
Germline Variants and Characteristic Features of Hereditary Hematological Malignancy Syndrome
 , , ,
, , ,
Abstract
:1. Introduction
2. Myeloid Neoplasms without a Preexisting Disorder or Organ Dysfunction
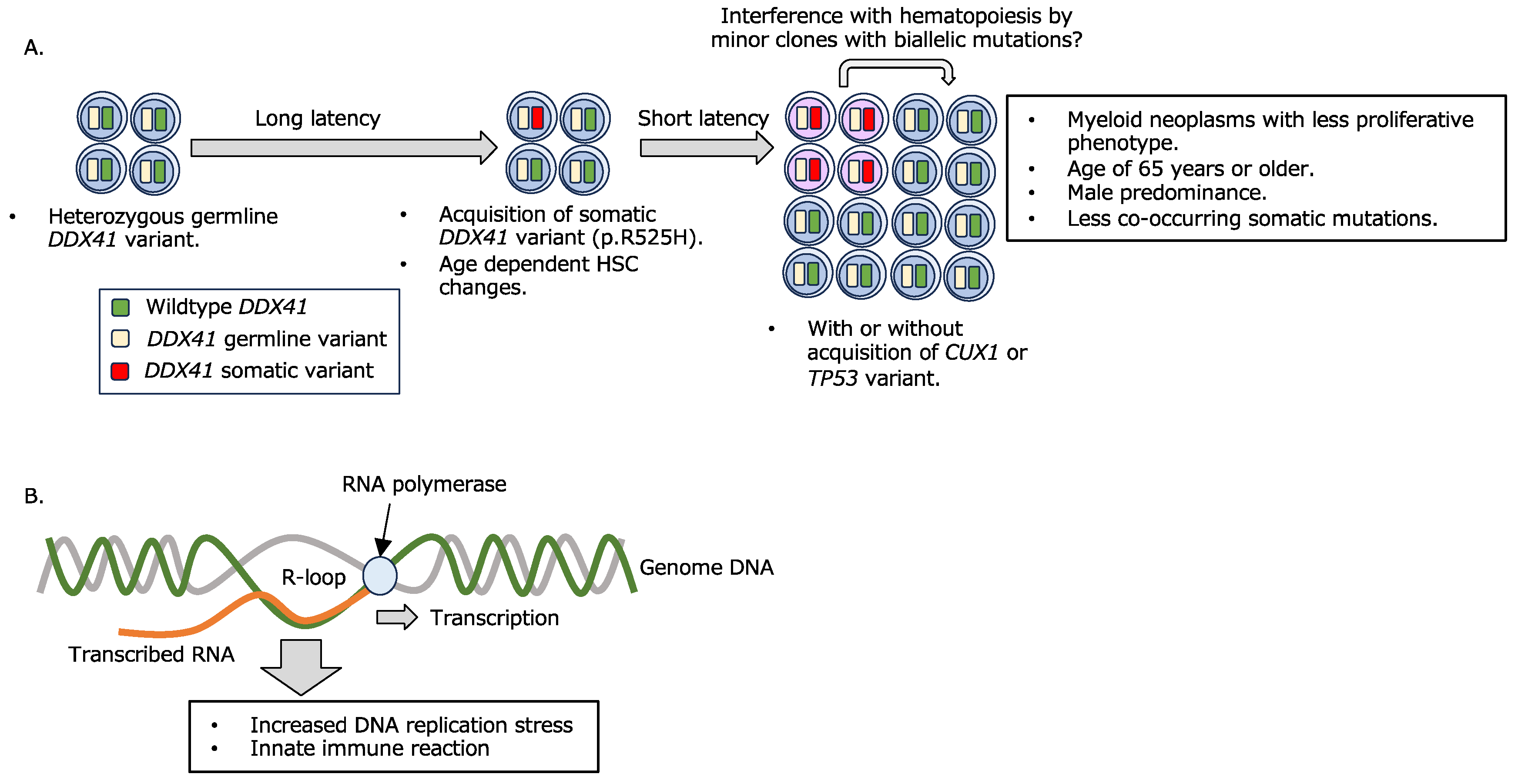
2.1. Myeloid Neoplasms with a Germline DDX41 Variant
- A combination of germline and somatic DDX41 variants confers myeloid disease development.
- B.
- R-loop formation and its consequence.
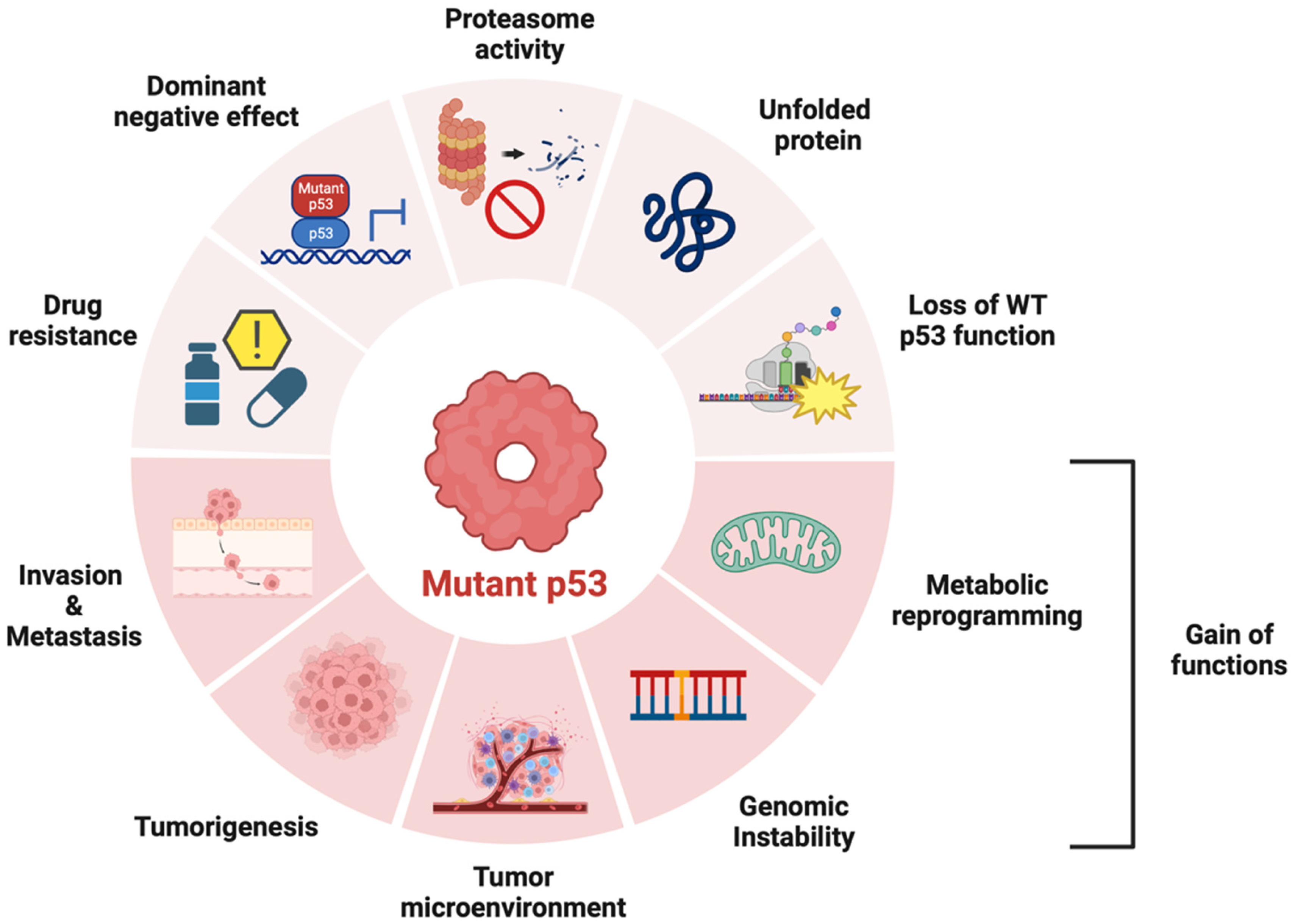
2.2. Li-Fraumeni Syndrome (LFS)
2.3. AML with a Germline CEBPA Variant
2.4. Myeloid Neoplasms with Other Germline Variants (ATM and CHEK2)
3. Myeloid Neoplasms with Preexisting Platelet Disorders
3.1. Myeloid and Lymphoid Neoplasms with a Germline RUNX1 Variant
3.2. Myeloid Neoplasms with a Germline ANKRD26 Variant
3.3. Myeloid and Lymphoid Neoplasms with a Germline ETV6 Variant
4. Myeloid Neoplasms with Other Organ Dysfunction
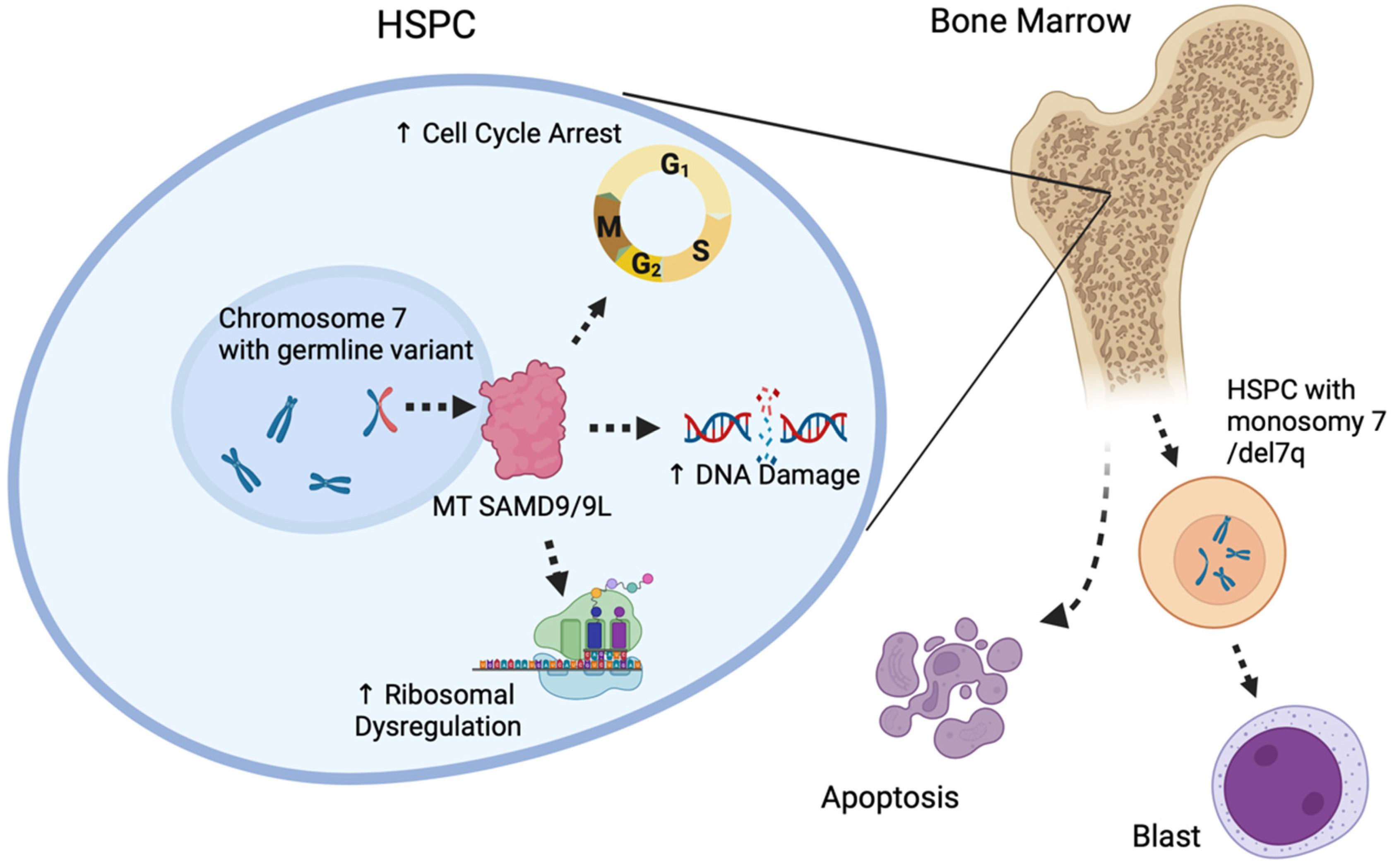
4.1. Myeloid Neoplasms with a Germline SAMD9/SAMD9L Variant
4.2. Myeloid Neoplasms with a Germline GATA2 Variant
5. IBMFS
6. Infant Leukemia with a Germline Predisposition
7. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A. Germline mutations in MDS/AML predisposition disorders. Curr. Opin. Hematol. 2021, 28, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Fenwarth, L.; Caulier, A.; Lachaier, E.; Goursaud, L.; Marceau-Renaut, A.; Fournier, E.; Lebon, D.; Boyer, T.; Berthon, C.; Marolleau, J.P.; et al. Hereditary Predisposition to Acute Myeloid Leukemia in Older Adults. Hemasphere 2021, 5, e552. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, F.; Lopez-Guerra, M.; Morata, J.; Bataller, A.; Paz, S.; Cornet-Masana, J.M.; Banus-Mulet, A.; Cuesta-Casanovas, L.; Carbo, J.M.; Castano-Diez, S.; et al. Germ line variants in patients with acute myeloid leukemia without a suspicion of hereditary hematologic malignancy syndrome. Blood Adv. 2023, 7, 5799–5811. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E. Familial myelodysplastic syndrome/acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2017, 30, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Roloff, G.W.; Shaw, R.; Acevedo, M.; Smith, S.; Drazer, M.W. Clinical guideline variability in the diagnosis of hereditary hematopoietic malignancy syndromes. Leuk. Lymphoma 2023, 64, 1562–1565. [Google Scholar] [CrossRef]
- Stieglitz, E.; Loh, M.L. Genetic predispositions to childhood leukemia. Ther. Adv. Hematol. 2013, 4, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Babushok, D.V.; Bessler, M. Genetic predisposition syndromes: When should they be considered in the work-up of MDS? Best Pract. Res. Clin. Haematol. 2015, 28, 55–68. [Google Scholar] [CrossRef]
- Kotmayer, L.; Kallay, K.; Bodor, C. Hereditary haematological malignancies. Magy. Onkol. 2020, 64, 43–55. [Google Scholar]
- Furutani, E.; Shimamura, A. Germline Genetic Predisposition to Hematologic Malignancy. J. Clin. Oncol. 2017, 35, 1018–1028. [Google Scholar] [CrossRef]
- Zahid, M.F.; Malik, U.A.; Sohail, M.; Hassan, I.N.; Ali, S.; Shaukat, M.H.S. Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 231–239. [Google Scholar] [PubMed]
- Yoshida, M.; Tanase-Nakao, K.; Shima, H.; Shirai, R.; Yoshida, K.; Osumi, T.; Deguchi, T.; Mori, M.; Arakawa, Y.; Takagi, M.; et al. Prevalence of germline GATA2 and SAMD9/9L variants in paediatric haematological disorders with monosomy 7. Br. J. Haematol. 2020, 191, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.S.; Kozyra, E.J.; Wlodarski, M.W. Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract. Res. Clin. Haematol. 2020, 33, 101197. [Google Scholar] [CrossRef] [PubMed]
- Rafei, H.; DiNardo, C.D. Hereditary myeloid malignancies. Best Pract. Res. Clin. Haematol. 2019, 32, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: Current strategies and challenges. Br. J. Haematol. 2022, 196, 1293–1310. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; van den Berg, E.; Sikkema-Raddatz, B.; Abbott, K.M.; Sinke, R.J.; Bungener, L.B.; Mulder, A.B.; Vellenga, E. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia 2017, 31, 520–522. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Lewinsohn, M.; Brown, A.L.; Weinel, L.M.; Phung, C.; Rafidi, G.; Lee, M.K.; Schreiber, A.W.; Feng, J.; Babic, M.; Chong, C.E.; et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood 2016, 127, 1017–1023. [Google Scholar] [CrossRef]
- Cheah, J.J.C.; Hahn, C.N.; Hiwase, D.K.; Scott, H.S.; Brown, A.L. Myeloid neoplasms with germline DDX41 mutation. Int. J. Hematol. 2017, 106, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Bowman, T.V.; Godley, L.A. DDX41-associated susceptibility to myeloid neoplasms. Blood 2023, 141, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Sébert, M.; Passet, M.; Raimbault, A.; Rahmé, R.; Raffoux, E.; Sicre de Fontbrune, F.; Cerrano, M.; Quentin, S.; Vasquez, N.; Da Costa, M.; et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood 2019, 134, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Lalloo, F.; Varley, J.; Ellis, D.; Moran, A.; O’Dair, L.; Pharoah, P.; Evans, D.G.; The Early Onset Breast Cancer Study Group. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 2003, 361, 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugieres, L.; et al. Revisiting Li-Fraumeni Syndrome from TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Dodd-Eaton, E.B.; Peng, G.; Bojadzieva, J.; Chen, J.; Amos, C.I.; Frone, M.N.; Khincha, P.P.; Mai, P.L.; Savage, S.A.; et al. Penetrance of Different Cancer Types in Families with Li-Fraumeni Syndrome: A Validation Study Using Multicenter Cohorts. Cancer Res. 2020, 80, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G.; European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; Escherich, G.; Moricke, A.; von Stackelberg, A.; Stanulla, M.; Bailey, S.; Richter, L.; Steinemann, D.; et al. Clinical and genetic characteristics of children with acute lymphoblastic leukemia and Li-Fraumeni syndrome. Leukemia 2021, 35, 1475–1479. [Google Scholar] [CrossRef]
- Swaminathan, M.; Bannon, S.A.; Routbort, M.; Naqvi, K.; Kadia, T.M.; Takahashi, K.; Alvarado, Y.; Ravandi-Kashani, F.; Patel, K.P.; Champlin, R.; et al. Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003210. [Google Scholar] [CrossRef]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef]
- Pathak, A.; Seipel, K.; Pemov, A.; Dewan, R.; Brown, C.; Ravichandran, S.; Luke, B.T.; Malasky, M.; Suman, S.; Yeager, M.; et al. Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica 2016, 101, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, H.; Chen, P.H.; Pine, A.B.; Siddon, A.J.; Bale, A.E.; Gowda, L.; Killie, A.; Richards, J.; Varin-Tremblay, C.; Kloss, R.; et al. A case of acute myeloid leukemia with unusual germline CEBPA mutation: Lessons learned about mutation detection, location, and penetrance. Leuk. Lymphoma 2021, 62, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Tawana, K.; Rio-Machin, A.; Preudhomme, C.; Fitzgibbon, J. Familial CEBPA-mutated acute myeloid leukemia. Semin. Hematol. 2017, 54, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, A.M.; Trottier, A.M. Hereditary acute myeloid leukemia associated with C-terminal CEBPA germline variants. Fam. Cancer 2023, 22, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.; Kuiper, R.P.; Carmichael, C.L.; Wilkins, E.J.; Dors, N.; Carmagnac, A.; Schouten-Van Meeteren, A.Y.; Li, X.; Stankovic, M.; Kamping, E.; et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: Clues for improved identification of the FPD/AML syndrome. Leukemia 2010, 24, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Renneville, A.; Bourdon, V.; Philippe, N.; Roche-Lestienne, C.; Boissel, N.; Dhedin, N.; Andre, J.M.; Cornillet-Lefebvre, P.; Baruchel, A.; et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009, 113, 5583–5587. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Hahn, C.N.; Scott, H.S. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood 2020, 136, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Arts, P.; Carmichael, C.L.; Babic, M.; Dobbins, J.; Chong, C.E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef]
- Homan, C.C.; King-Smith, S.L.; Lawrence, D.M.; Arts, P.; Feng, J.; Andrews, J.; Armstrong, M.; Ha, T.; Dobbins, J.; Drazer, M.W.; et al. The RUNX1 database (RUNX1db): Establishment of an expert curated RUNX1 registry and genomics database as a public resource for familial platelet disorder with myeloid malignancy. Haematologica 2021, 106, 3004–3007. [Google Scholar] [CrossRef]
- Forster, A.; Decker, M.; Schlegelberger, B.; Ripperger, T. Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies. Cancers 2022, 14, 3431. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Boutroux, H.; Petit, A.; Auvrignon, A.; Lapillonne, H.; Ballerini, P.; Favier, R.; Leverger, G. Childhood diagnosis of genetic thrombocytopenia with mutation in the ankyrine repeat domain 26 gene. Eur. J. Pediatr. 2015, 174, 1399–1403. [Google Scholar] [CrossRef]
- Feurstein, S.; Godley, L.A. Germline ETV6 mutations and predisposition to hematological malignancies. Int. J. Hematol. 2017, 106, 189–195. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Haydu, J.E.; Rigo, I.; Hadler, M.; Tosello, V.; Della Gatta, G.; Paietta, E.; Racevskis, J.; et al. ETV6 mutations in early immature human T cell leukemias. J. Exp. Med. 2011, 208, 2571–2579. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.; et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef]
- Moriyama, T.; Metzger, M.L.; Wu, G.; Nishii, R.; Qian, M.; Devidas, M.; Yang, W.; Cheng, C.; Cao, X.; Quinn, E.; et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol. 2015, 16, 1659–1666. [Google Scholar] [CrossRef]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Below, J.E.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef]
- Davidsson, J.; Puschmann, A.; Tedgard, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, F.; Kuhnen, P.; Suntharalingham, J.P.; Del Valle, I.; Digweed, M.; Stachelscheid, H.; Khajavi, N.; Didi, M.; Brady, A.F.; Blankenstein, O.; et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J. Clin. Investig. 2017, 127, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Bryant, V.; Lamprecht, T.; Ma, J.; Walsh, M.; Schwartz, J.; Del Pilar Alzamora, M.; Mullighan, C.G.; Loh, M.L.; Ribeiro, R.; et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 2018, 3, e121086. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef]
- Homan, C.C.; Venugopal, P.; Arts, P.; Shahrin, N.H.; Feurstein, S.; Rawlings, L.; Lawrence, D.M.; Andrews, J.; King-Smith, S.L.; Harvey, N.L.; et al. GATA2 deficiency syndrome: A decade of discovery. Hum. Mutat. 2021, 42, 1399–1421. [Google Scholar] [CrossRef]
- Al Seraihi, A.F.; Rio-Machin, A.; Tawana, K.; Bodor, C.; Wang, J.; Nagano, A.; Heward, J.A.; Iqbal, S.; Best, S.; Lea, N.; et al. GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2-mutated MDS/AML. Leukemia 2018, 32, 2502–2507. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Yi, S.; Venus, S.; Jankowsky, E.; Bohnsack, M.T. Cellular functions of eukaryotic RNA helicases and their links to human diseases. Nat. Rev. Mol. Cell. Biol. 2023, 24, 749–769. [Google Scholar] [CrossRef]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef]
- Makishima, H.; Saiki, R.; Nannya, Y.; Korotev, S.; Gurnari, C.; Takeda, J.; Momozawa, Y.; Best, S.; Krishnamurthy, P.; Yoshizato, T.; et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood 2023, 141, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Long, N.; Anekpuritanang, T.; Bottomly, D.; Savage, J.C.; Lee, T.; Solis-Ruiz, J.; Borate, U.; Wilmot, B.; Tognon, C.; et al. Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML. Blood 2022, 139, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Brown, S.; Williams, M.; White, T.; Xie, W.; Cui, W.; Peker, D.; Lei, L.; Kunder, C.A.; Wang, H.Y.; et al. The genetic landscape of germline DDX41 variants predisposing to myeloid neoplasms. Blood 2022, 140, 716–755. [Google Scholar] [CrossRef] [PubMed]
- Cheloor Kovilakam, S.; Gu, M.; Dunn, W.G.; Marando, L.; Barcena, C.; Nik-Zainal, S.; Mohorianu, I.; Kar, S.; Fabre, M.A.; Quiros, P.M.; et al. Prevalence and significance of DDX41 gene variants in the general population. Blood 2023, 142, 1185–1192. [Google Scholar] [CrossRef]
- Molteni, E.; Bono, E.; Galli, A.; Elena, C.; Ferrari, J.; Fiorelli, N.; Pozzi, S.; Ferretti, V.V.; Sarchi, M.; Rizzo, E.; et al. Prevalence and clinical expression of germline predisposition to myeloid neoplasms in adults with marrow hypocellularity. Blood 2023, 142, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Kadono, M.; Kanai, A.; Nagamachi, A.; Shinriki, S.; Kawata, J.; Iwato, K.; Kyo, T.; Oshima, K.; Yokoyama, A.; Kawamura, T.; et al. Biological implications of somatic DDX41 p.R525H mutation in acute myeloid leukemia. Exp. Hematol. 2016, 44, 745–754.e4. [Google Scholar] [CrossRef]
- Tierens, A.; Kagotho, E.; Shinriki, S.; Seto, A.; Smith, A.C.; Care, M.; Maze, D.; Sibai, H.; Yee, K.W.; Schuh, A.C.; et al. Biallelic disruption of DDX41 activity is associated with distinct genomic and immunophenotypic hallmarks in acute leukemia. Front. Oncol. 2023, 13, 1153082. [Google Scholar] [CrossRef]
- Badar, T.; Nanaa, A.; Foran, J.M.; Viswanatha, D.; Al-Kali, A.; Lasho, T.; Finke, C.; Alkhateeb, H.B.; He, R.; Gangat, N.; et al. Clinical and molecular correlates of somatic and germline DDX41 variants in patients and families with myeloid neoplasms. Haematologica 2023, 108, 3033–3043. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kobayashi, A.; Osawa, Y.; Nagao, S.; Takano, K.; Okada, Y.; Tachi, N.; Teramoto, M.; Kawamura, T.; Horiuchi, T.; et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia 2017, 31, 1020–1022. [Google Scholar] [CrossRef]
- Rolles, B.; Meyer, R.; Begemann, M.; Elbracht, M.; Jost, E.; Stelljes, M.; Kurth, I.; Brummendorf, T.H.; Silling, G. DDX41 germline variants causing donor cell leukemia indicate a need for further genetic workup in the context of hematopoietic stem cell transplantation. Blood Cancer J. 2023, 13, 73. [Google Scholar] [CrossRef]
- Hirsch, P.; Bories, D.; Chapiro, E.; Nguyen-Khac, F.; Benusiglio, P.R.; Norol, F.; Nguyen, S. Successive relapses from donor and host cells in a patient with DEAD-box helicase 41 (DDX41)-associated myelodysplastic syndrome: The lessons to be learned. Br. J. Haematol. 2022, 199, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Zhang, Z.; Zhou, H.; Xie, J.; Jiang, A.; Wang, Q.; Ding, Z.; Dai, H.; Liu, D.; Wu, N.; et al. Causative germline variant p.Y259C of DDX41 recurrently identified in acute lymphoblastic leukaemia. Br. J. Haematol. 2023, 202, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Jelloul, F.Z.; Routbort, M.J.; DiNardo, C.D.; Bueso-Ramos, C.E.; Kanagal-Shamanna, R.; Thakral, B.; Zuo, Z.; Yin, C.C.; Loghavi, S.; Ok, C.Y.; et al. DDX41 mutations in patients with non-myeloid hematologic neoplasms. Am. J. Hematol. 2023, 98, E193–E196. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Mahmud, N.; Bosland, M.C.; Ross, S.R. DDX41 is needed for pre- and postnatal hematopoietic stem cell differentiation in mice. Stem Cell Rep. 2022, 17, 879–893. [Google Scholar] [CrossRef]
- Chlon, T.M.; Stepanchick, E.; Hershberger, C.E.; Daniels, N.J.; Hueneman, K.M.; Kuenzi Davis, A.; Choi, K.; Zheng, Y.; Gurnari, C.; Haferlach, T.; et al. Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell 2021, 28, 1966–1981.e6. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Cusan, M.; Shen, H.; Zhang, B.; Liao, A.; Yang, L.; Jin, M.; Fernandez, M.; Iyer, P.; Wu, Y.; Hart, K.L.; et al. SF3B1 mutation and ATM deletion co-drive leukemogenesis via centromeric R-loop dysregulation. J. Clin. Investig. 2023, 133, e163325. [Google Scholar] [CrossRef]
- Weinreb, J.T.; Ghazale, N.; Pradhan, K.; Gupta, V.; Potts, K.S.; Tricomi, B.; Daniels, N.J.; Padgett, R.A.; De Oliveira, S.; Verma, A.; et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 2021, 56, 627–640.e5. [Google Scholar] [CrossRef]
- Mosler, T.; Conte, F.; Longo, G.M.C.; Mikicic, I.; Kreim, N.; Möckel, M.M.; Petrosino, G.; Flach, J.; Barau, J.; Luke, B.; et al. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat. Commun. 2021, 12, 7314. [Google Scholar] [CrossRef] [PubMed]
- Shinriki, S.; Hirayama, M.; Nagamachi, A.; Yokoyama, A.; Kawamura, T.; Kanai, A.; Kawai, H.; Iwakiri, J.; Liu, R.; Maeshiro, M.; et al. DDX41 coordinates RNA splicing and transcriptional elongation to prevent DNA replication stress in hematopoietic cells. Leukemia 2022, 36, 2605–2620. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Cvitkovic, I.; Jurica, M.S. Spliceosome database: A tool for tracking components of the spliceosome. Nucleic Acids Res. 2013, 41, D132–D141. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.S.; Vidhyasagar, V.; Yang, S.; Arna, A.B.; Yadav, M.; Aggarwal, A.; Aguilera, A.N.; Shinriki, S.; Bhanumathy, K.K.; Pandey, K.; et al. DDX41 is required for cGAS-STING activation against DNA virus infection. Cell Rep. 2022, 39, 110856. [Google Scholar] [CrossRef]
- Crossley, M.P.; Song, C.; Bocek, M.J.; Choi, J.H.; Kousorous, J.; Sathirachinda, A.; Lin, C.; Brickner, J.R.; Bai, G.; Lans, H.; et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature 2023, 613, 187–194. [Google Scholar] [CrossRef]
- Challakkara, M.F.; Chhabra, R. snoRNAs in hematopoiesis and blood malignancies: A comprehensive review. J. Cell. Physiol. 2023, 238, 1207–1225. [Google Scholar] [CrossRef]
- Dong, J.; Wang, H.; Zhang, Z.; Yang, L.; Qian, X.; Qian, W.; Han, Y.; Huang, H.; Qian, P. Small but strong: Pivotal roles and potential applications of snoRNAs in hematopoietic malignancies. Front. Oncol. 2022, 12, 939465. [Google Scholar] [CrossRef]
- Tungalag, S.; Shinriki, S.; Hirayama, M.; Nagamachi, A.; Kanai, A.; Inaba, T.; Matsui, H. Ribosome profiling analysis reveals the roles of DDX41 in translational regulation. Int. J. Hematol. 2023, 117, 876–888. [Google Scholar] [CrossRef]
- Ramdzan, Z.M.; Nepveu, A. CUX1, a haploinsufficient tumour suppressor gene overexpressed in advanced cancers. Nat. Rev. Cancer 2014, 14, 673–682. [Google Scholar] [CrossRef]
- Imgruet, M.K.; Lutze, J.; An, N.; Hu, B.; Khan, S.; Kurkewich, J.; Martinez, T.C.; Wolfgeher, D.; Gurbuxani, S.K.; Kron, S.J.; et al. Loss of a 7q gene, CUX1, disrupts epigenetically driven DNA repair and drives therapy-related myeloid neoplasms. Blood 2021, 138, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. p53: An anticancer protein’s chequered past and promising future. Nature 2022, 603, S1. [Google Scholar] [CrossRef] [PubMed]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Lapke, N.; Lu, Y.J.; Liao, C.T.; Lee, L.Y.; Lin, C.Y.; Wang, H.M.; Ng, S.H.; Chen, S.J.; Yen, T.C. Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 44194–44210. [Google Scholar] [CrossRef]
- Hansen, S.; Hupp, T.R.; Lane, D.P. Allosteric regulation of the thermostability and DNA binding activity of human p53 by specific interacting proteins. CRC Cell Transformation Group. J. Biol. Chem. 1996, 271, 3917–3924. [Google Scholar] [CrossRef]
- Alvarado-Ortiz, E.; de la Cruz-Lopez, K.G.; Becerril-Rico, J.; Sarabia-Sanchez, M.A.; Ortiz-Sanchez, E.; Garcia-Carranca, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Keymling, M.; Schlemmer, H.P.; Kratz, C.; Pfeil, A.; Bickelhaupt, S.; Alsady, T.M.; Renz, D.M. Li-Fraumeni syndrome. Radiologie 2022, 62, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Sejben, A.; Tiszlavicz, L.; Polyak, K.; Kovacs, L.; Maraz, A.; Torok, D.; Lepran, A.; Ottlakan, A.; Furak, J. Li-Fraumeni syndrome. Orvosi Hetil. 2019, 160, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T.; et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J. Clin. Oncol. 2018, 36, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Comeaux, E.Q.; Mullighan, C.G. TP53 Mutations in Hypodiploid Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026286. [Google Scholar] [CrossRef]
- Preudhomme, C.; Sagot, C.; Boissel, N.; Cayuela, J.M.; Tigaud, I.; de Botton, S.; Thomas, X.; Raffoux, E.; Lamandin, C.; Castaigne, S.; et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: A study from the Acute Leukemia French Association (ALFA). Blood 2002, 100, 2717–2723. [Google Scholar] [CrossRef]
- Avellino, R.; Delwel, R. Expression and regulation of C/EBPalpha in normal myelopoiesis and in malignant transformation. Blood 2017, 129, 2083–2091. [Google Scholar] [CrossRef]
- Pabst, T.; Mueller, B.U. Complexity of CEBPA dysregulation in human acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 5303–5307. [Google Scholar] [CrossRef]
- Tsukada, J.; Yoshida, Y.; Kominato, Y.; Auron, P.E. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 2011, 54, 6–19. [Google Scholar] [CrossRef]
- Pulikkan, J.A.; Tenen, D.G.; Behre, G. C/EBPalpha deregulation as a paradigm for leukemogenesis. Leukemia 2017, 31, 2279–2285. [Google Scholar] [CrossRef]
- Tawana, K.; Fitzgibbon, J. CEBPA-Associated Familial Acute Myeloid Leukemia (AML). In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- West, A.H.; Godley, L.A.; Churpek, J.E. Familial myelodysplastic syndrome/acute leukemia syndromes: A review and utility for translational investigations. Ann. N. Y Acad. Sci. 2014, 1310, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.; Mueller, B.U.; Zhang, P.; Radomska, H.S.; Narravula, S.; Schnittger, S.; Behre, G.; Hiddemann, W.; Tenen, D.G. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat. Genet. 2001, 27, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Frohling, S.; Schlenk, R.F.; Stolze, I.; Bihlmayr, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Dohner, H.; Dohner, K. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: Prognostic relevance and analysis of cooperating mutations. J. Clin. Oncol. 2004, 22, 624–633. [Google Scholar] [CrossRef]
- Xiao, H.; Shi, J.; Luo, Y.; Tan, Y.; He, J.; Xie, W.; Zhang, L.; Wang, Y.; Liu, L.; Wu, K.; et al. First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood 2011, 117, 5257–5260. [Google Scholar] [CrossRef]
- Vitor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef]
- Gelot, C.; Magdalou, I.; Lopez, B.S. Replication stress in Mammalian cells and its consequences for mitosis. Genes 2015, 6, 267–298. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J. Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef] [PubMed]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020, 69, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Kostantopoulou, I.; Apostolou, P.; Papamentzelopoulou, M.S.; Papadimitriou, C.; Faliakou, E.; Christodoulou, C.; Boukovinas, I.; Razis, E.; Tryfonopoulos, D.; et al. One in three highly selected Greek patients with breast cancer carries a loss-of-function variant in a cancer susceptibility gene. J. Med. Genet. 2020, 57, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.C.; et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients with Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228–1235. [Google Scholar] [CrossRef]
- Kaczmarek-Rys, M.; Ziemnicka, K.; Hryhorowicz, S.T.; Gorczak, K.; Hoppe-Golebiewska, J.; Skrzypczak-Zielinska, M.; Tomys, M.; Golab, M.; Szkudlarek, M.; Budny, B.; et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered. Cancer Clin. Pract. 2015, 13, 8. [Google Scholar] [CrossRef]
- Katona, B.W.; Yang, Y.X. Colorectal cancer risk associated with the CHEK2 1100delC variant. Eur. J. Cancer 2017, 83, 103–105. [Google Scholar] [CrossRef]
- Laitman, Y.; Nielsen, S.M.; Hatchell, K.E.; Truty, R.; Bernstein-Molho, R.; Esplin, E.D.; Friedman, E. Re-evaluating cancer risks associated with the CHEK2 p.Ser428Phe Ashkenazi Jewish founder pathogenic variant. Fam. Cancer 2022, 21, 305–308. [Google Scholar] [CrossRef]
- Janiszewska, H.; Bak, A.; Pilarska, M.; Heise, M.; Junkiert-Czarnecka, A.; Kuliszkiewicz-Janus, M.; Calbecka, M.; Jazwiec, B.; Wolowiec, D.; Kuliczkowski, K.; et al. A risk of essential thrombocythemia in carriers of constitutional CHEK2 gene mutations. Haematologica 2012, 97, 366–370. [Google Scholar] [CrossRef]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, H.; Bak, A.; Hartwig, M.; Kuliszkiewicz-Janus, M.; Calbecka, M.; Jazwiec, B.; Kuliczkowski, K.; Haus, O. The germline mutations of the CHEK2 gene are associated with an increased risk of polycythaemia vera. Br. J. Haematol. 2016, 173, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, H.; Bak, A.; Skonieczka, K.; Jaskowiec, A.; Kielbinski, M.; Jachalska, A.; Czyzewska, M.; Jazwiec, B.; Kuliszkiewicz-Janus, M.; Czyz, J.; et al. Constitutional mutations of the CHEK2 gene are a risk factor for MDS, but not for de novo AML. Leuk. Res. 2018, 70, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.J.; Zaker, F.; Nasiri, N.; Yaghmaie, M. Epigenetic changes in FOXO3 and CHEK2 genes and their correlation with clinicopathological findings in myelodysplastic syndromes. Hematol. Oncol. Stem Cell Ther. 2020, 13, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; van den Berg, E.; Smetsers, S.; Leegte, B.K.; Sijmons, R.H.; Abbott, K.M.; Mulder, A.B.; Vellenga, E. Fanconi anaemia presenting as acute myeloid leukaemia and myelodysplastic syndrome in adulthood: A family report on co-occurring FANCC and CHEK2 mutations. Br. J. Haematol. 2019, 184, 1071–1073. [Google Scholar] [CrossRef]
- Paperna, T.; Sharon-Shwartzman, N.; Kurolap, A.; Goldberg, Y.; Moustafa, N.; Carasso, Y.; Feinstien, M.; Mory, A.; Reznick-Levi, G.; Gonzaga-Jauregui, C.; et al. Homozygosity for CHEK2 p.Gly167Arg leads to a unique cancer syndrome with multiple complex chromosomal translocations in peripheral blood karyotype. J. Med. Genet. 2020, 57, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.K.; Miller, C.W.; Tsukasaki, K.; Tavor, S.; Ikezoe, T.; Hoelzer, D.; Takeuchi, S.; Koeffler, H.P. Mutation analysis of the DNA-damage checkpoint gene CHK2 in myelodysplastic syndromes and acute myeloid leukemias. Leuk. Res. 2001, 25, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Chen, H.; Wen, Z.; Guo, W.; Huang, Y.; Mo, X. Abnormal expression of p-ATM/CHK2 in nasal extranodal NK/T cell lymphoma, nasal type, is correlated with poor prognosis. J. Clin. Pathol. 2021, 74, 223–227. [Google Scholar] [CrossRef]
- Ueno, S.; Sudo, T.; Hirasawa, A. ATM: Functions of ATM Kinase and Its Relevance to Hereditary Tumors. Int. J. Mol. Sci. 2022, 23, 523. [Google Scholar] [CrossRef]
- Nakajima, H. Genetic abnormalities in AML. Rinsho Ketsueki 2019, 60, 584–593. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.; Chase, C.L.; Morrell, D. Cancer predisposition of ataxia-telangiectasia heterozygotes. Cancer Genet. Cytogenet. 1990, 46, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liao, W.; Peng, H.; Luo, X.; Luo, Z.; Jiang, H.; Xu, L. miR-181a promotes G1/S transition and cell proliferation in pediatric acute myeloid leukemia by targeting ATM. J. Cancer Res. Clin. Oncol. 2016, 142, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Putzer, S.; von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, Y.; Chen, W.; Ding, N.; Liu, W.; Xie, Y.; Wang, Y.; Zhu, J.; Zeng, C. Germline variants of DNA repair genes in early onset mantle cell lymphoma. Oncogene 2021, 40, 551–563. [Google Scholar] [CrossRef]
- Tiao, G.; Improgo, M.R.; Kasar, S.; Poh, W.; Kamburov, A.; Landau, D.A.; Tausch, E.; Taylor-Weiner, A.; Cibulskis, C.; Bahl, S.; et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017, 31, 2244–2247. [Google Scholar] [CrossRef]
- Homan, C.C.; Scott, H.S.; Brown, A.L. Hereditary platelet disorders associated with germ line variants in RUNX1, ETV6, and ANKRD26. Blood 2023, 141, 1533–1543. [Google Scholar] [CrossRef]
- Galera, P.; Dulau-Florea, A.; Calvo, K.R. Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. Int. J. Lab. Hematol. 2019, 41 (Suppl. 1), 131–141. [Google Scholar] [CrossRef] [PubMed]
- Asou, N. The role of a Runt domain transcription factor AML1/RUNX1 in leukemogenesis and its clinical implications. Crit. Rev. Oncol. Hematol. 2003, 45, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.J.; Peterson, L.F.; Okumura, F.; Boyapati, A.; Zhang, D.E. t(8;21)(q22;q22) Fusion proteins preferentially bind to duplicated AML1/RUNX1 DNA-binding sequences to differentially regulate gene expression. Blood 2008, 112, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Kamath-Loeb, A.S.; Shen, J.C.; Schmitt, M.W.; Kohrn, B.F.; Loeb, K.R.; Estey, E.H.; Dai, J.; Chien, S.; Loeb, L.A.; Becker, P.S. Accurate detection of subclonal variants in paired diagnosis-relapse acute myeloid leukemia samples by next generation Duplex Sequencing. Leuk. Res. 2022, 115, 106822. [Google Scholar] [CrossRef] [PubMed]
- Zharlyganova, D.; Harada, H.; Harada, Y.; Shinkarev, S.; Zhumadilov, Z.; Zhunusova, A.; Tchaizhunusova, N.J.; Apsalikov, K.N.; Kemaikin, V.; Zhumadilov, K.; et al. High frequency of AML1/RUNX1 point mutations in radiation-associated myelodysplastic syndrome around Semipalatinsk nuclear test site. J. Radiat. Res. 2008, 49, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Sendker, S.; Awada, A.; Domagalla, S.; Sendker, M.; Orhan, E.; Hoffmeister, L.M.; Antoniou, E.; Niktoreh, N.; Reinhardt, D.; von Neuhoff, N.; et al. RUNX1 mutation has no prognostic significance in paediatric AML: A retrospective study of the AML-BFM study group. Leukemia 2023, 37, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Harada, Y.; Huang, G.; Harada, H. Myeloid neoplasms with germ line RUNX1 mutation. Int. J. Hematol. 2017, 106, 183–188. [Google Scholar] [CrossRef]
- Bellissimo, D.C.; Speck, N.A. RUNX1 Mutations in Inherited and Sporadic Leukemia. Front. Cell Dev. Biol. 2017, 5, 111. [Google Scholar] [CrossRef]
- Ng, I.K.; Lee, J.; Ng, C.; Kosmo, B.; Chiu, L.; Seah, E.; Mok, M.M.H.; Tan, K.; Osato, M.; Chng, W.J.; et al. Preleukemic and second-hit mutational events in an acute myeloid leukemia patient with a novel germline RUNX1 mutation. Biomark. Res. 2018, 6, 16. [Google Scholar] [CrossRef]
- Hong, D.; Fritz, A.J.; Gordon, J.A.; Tye, C.E.; Boyd, J.R.; Tracy, K.M.; Frietze, S.E.; Carr, F.E.; Nickerson, J.A.; Van Wijnen, A.J.; et al. RUNX1-dependent mechanisms in biological control and dysregulation in cancer. J. Cell. Physiol. 2019, 234, 8597–8609. [Google Scholar] [CrossRef]
- Deuitch, N.; Broadbridge, E.; Cunningham, L.; Liu, P. RUNX1 Familial Platelet Disorder with Associated Myeloid Malignancies. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Homan, C.C.; Drazer, M.W.; Yu, K.; Lawrence, D.M.; Feng, J.; Arriola-Martinez, L.; Pozsgai, M.J.; McNeely, K.E.; Ha, T.; Venugopal, P.; et al. Somatic mutational landscape of hereditary hematopoietic malignancies caused by germline variants in RUNX1, GATA2, and DDX41. Blood Adv. 2023, 7, 6092–6107. [Google Scholar] [CrossRef] [PubMed]
- Vyas, H.; Alcheikh, A.; Lowe, G.; Stevenson, W.S.; Morgan, N.V.; Rabbolini, D.J. Prevalence and natural history of variants in the ANKRD26 gene: A short review and update of reported cases. Platelets 2022, 33, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Regazzo, D.; Omenetto, E.; Scaroni, C.; Semenzato, G.; Fabris, F.; Vianello, F. A novel RUNX1 mutation with ANKRD26 dysregulation is related to thrombocytopenia in a sporadic form of myelodysplastic syndrome. Aging Clin. Exp. Res. 2021, 33, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Palmer, E.L.; Botero, J.P. ANKRD26-Related Thrombocytopenia and Predisposition to Myeloid Neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Melazzini, F.; Palombo, F.; Balduini, A.; De Rocco, D.; Marconi, C.; Noris, P.; Gnan, C.; Pippucci, T.; Bozzi, V.; Faleschini, M.; et al. Clinical and pathogenic features of ETV6-related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica 2016, 101, 1333–1342. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, S.; Yao, H.; Wen, L.; Qiu, H.; Qin, L.; Ma, L.; Chen, S. ETV6 mutation in a cohort of 970 patients with hematologic malignancies. Haematologica 2014, 99, e176–e178. [Google Scholar] [CrossRef]
- Di Paola, J.; Porter, C.C. ETV6-related thrombocytopenia and leukemia predisposition. Blood 2019, 134, 663–667. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, G.; Casado-Garcia, A.; Isidro-Hernandez, M.; Picard, D.; Raboso-Gallego, J.; Aleman-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The Second Oncogenic Hit Determines the Cell Fate of ETV6-RUNX1 Positive Leukemia. Front. Cell Dev. Biol. 2021, 9, 704591. [Google Scholar] [CrossRef]
- Filipiuk, A.; Kozakiewicz, A.; Kosmider, K.; Lejman, M.; Zawitkowska, J. Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies-A Review. Cancers 2022, 14, 3569. [Google Scholar] [CrossRef]
- Schafer, D.; Olsen, M.; Lahnemann, D.; Stanulla, M.; Slany, R.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 2018, 131, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Rundberg Nilsson, A.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Pastor, V.B.; Sahoo, S.S.; Boklan, J.; Schwabe, G.C.; Saribeyoglu, E.; Strahm, B.; Lebrecht, D.; Voss, M.; Bryceson, Y.T.; Erlacher, M.; et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica 2018, 103, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Mekhedov, S.L.; Makarova, K.S.; Koonin, E.V. The complex domain architecture of SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol. Direct 2017, 12, 13. [Google Scholar] [CrossRef]
- Nagamachi, A.; Matsui, H.; Asou, H.; Ozaki, Y.; Aki, D.; Kanai, A.; Takubo, K.; Suda, T.; Nakamura, T.; Wolff, L.; et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 2013, 24, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Nagamachi, A.; Kanai, A.; Nakamura, M.; Okuda, H.; Yokoyama, A.; Shinriki, S.; Matsui, H.; Inaba, T. Multiorgan failure with abnormal receptor metabolism in mice mimicking Samd9/9L syndromes. J. Clin. Investig. 2021, 131, e140147. [Google Scholar] [CrossRef]
- Meng, X.; Xiang, Y. RNA granules associated with SAMD9-mediated poxvirus restriction are similar to antiviral granules in composition but do not require TIA1 for poxvirus restriction. Virology 2019, 529, 16–22. [Google Scholar] [CrossRef]
- Zhang, F.; Ji, Q.; Chaturvedi, J.; Morales, M.; Mao, Y.; Meng, X.; Dong, L.; Deng, J.; Qian, S.B.; Xiang, Y. Human SAMD9 is a poxvirus-activatable anticodon nuclease inhibiting codon-specific protein synthesis. Sci. Adv. 2023, 9, eadh8502. [Google Scholar] [CrossRef]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef]
- Yahata, T.; Takanashi, T.; Muguruma, Y.; Ibrahim, A.A.; Matsuzawa, H.; Uno, T.; Sheng, Y.; Onizuka, M.; Ito, M.; Kato, S.; et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011, 118, 2941–2950. [Google Scholar] [CrossRef]
- Zhou, T.; Hasty, P.; Walter, C.A.; Bishop, A.J.; Scott, L.M.; Rebel, V.I. Myelodysplastic syndrome: An inability to appropriately respond to damaged DNA? Exp. Hematol. 2013, 41, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.E., 3rd; Abdelhamed, S.; Hiltenbrand, R.; Schwartz, J.R.; Sakurada, S.M.; Walsh, M.; Song, G.; Ma, J.; Pruett-Miller, S.M.; Klco, J.M. Pediatric MDS and bone marrow failure-associated germline mutations in SAMD9 and SAMD9L impair multiple pathways in primary hematopoietic cells. Leukemia 2021, 35, 3232–3244. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Mufti, G.J.; Rasool, F.; Mijovic, A.; Devereux, S.; Pagliuca, A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000, 96, 3932–3938. [Google Scholar] [CrossRef] [PubMed]
- Tanase-Nakao, K.; Olson, T.S.; Narumi, S. MIRAGE Syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Yoshizaki, K.; Hachiya, R.; Tomobe, Y.; Kaku, U.; Akiba, K.; Shima, H.; Narumi, S.; Hasegawa, Y. MIRAGE syndrome with recurrent pneumonia probably associated with gastroesophageal reflux and achalasia: A case report. Clin. Pediatr. Endocrinol. 2019, 28, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Viaene, A.N.; Harding, B.N. The Neuropathology of MIRAGE Syndrome. J. Neuropathol. Exp. Neurol. 2020, 79, 458–462. [Google Scholar] [CrossRef]
- Basilious, A.; Basilious, A.; ElJalbout, R.; Robert, M.C. Lacrimal Gland Hypoplasia and Corneal Anesthesia in MIRAGE Syndrome: A Case Report and Literature Review. Cornea 2022, 41, 1041–1044. [Google Scholar] [CrossRef]
- Janjua, D.; Shankar, S.; AlMaazmi, M.; Jadhav, D.V. MIRAGE Syndrome Enteropathy Responding to Pancrelipase Despite Normal Pancreatic Fecal Elastase: A Case Report. Am. J. Case Rep. 2022, 23, e937057. [Google Scholar] [CrossRef]
- Gorcenco, S.; Komulainen-Ebrahim, J.; Nordborg, K.; Suo-Palosaari, M.; Andreasson, S.; Kruger, J.; Nilsson, C.; Kjellstrom, U.; Rahikkala, E.; Turkiewicz, D.; et al. Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol. Genet. 2017, 3, e183. [Google Scholar] [CrossRef]
- Vaughan, D.; Bogdanova-Mihaylova, P.; Costello, D.J.; Sweeney, B.J.; McNamara, B.; Walsh, R.A.; Murphy, S.M. Ataxia pancytopenia syndrome due to SAMD9L mutation presenting as demyelinating neuropathy. J. Peripher. Nerv. Syst. 2020, 25, 433–437. [Google Scholar] [CrossRef]
- King-Robson, J.; Marshall, J.; Smith, F.; Willoughby, L.; Mansour, S.; Sztriha, L. Ataxia-Pancytopenia Syndrome due to a de Novo SAMD9L Mutation. Neurol. Genet. 2021, 7, e580. [Google Scholar] [CrossRef] [PubMed]
- Raskind, W.H.; Chen, D.H.; Bird, T. SAMD9L Ataxia-Pancytopenia Syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Smeenk, L.; Ottema, S.; Mulet-Lazaro, R.; Ebert, A.; Havermans, M.; Varea, A.A.; Fellner, M.; Pastoors, D.; van Herk, S.; Erpelinck-Verschueren, C.; et al. Selective Requirement of MYB for Oncogenic Hyperactivation of a Translocated Enhancer in Leukemia. Cancer Discov. 2021, 11, 2868–2883. [Google Scholar] [CrossRef] [PubMed]
- Belloucif, Y.; Lobry, C. Super-Enhancers Dysregulations in Hematological Malignancies. Cells 2022, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- de Pater, E.; Kaimakis, P.; Vink, C.S.; Yokomizo, T.; Yamada-Inagawa, T.; van der Linden, R.; Kartalaei, P.S.; Camper, S.A.; Speck, N.; Dzierzak, E. Gata2 is required for HSC generation and survival. J. Exp. Med. 2013, 210, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Liquori, A.; Such, E.; Zuniga, A.; Cervera, J. The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers 2023, 15, 1590. [Google Scholar] [CrossRef]
- Wehr, C.; Grotius, K.; Casadei, S.; Bleckmann, D.; Bode, S.F.N.; Frye, B.C.; Seidl, M.; Gulsuner, S.; King, M.C.; Percival, M.B.; et al. A novel disease-causing synonymous exonic mutation in GATA2 affecting RNA splicing. Blood 2018, 132, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Oleaga-Quintas, C.; de Oliveira-Junior, E.B.; Rosain, J.; Rapaport, F.; Deswarte, C.; Guerin, A.; Sajjath, S.M.; Zhou, Y.J.; Marot, S.; Lozano, C.; et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J. Clin. Immunol. 2021, 41, 639–657. [Google Scholar] [CrossRef]
- Calvo, K.R.; Hickstein, D.D. The spectrum of GATA2 deficiency syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Calvo, K.R.; Holland, S.M. Germline GATA2 Mutation and Bone Marrow Failure. Hematol. Oncol. Clin. N. Am. 2018, 32, 713–728. [Google Scholar] [CrossRef]
- Mir, M.A.; Kochuparambil, S.T.; Abraham, R.S.; Rodriguez, V.; Howard, M.; Hsu, A.P.; Jackson, A.E.; Holland, S.M.; Patnaik, M.M. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med. 2015, 4, 490–499. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Collin, M.; Horwitz, M.S. GATA2 deficiency and related myeloid neoplasms. Semin. Hematol. 2017, 54, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.P.; McReynolds, L.J.; Holland, S.M. GATA2 deficiency. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, A. Aplastic anemia and clonal evolution: Germ line and somatic genetics. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Stary, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Park, M. Overview of inherited bone marrow failure syndromes. Blood Res. 2022, 57, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bledsoe, J.R. Inherited bone marrow failure syndromes and germline predisposition to myeloid neoplasia: A practical approach for the pathologist. Semin. Diagn. Pathol. 2023, 40, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; McReynolds, L.J. Inherited bone marrow failure syndromes: A review of current practices and potential future research directions. Curr. Opin. Pediatr. 2023, 35, 75–83. [Google Scholar] [CrossRef]
- Dokal, I.; Vulliamy, T. Inherited bone marrow failure syndromes. Haematologica 2010, 95, 1236–1240. [Google Scholar] [CrossRef]
- Bhandari, J.; Thada, P.K.; Puckett, Y. Fanconi Anemia. In StatPearls; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Dufour, C.; Pierri, F. Modern management of Fanconi anemia. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 649–657. [Google Scholar] [CrossRef]
- Thakur, B.; Hiwale, K.M. Fanconi Anemia: A Rare Genetic Disorder. Cureus 2023, 15, e38899. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Leblanc, T.; Mohandas, N. Diamond-Blackfan anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.M.; Marie, I.; Leblanc, T.M. Diamond-Blackfan anemia. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Gadhiya, K.; Wills, C. Diamond Blackfan Anemia. In StatPearls; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- AlSabbagh, M.M. Dyskeratosis congenita: A literature review. J. Dtsch. Dermatol. Ges. 2020, 18, 943–967. [Google Scholar] [CrossRef] [PubMed]
- Gitto, L.; Stoppacher, R.; Richardson, T.E.; Serinelli, S. Dyskeratosis congenita. Autops. Case Rep. 2020, 10, e2020203. [Google Scholar] [CrossRef] [PubMed]
- Garofola, C.; Nassereddin, A.; Gross, G.P. Dyskeratosis Congenita. In StatPearls; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nelson, N.; Feurstein, S.; Niaz, A.; Truong, J.; Holien, J.K.; Lucas, S.; Fairfax, K.; Dickinson, J.; Bryan, T.M. Functional genomics for curation of variants in telomere biology disorder associated genes: A systematic review. Genet. Med. 2023, 25, 100354. [Google Scholar] [CrossRef]
- Cordell, V.; Osoba, L. Pregnancy in a patient with Schwachman-Diamond syndrome. BMJ Case Rep. 2015, 2015, bcr2015209644. [Google Scholar] [CrossRef]
- Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes 2021, 12, 1520. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Peters, J.A.; Loud, J.T.; Leathwood, L.; Carr, A.G.; Greene, M.H.; Rosenberg, P.S. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 2010, 150, 179–188. [Google Scholar] [CrossRef]
- van Dooijeweert, B.; Kia, S.K.; Dahl, N.; Fenneteau, O.; Leguit, R.; Nieuwenhuis, E.; van Solinge, W.; van Wijk, R.; Da Costa, L.; Bartels, M. GATA-1 Defects in Diamond-Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease. Genes 2022, 13, 447. [Google Scholar] [CrossRef]
- Mello, F.V.; Land, M.G.P.; Costa, E.S.; Teodosio, C.; Sanchez, M.L.; Barcena, P.; Peres, R.T.; Pedreira, C.E.; Alves, L.R.; Orfao, A. Maturation-associated gene expression profiles during normal human bone marrow erythropoiesis. Cell Death Discov. 2019, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Dufour, C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin. Hematol. 2017, 54, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Adegunsoye, A.; Mojsilovic, D.; Vij, R.; West DePersia, A.H.; Rajagopal, P.S.; Osman, A.; Collins, R.H.; Kim, R.H.; Gore, S.D.; et al. Telomere biology disorder prevalence and phenotypes in adults with familial hematologic and/or pulmonary presentations. Blood Adv. 2020, 4, 4873–4886. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. eBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.C.; Davies, S.M.; Shimamura, A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: An update. Hematol. Oncol. Clin. N. Am. 2013, 27, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Kermasson, L.; Hoslin, A.; Jaako, P.; Faille, A.; Acevedo-Arozena, A.; Lengline, E.; Ranta, D.; Poiree, M.; Fenneteau, O.; et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 2019, 134, 277–290. [Google Scholar] [CrossRef]
- Godley, L.A. DNAJC21: The new kid on the SDS block. Blood 2017, 129, 1413–1414. [Google Scholar] [CrossRef]
- Federico, S.; Brennan, R.; Dyer, M.A. Childhood cancer and developmental biology a crucial partnership. Curr. Top. Dev. Biol. 2011, 94, 1–13. [Google Scholar] [CrossRef]
- Felix, C.A.; Lange, B.J. Leukemia in infants. Oncologist 1999, 4, 225–240. [Google Scholar] [CrossRef]
- Montes, R.; Ayllon, V.; Gutierrez-Aranda, I.; Prat, I.; Hernandez-Lamas, M.C.; Ponce, L.; Bresolin, S.; Te Kronnie, G.; Greaves, M.; Bueno, C.; et al. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood 2011, 117, 4746–4758. [Google Scholar] [CrossRef]
- Bursen, A.; Schwabe, K.; Ruster, B.; Henschler, R.; Ruthardt, M.; Dingermann, T.; Marschalek, R. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood 2010, 115, 3570–3579. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Ayllon, V.; Montes, R.; Navarro-Montero, O.; Ramos-Mejia, V.; Real, P.J.; Romero-Moya, D.; Arauzo-Bravo, M.J.; Menendez, P. FLT3 activation cooperates with MLL-AF4 fusion protein to abrogate the hematopoietic specification of human ESCs. Blood 2013, 121, 3867–3878. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Valekunja, U.K.; Edgar, R.S.; Oklejewicz, M.; van der Horst, G.T.; O’Neill, J.S.; Tamanini, F.; Turner, D.J.; Reddy, A.B. Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, D.H.; Lee, S.; Yang, Q.H.; Lee, D.K.; Lee, S.K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef]
- Eguchi, M.; Eguchi-Ishimae, M.; Greaves, M. The role of the MLL gene in infant leukemia. Int. J. Hematol. 2003, 78, 390–401. [Google Scholar] [CrossRef]
- Valentine, M.C.; Linabery, A.M.; Chasnoff, S.; Hughes, A.E.; Mallaney, C.; Sanchez, N.; Giacalone, J.; Heerema, N.A.; Hilden, J.M.; Spector, L.G.; et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL-infant leukemia: A Children’s Oncology Group report. Leukemia 2014, 28, 1235–1241. [Google Scholar] [CrossRef]
- Arcipowski, K.M.; Bulic, M.; Gurbuxani, S.; Licht, J.D. Loss of Mll3 Catalytic Function Promotes Aberrant Myelopoiesis. PLoS ONE 2016, 11, e0162515. [Google Scholar] [CrossRef]
- Tawana, K.; Drazer, M.W.; Churpek, J.E. Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: Are we there yet? Leukemia 2018, 32, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Roloff, G.W.; Griffiths, E.A. When to obtain genomic data in acute myeloid leukemia (AML) and which mutations matter. Blood Adv. 2018, 2, 3070–3080. [Google Scholar] [CrossRef]
- Padella, A.; Ghelli Luserna Di Rora, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Roloff, G.W.; Shaw, R.; O’Connor, T.E.; Hathaway, F.; Drazer, M.W. Stagnation in quality of next-generation sequencing assays for the diagnosis of hereditary hematopoietic malignancies. J. Genet. Couns. 2023, 32, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A.; Shimamura, A. Genetic predisposition to hematologic malignancies: Management and surveillance. Blood 2017, 130, 424–432. [Google Scholar] [CrossRef]
- Carraway, H.E.; LaFramboise, T. Myeloid neoplasms with germline predisposition: Practical considerations and complications in the search for new susceptibility loci. Best Pract. Res. Clin. Haematol. 2020, 33, 101191. [Google Scholar] [CrossRef] [PubMed]
- Mujahed, H.; Jansson, M.; Bengtzen, S.; Lehamnn, S. Bone marrow stroma cells derived from mononuclear cells at diagnosis as a source of germline control DNA for determination of somatic mutations in acute myeloid leukemia. Blood Cancer J. 2017, 7, e616. [Google Scholar] [CrossRef] [PubMed]
- Kraft, I.L.; Godley, L.A. Identifying potential germline variants from sequencing hematopoietic malignancies. Blood 2020, 136, 2498–2506. [Google Scholar] [CrossRef]
- Saygin, C.; Roloff, G.; Hahn, C.N.; Chhetri, R.; Gill, S.; Elmariah, H.; Talati, C.; Nunley, E.; Gao, G.; Kim, A.; et al. Allogeneic hematopoietic stem cell transplant outcomes in adults with inherited myeloid malignancies. Blood Adv. 2023, 7, 549–554. [Google Scholar] [CrossRef]
- Williams, L.; Doucette, K.; Karp, J.E.; Lai, C. Genetics of donor cell leukemia in acute myelogenous leukemia and myelodysplastic syndrome. Bone Marrow Transplant. 2021, 56, 1535–1549. [Google Scholar] [CrossRef]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes after Transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Altman, J.K.; Assi, R.; Bixby, D.; Fathi, A.T.; Foran, J.M.; Gojo, I.; Hall, A.C.; Jonas, B.A.; Kishtagari, A.; et al. Acute Myeloid Leukemia, Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- de Haas, V.; Ismaila, N.; Advani, A.; Arber, D.A.; Dabney, R.S.; Patel-Donelly, D.; Kitlas, E.; Pieters, R.; Pui, C.H.; Sweet, K.; et al. Initial Diagnostic Work-Up of Acute Leukemia: ASCO Clinical Practice Guideline Endorsement of the College of American Pathologists and American Society of Hematology Guideline. J. Clin. Oncol. 2019, 37, 239–253. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome Location | Disorder Name | Penetrance and Lifetime Risk of HM | Prevalence | Age of MN Onset, Years | Malignancy Types | Other Manifestations | Citations |
|---|---|---|---|---|---|---|---|---|
| DDX41 | 5q35.3 | Familial MDS/AML with mutated DDX41 | incomplete penetrance | Up to 13% of myeloid neoplasms have a genetic background, of which DDX41 variants account for about 80% of cases. | Median age is 65 years, ranging from 44 to 88 years, which notably overlaps with the average age of sporadic myeloid malignancies. | MDS, AML, t-MN, solid tumors, especially colon and prostate cancer and melanoma, but not yet definitively linked | cytopenia, macrocytosis, autoimmune diseases | [13,14,18,19,20,21,22,23] |
| TP53 | 17p13.1 | Li-Fraumeni syndrome (LFS) | lifetime risk of HM is about 6% | LFS affects all ethnicities and has an estimated incidence of 1:5000. | Nearly 100% of individuals develop cancer by the age of 70, with the median age of first cancer at 20 to 30 years. | MDS, AML, ALL, t-MN, lymphoma, MM, osteosarcoma, breast cancer, brain tumors, soft tissue sarcoma, adrenocortical carcinoma and other solid tumors | none | [7,24,25,26,27,28,29] |
| CEBPA | 19q13.1 | Familial AML with mutated CEBPA | >80% lifetime risk of AML | <20 families reported | Median age is 24.5 years, ranging from 2 to 50 years. | AML | none | [13,30,31,32,33,34] |
| RUNX1 | 21q22.12 | Familial platelet disorder with propensity to myeloid malignancy | unknown | >250 families reported | Median age is 33 years, ranging from 6 to 76 years. | MDS, AML, ALL, other lymphoid malignancies | thrombocytopenia, platelet dysfunction, atopic and autoimmune disorders | [13,35,36,37,38,39,40,41] |
| ANKRD26 | 10p12.1 | Thrombocytopenia 2 | penetrance for thrombocytopenia is near complete, lifetime risk of HM is about 8% | Unknown | Median age is over 30 years, ranging from 20s to 70s years. | MDS, AML, CML, MPN, ALL, CLL, MM | thrombocytopenia, leukocytosis, erythrocytosis, mild bleeding tendency | [14,42,43,44] |
| ETV6 | 12p13.2 | Thrombocytopenia 5 | penetrance for thrombocytopenia is near complete | ALL is more frequent, especially in B-ALL (0.8% of unselected childhood B-cell ALL). The ratio of lymphoid versus myeloid malignancies is roughly 2:1. | Age ranges from 8 to 82 years and seem to occur at a younger age than usual but is not yet defined. | ALL, MDS, AML, CMML, MM, GI cancers | thrombocytopenia, macrocytosis, platelet dysfunction | [13,15,45,46,47,48,49,50] |
| SAMD9 | 7q21.2 | MIRAGE Syndrome | unknown | 8–17% of childhood onset MDS >110 individuals reported | Pediatric age, not yet defined. | MDS, AML, CMML | bone marrow failure, cytopenia, infections, growth restriction, adrenal hypoplasia, enteropathy, genital abnormalities | [13,51,52,53,54,55] |
| SAMD9L | 7q21.2 | Ataxia, Pancytopenia Syndrome | systemic autoinflammatory disease, bone marrow failure, ataxia | |||||
| GATA2 | 3q21.3 | GATA2 deficiency syndrome | incomplete penetrance | >480 individuals reported, with 240 of these confirmed to be familial and 24 de novo | Median age is 17 years, ranging from 0 to 78 years. | MDS, AML, CMML, ALL | immunodeficiency, bone marrow failure, monocytopenia, lymphopenia, neutropenia, other cytopenia, infections, lymphedema, congenital deafness, pulmonary alveolar proteinosis, venous and arterial thrombosis | [13,37,56,57,58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arai, H.; Matsui, H.; Chi, S.; Utsu, Y.; Masuda, S.; Aotsuka, N.; Minami, Y. Germline Variants and Characteristic Features of Hereditary Hematological Malignancy Syndrome. Int. J. Mol. Sci. 2024, 25, 652. https://doi.org/10.3390/ijms25010652
Arai H, Matsui H, Chi S, Utsu Y, Masuda S, Aotsuka N, Minami Y. Germline Variants and Characteristic Features of Hereditary Hematological Malignancy Syndrome. International Journal of Molecular Sciences. 2024; 25(1):652. https://doi.org/10.3390/ijms25010652
Chicago/Turabian StyleArai, Hironori, Hirotaka Matsui, SungGi Chi, Yoshikazu Utsu, Shinichi Masuda, Nobuyuki Aotsuka, and Yosuke Minami. 2024. "Germline Variants and Characteristic Features of Hereditary Hematological Malignancy Syndrome" International Journal of Molecular Sciences 25, no. 1: 652. https://doi.org/10.3390/ijms25010652