castro, P.H.; Bozza, F.A.; Souza Rodrigues, R.; et al. Persisting Platelet Activation and Hyperactivity in COVID-19 Survivors. Circ. Res. 2022, 131, 944–947. [Google Scholar] [CrossRef]
- Turner, S.; Khan, M.A.; Putrino, D.; Woodcock, A.; Kell, D.B.; Pretorius, E. Long COVID: Pathophysiological factors and abnormalities of coagulation. Trends Endocrinol. Metab. 2023, 34, 321–344. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, X.; Gao, Y.; Yu, Y.; Li, C. Crosstalk between Platelets and SARS-CoV-2: Implications in Thrombo-Inflammatory Complications in COVID-19. Int. J. Mol. Sci. 2023, 24, 14133. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.H.; Khanna, A.K.; Kethireddy, S.; Yamane, D.; Levine, A.; Jackson, A.M.; McCurdy, M.T.; Tabatabai, A.; Kumar, G.; Park, P.; et al. Aspirin Use Is Associated With Decreased Mechanical Ventilation, Intensive Care Unit Admission, and In-Hospital Mortality in Hospitalized Patients With Coronavirus Disease 2019. Anesth. Analg. 2021, 132, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.L.; Goshua, G.; Liu, Y.; Fine, R.; Amin, K.; Chang, E.; DeFilippo, N.; Keating, C.; Liu, Y.; Mankbadi, M.; et al. Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: A propensity score-matched analysis. Am. J. Hematol. 2021, 96, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Kaiser, R.; Stark, K. Thromboinflammation in long COVID—the elusive key to postinfection sequelae? J. Thromb. Haemost. 2023, 21, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Mereweather, L.J.; Constantinescu-Bercu, A.; Crawley, J.T.B.; Salles-Crawley, I.I. Platelet–Neutrophil Crosstalk in Thrombosis. Int. J. Mol. Sci. 2023, 24, 1266. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.; Uzun, G.; Zlamal, J.; Singh, A.; Bakchoul, T. Platelet-neutrophil interaction in COVID-19 and vaccine-induced thrombotic thrombocytopenia. Front. Immunol. 2023, 14, 1186000. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic. Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2975–2989. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate With SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Gervaise, A.; Bouzad, C.; Peroux, E.; Helissey, C. Acute pulmonary embolism in non-hospitalized COVID-19 patients referred to CTPA by emergency department. Eur. Radiol. 2020, 30, 6170–6177. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.E.; Wong, S.W.; Sum, C.L.L.; Lim, G.H.; Leung, B.P.; Tan, C.W.; Ramanathan, K.; Dalan, R.; Cheung, C.; Lim, X.R.; et al. Hypercoagulability, endotheliopathy, and inflammation approximating 1 year after recovery: Assessing the long-term outcomes in COVID -19 patients. Am. J. Hematol. 2022, 97, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, M.; Baryshnikova, E.; Anguissola, M.; Pugliese, S.; Falco, M.; Menicanti, L. The Long Term Residual Effects of COVID-Associated Coagulopathy. Int. J. Mol. Sci. 2023, 24, 5514. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; Van De Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Karampini, E.; O’Donnell, A.S.; Ward, S.E.; O’Sullivan, J.M.; O’Donnell, J.S. The Irish COVID-19 Vasculopathy Study (iCVS) investigators Persistent endotheliopathy in the pathogenesis of long COVID syndrome—Reply to comment from von Meijenfeldt et al. J. Thromb. Haemost. 2021, 20, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Bercu, A.; Kessler, A.; Groot, R.; Dragunaite, B.; Heightman, M.; Hillman, T.; Price, L.C.; Brennan, E.; Sivera, R.; Vanhoorelbeke, K. Analysis of thrombogenicity under flow reveals new insights into the prothrombotic state of patients with post-COVID syndrome. J. Thromb. Haemost. 2023, 21, 94–100. [Google Scholar] [CrossRef]
- Fogarty, H.; Ward, S.E.; Townsend, L.; Karampini, E.; Elliott, S.; Conlon, N.; Dunne, J.; Kiersey, R.; Naughton, A.; Gardiner, M.; et al. Sustained VWF-ADAMTS-13 axis imbalance and endotheliopathy in long COVID syndrome is related to immune dysfunction. J. Thromb. Haemost. 2022, 20, 2429–2438. [Google Scholar] [CrossRef]
- Zheng, X.L. ADAMTS13 and von Willebrand Factor in Thrombotic Thrombocytopenic Purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef]
- Prasannan, N.; Heightman, M.; Hillman, T.; Wall, E.; Bell, R.; Kessler, A.; Neave, L.; Doyle, A.; Devaraj, A.; Singh, D.; et al. Impaired exercise capacity in post–COVID-19 syndrome: The role of VWF-ADAMTS13 axis. Blood Adv. 2022, 6, 4041–4048. [Google Scholar] [CrossRef]
- Von Meijenfeldt, F.A.; Havervall, S.; Adelmeijer, J.; Lundström, A.; Magnusson, M.; Mackman, N.; Thalin, C.; Lisman, T. Sustained prothrombotic changes in COVID-19 patients 4 months after hospital discharge. Blood Adv. 2021, 5, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.; Fogarty, H.; Dyer, A.; Martin-Loeches, I.; Bannan, C.; Nadarajan, P.; Bergin, C.; O’Farrelly, C.; Conlon, N.; Bourke, N.M.; et al. Prolonged elevation of D-dimer levels in convalescent COVID-19 patients is independent of the acute phase response. J. Thromb. Haemost. 2021, 19, 1064–1070. [Google Scholar] [CrossRef]
- Sunder, A.; Saha, S.; Kamath, S.; Kumar, M. Vaccine-induced thrombosis and thrombocytopenia (VITT); Exploring the unknown. J. Fam. Med. Prim. Care 2022, 11, 2231. [Google Scholar] [CrossRef] [PubMed]
- Thiele, T.; Ulm, L.; Holtfreter, S.; Schönborn, L.; Kuhn, S.O.; Scheer, C.; Warkentin, T.E.; Bröker, B.M.; Becker, K.; Aurich, K.; et al. Frequency of positive anti-PF4/polyanion antibody tests after COVID-19 vaccination with ChAdOx1 nCoV-19 and BNT162b2. Blood 2021, 138, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.J.; Hope, C.M.; Masavuli, M.G.; Lynn, M.A.; Mekonnen, Z.A.; Yeow, A.E.L.; Garcia-Valtanen, P.; Al-Delfi, Z.; Gummow, J.; Ferguson, C.; et al. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. 2022, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef]
- Ragnoli, B.; Da Re, B.; Galantino, A.; Kette, S.; Salotti, A.; Malerba, M. Interrelationship between COVID-19 and Coagulopathy: Pathophysiological and Clinical Evidence. Int. J. Mol. Sci. 2023, 24, 8945. [Google Scholar] [CrossRef]
- Kumar, R.; Aktay-Cetin, Ö.; Craddock, V.; Morales-Cano, D.; Kosanovic, D.; Cogolludo, A.; Perez-Vizcaino, F.; Avdeev, S.; Kumar, A.; Ram, A.K.; et al. Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: Multilayered cross-talks in the setting of coinfections and comorbidities. PLoS Pathog. 2023, 19, e1011063. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Reed, A.; Desai, S.R.; Devaraj, A.; Faiez, T.S.; Laverty, S.; Kanwal, A.; Esneau, C.; Liu, M.K.; Kamal, F. A persistent neutrophil-associated immune signature characterizes post–COVID-19 pulmonary sequelae. Sci. Transl. Med. 2022, 14, eabo5795. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022, 28, 2406–2415. [Google Scholar] [CrossRef]
- Campbell, M.; Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O’Keeffe, E.; Zaporojan, L.; Connolly, E.; Cheallaigh, C.N.; Conlon, N.; et al. Blood-brain barrier disruption in Long COVID-associated cognitive impairment. Res. Sq. 2022; preprint. [Google Scholar]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef]
- Mak, G.C.; Cheng, P.K.; Lau, S.S.; Wong, K.K.; Lau, C.; Lam, E.T.; Chan, R.C.; Tsang, D.N. Evaluation of rapid antigen test for detection of SARS-CoV-2 virus. J. Clin. Virol. 2020, 129, 104500. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yamahara, K.M.; Cao, Y.; Boehm, A.B. Absolute Quantification of Enterococcal 23S rRNA Gene Using Digital PCR. Environ. Sci. Technol. 2016, 50, 3399–3408. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. JALA J. Assoc. Lab. Autom. 2017, 22, 369–386. [Google Scholar] [CrossRef]
- Pavšič, J.; Žel, J.; Milavec, M. Assessment of the real-time PCR and different digital PCR platforms for DNA quantification. Anal. Bioanal. Chem. 2016, 408, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Sedlak, R.H.; Cook, L.; Jerome, K.R. Tolerance of Droplet-Digital PCR vs Real-Time Quantitative PCR to Inhibitory Substances. Clin. Chem. 2013, 59, 1670–1672. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Cai, Y.; Li, Z.; Shen, S.; Sha, M.; Head, S.R.; Wang, Y. A digital PCR assay development to detect EGFR T790M mutation in NSCLC patients. Front. Lab. Med. 2018, 2, 89–96. [Google Scholar] [CrossRef]
- Zhou, R.; Cai, Y.; Shen, S.; Sha, M.; Li, Z.; Head, S.R.; Wang, Y. A digital PCR based assay to detect all ALK fusion species. Front. Lab. Med. 2018, 2, 49–54. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Xu, P.; Servellita, V.; Miller, S.; Liu, L.; Gopez, A.; Chiu, C.Y.; Abate, A.R. Digital droplet PCR accurately quantifies SARS-CoV-2 viral load from crude lysate without nucleic acid purification. Sci. Rep. 2021, 11, 780. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Antonello, M.; Colagrossi, L.; Merli, M.; Ughi, N.; Renica, S.; Matarazzo, E.; Di Ruscio, F.; Tartaglione, L.; et al. Detection and quantification of SARS-CoV-2 by droplet digital PCR in real-time PCR negative nasopharyngeal swabs from suspected COVID-19 patients. PLoS ONE 2020, 15, e0236311. [Google Scholar] [CrossRef]
- Falzone, L.; Musso, N.; Gattuso, G.; Bongiorno, D.; Palermo, C.; Scalia, G.; Libra, M.; Stefani, S. Sensitivity assessment of droplet digital PCR for SARS-CoV-2 detection. Int. J. Mol. Med. 2020, 46, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Zhao, A.; Fredengren, E.; Fedorowski, A.; Braunschweig, F.; Nygren-Bonnier, M.; Runold, M.; Bruchfeld, J.; Nickander, J.; Deng, Q.; et al. Dysregulations in hemostasis, metabolism, immune response, and angiogenesis in post-acute COVID-19 syndrome with and without postural orthostatic tachycardia syndrome: A multi-omic profiling study. Sci. Rep. 2023, 13, 20230. [Google Scholar] [CrossRef] [PubMed]
- Longworth, J.; Dittmar, G. An antigen microarray protocol for COVID-19 serological analysis. STAR Protoc. 2021, 2, 100815. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, Y.; Zhang, H.; Wang, W.; Yang, X.; Qi, H.; Li, H.; Men, D.; Zhou, J.; Tao, S. SARS-CoV-2 proteome microarray for global profiling of COVID-19 specific IgG and IgM responses. Nat. Commun. 2020, 11, 3581. [Google Scholar] [CrossRef] [PubMed]
- Omer, A. MicroRNAs as powerful tool against COVID-19: Computational perspective. Wiley Interdiscip. Rev. Syst. Biol. Med. 2023, 15, e1621. [Google Scholar] [CrossRef] [PubMed]
- Tribolet, L.; Kerr, E.; Cowled, C.; Bean, A.G.D.; Stewart, C.R.; Dearnley, M.; Farr, R.J. MicroRNA Biomarkers for Infectious Diseases: From Basic Research to Biosensing. Front. Microbiol. 2020, 11, 1197. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; Benítez, I.D.; Pinilla, L.; Carratalá, A.; Moncusí-Moix, A.; Gort-Paniello, C.; Molinero, M.; González, J.; Torres, G.; Bernal, M.; et al. Circulating microRNA profiles predict the severity of COVID-19 in hospitalized patients. Transl. Res. 2021, 236, 147–159. [Google Scholar] [CrossRef]
- Hum, C.; Loiselle, J.; Ahmed, N.; Shaw, T.A.; Toudic, C.; Pezacki, J.P. MicroRNA Mimics or Inhibitors as Antiviral Therapeutic Approaches Against COVID-19. Drugs 2021, 81, 517–531. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constantinescu-Bercu, A.; Lobiuc, A.; Căliman-Sturdza, O.A.; Oiţă, R.C.; Iavorschi, M.; Pavăl, N.-E.; Șoldănescu, I.; Dimian, M.; Covasa, M. Long COVID: Molecular Mechanisms and Detection Techniques. Int. J. Mol. Sci. 2024, 25, 408. https://doi.org/10.3390/ijms25010408
Constantinescu-Bercu A, Lobiuc A, Căliman-Sturdza OA, Oiţă RC, Iavorschi M, Pavăl N-E, Șoldănescu I, Dimian M, Covasa M. Long COVID: Molecular Mechanisms and Detection Techniques. International Journal of Molecular Sciences. 2024; 25(1):408. https://doi.org/10.3390/ijms25010408
Chicago/Turabian StyleConstantinescu-Bercu, Adela, Andrei Lobiuc, Olga Adriana Căliman-Sturdza, Radu Cristian Oiţă, Monica Iavorschi, Naomi-Eunicia Pavăl, Iuliana Șoldănescu, Mihai Dimian, and Mihai Covasa. 2024. "Long COVID: Molecular Mechanisms and Detection Techniques" International Journal of Molecular Sciences 25, no. 1: 408. https://doi.org/10.3390/ijms25010408
APA StyleConstantinescu-Bercu, A., Lobiuc, A., Căliman-Sturdza, O. A., Oiţă, R. C., Iavorschi, M., Pavăl, N.-E., Șoldănescu, I., Dimian, M., & Covasa, M. (2024). Long COVID: Molecular Mechanisms and Detection Techniques. International Journal of Molecular Sciences, 25(1), 408. https://doi.org/10.3390/ijms25010408