Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors

 ,
,
Abstract
:1. Introduction
2. Results
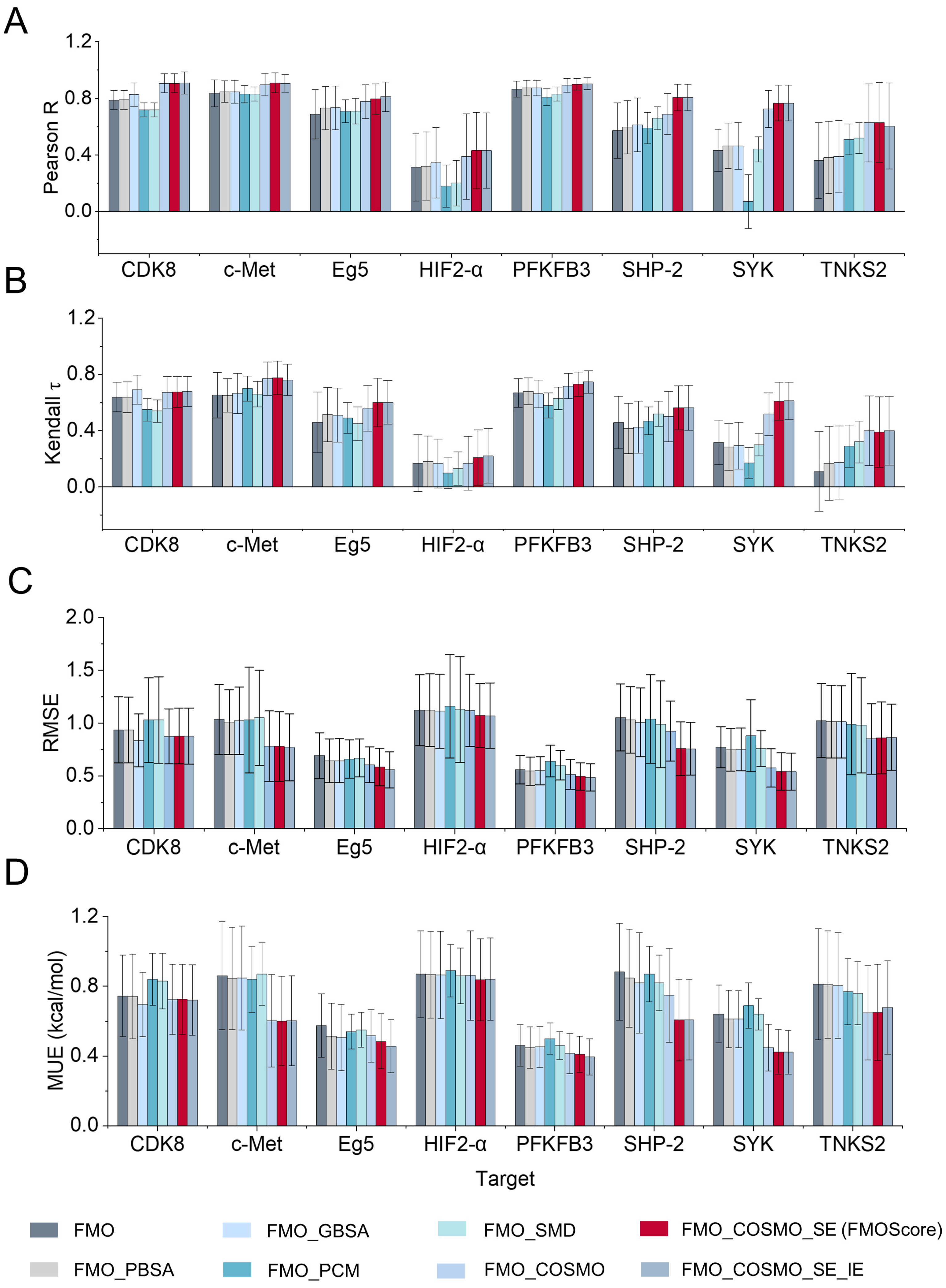
2.1. Influence of Energy Terms on the Performance of FMOScore
2.2. Prediction Performance of the FMOScore Approach
2.3. Comparison with Traditional Methods for Calculating the Binding Free Energy
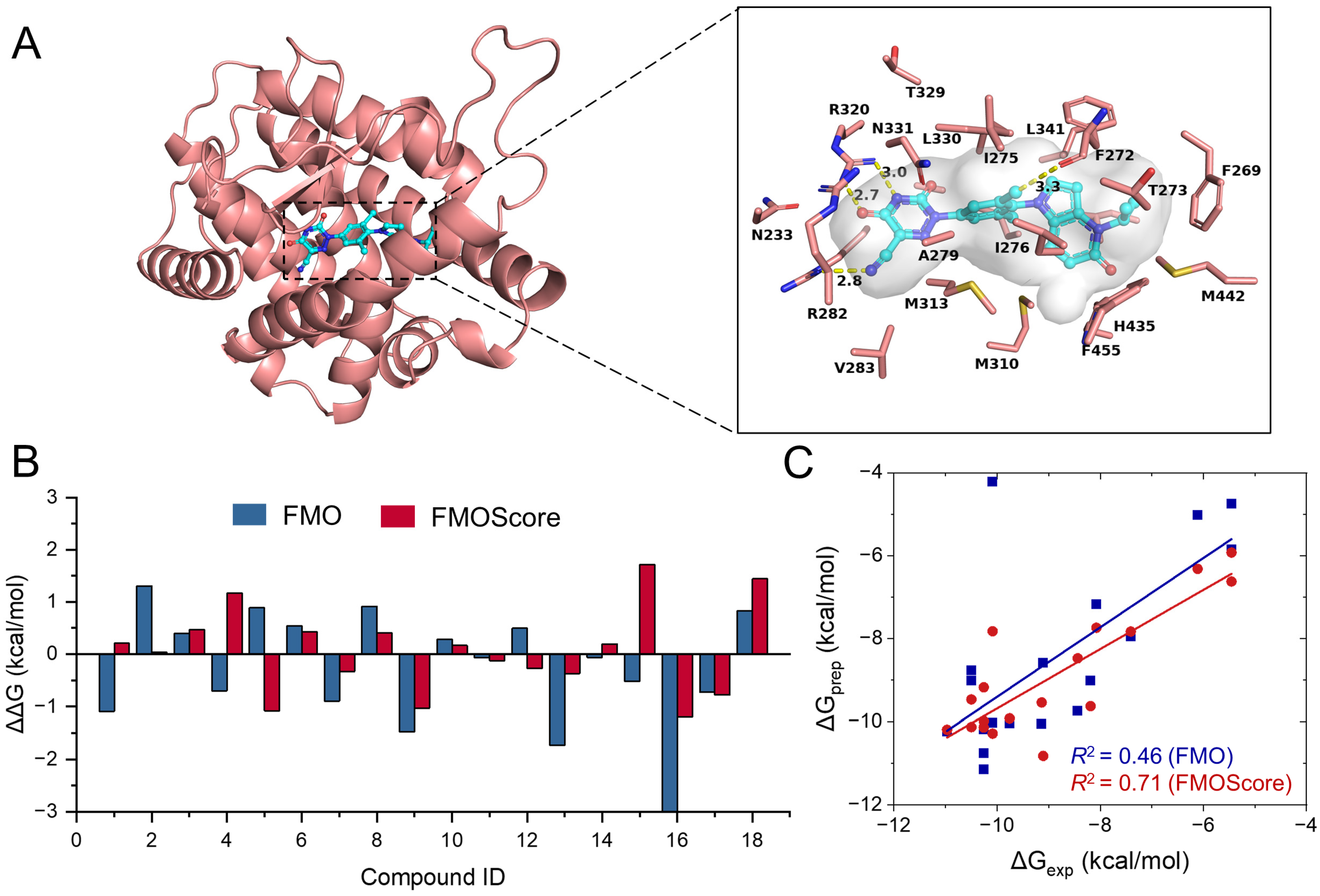
2.4. Retrospective Application of FMOScore to Predict Binding Free Energy
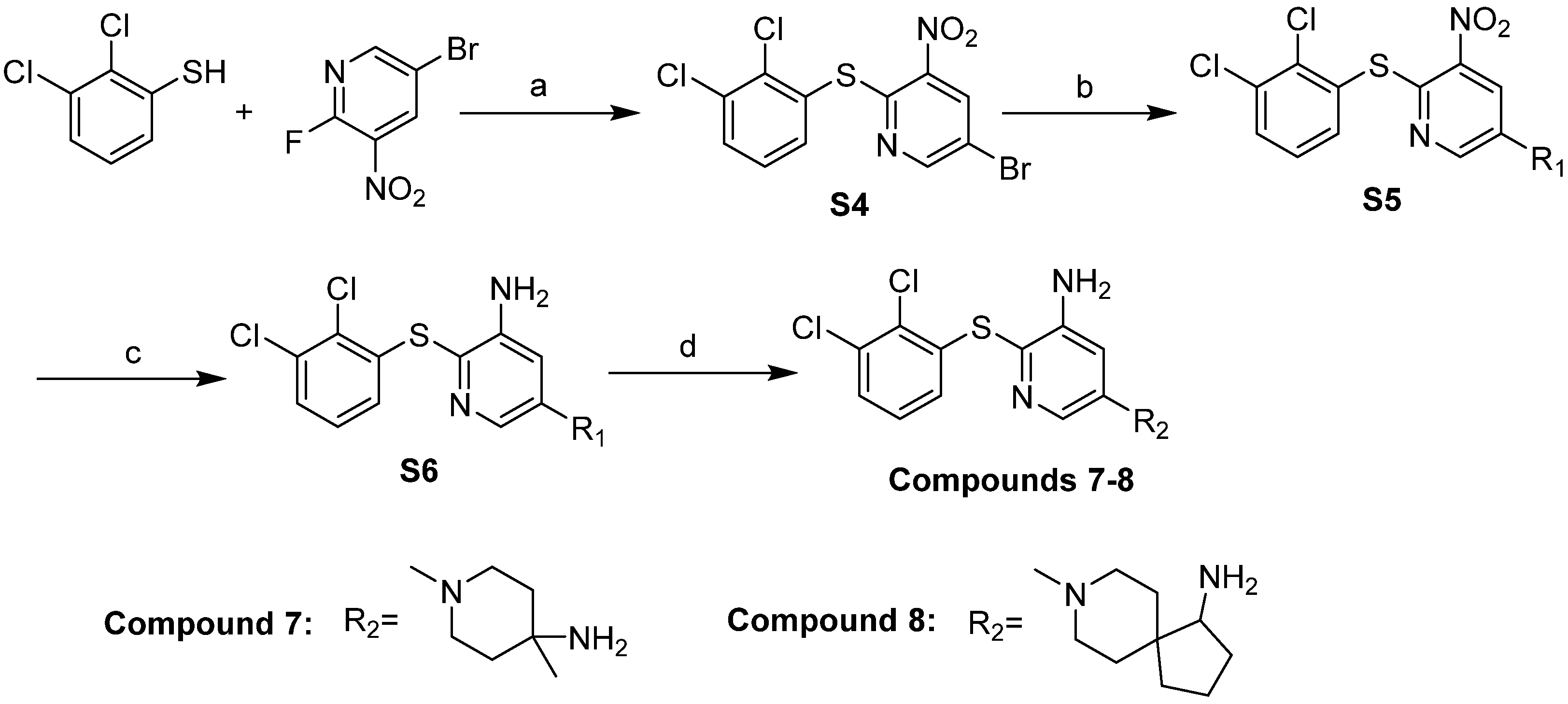
2.5. Design and Synthesis of Potent and Novel SHP-2 Inhibitors
3. Materials and Methods
3.1. FMOScore Calculations
3.2. Comparison with Traditional Methods
3.3. Statistical Analyses
3.4. Protein Expression, Purification, and Biochemical Assay
3.5. Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cavasotto, C.N.; Aucar, M.G.; Adler, N.S. Computational chemistry in drug lead discovery and design. Int. J. Quantum Chem. 2019, 119, e25678. [Google Scholar] [CrossRef]
- Deng, Y.; Roux, B. Computations of Standard Binding Free Energies with Molecular Dynamics Simulations. J. Phys. Chem. B 2009, 113, 2234–2246. [Google Scholar] [CrossRef]
- McCammon, J.A. Computer-aided molecular design. Science 1987, 238, 486–491. [Google Scholar] [CrossRef]
- Nascimento, I.J.d.S.; de Aquino, T.M.; da Silva-Júnior, E.F. The New Era of Drug Discovery: The Power of Computer-aided Drug Design (CADD). Lett. Drug Des. Discov. 2022, 19, 951–955. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2017, 4, 87. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MM/GBSA As a Tool to Understand the Binding Affinities of Filamin–Peptide Interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Liu, P.; Dehez, F.; Cai, W.; Chipot, C. A Toolkit for the Analysis of Free-energy Perturbation Calculations. J. Chem. Theory Comput. 2012, 8, 2606–2616. [Google Scholar] [CrossRef]
- Mitchell, M.J.; McCammon, J.A. Free Energy Difference Calculations by Thermodynamic Integration: Difficulties in Obtaining a Precise Value. J. Comput. Chem. 1991, 12, 271–275. [Google Scholar] [CrossRef]
- Straatsma, T.P.; Berendsen, H.J.C. Free Energy of Ionic Hydration: Analysis of a Thermodynamic Integration Technique to Evaluate Free Energy Differences by Molecular Dynamics Simulations. J. Chem. Phys. 1988, 89, 5876–5886. [Google Scholar] [CrossRef]
- Muñoz-García, A.B.; Ritzmann, A.M.; Pavone, M.; Keith, J.A.; Carter, E.A. Oxygen Transport in Perovskite-type Solid Oxide Fuel Cell Materials: Insights from Quantum Mechanics. Acc. Chem. Res. 2014, 47, 3340–3348. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment Molecular Orbital Method: An Approximate Computational Method for Large Molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Kitaura, K.; Sugiki, S.-I.; Nakano, T.; Komeiji, Y.; Uebayasi, M. Fragment Molecular Orbital Method: Analytical Energy Gradients. Chem. Phys. Lett. 2001, 336, 163–170. [Google Scholar] [CrossRef]
- Fedorov, D.G. The Fragment Molecular Orbital Method: Theoretical Development, Implementation in GAMESS, and Applications. Wires. Comput. Mol. Sci. 2017, 7, e1322. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Ishida, T.; Kitaura, K. Multilayer Formulation of the Fragment Molecular Orbital Method (FMO). J. Phys. Chem. A 2005, 109, 2638–2646. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nakano, S.; Seki, F.; Shigeta, Y.; Ito, S.; Tokiwa, H.; Takeda, M. Computational Analysis Reveals a Critical Point Mutation in the N-Terminal Region of the Signaling Lymphocytic Activation Molecule Responsible for the Cross-Species Infection with Canine Distemper Virus. Molecules 2021, 26, 1262. [Google Scholar] [CrossRef]
- Takaya, D.; Niwa, H.; Mikuni, J.; Nakamura, K.; Handa, N.; Tanaka, A.; Yokoyama, S.; Honma, T. Protein Ligand Interaction Analysis Against New CaMKK2 Inhibitors by Use of X-ray Crystallography and the Fragment Molecular Orbital (FMO) method. J. Mol. Graph. Model. 2020, 99, 107599. [Google Scholar] [CrossRef]
- Nishigaya, Y.; Takase, S.; Sumiya, T.; Kikuzato, K.; Sato, T.; Niwa, H.; Sato, S.; Nakata, A.; Sonoda, T.; Hashimoto, N.; et al. Discovery of Novel Substrate-Competitive lysine Methyltransferase G9a Inhibitors as Anticancer Agents. J. Med. Chem. 2023, 66, 4059–4085. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment Molecular Orbital Based Interaction Analyses on COVID-19 Main Protease—Inhibitor N3 Complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef]
- Ichihara, O.; Barker, J.; Law, R.J.; Whittaker, M. Compound Design by Fragment-Linking. Mol. Inform. 2011, 30, 298–306. [Google Scholar] [CrossRef]
- Watanabe, C.; Okiyama, Y.; Tanaka, S.; Fukuzawa, K.; Honma, T. Molecular Recognition of SARS-CoV-2 Spike Glycoprotein: Quantum Chemical Hot Spot and Epitope Analyses. Chem. Sci. 2021, 12, 4722–4739. [Google Scholar] [CrossRef]
- Kim, J.; Lim, H.; Moon, S.; Cho, S.Y.; Kim, M.; Park, J.H.; Park, H.W.; No, K.T. Hot Spot Snalysis of YAP-TEAD Protein-Protein Interaction Using the Fragment Molecular Orbital Method and Its Application for Inhibitor Discovery. Cancers 2021, 13, 4246. [Google Scholar] [CrossRef]
- Monteleone, S.; Fedorov, D.G.; Townsend-Nicholson, A.; Southey, M.; Bodkin, M.; Heifetz, A. Hotspot Identification and Drug Design of Protein–Protein Interaction Modulators Using the Fragment Molecular Orbital Method. J. Chem. Inf. Model. 2022, 62, 3784–3799. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ozono, H.; Akisawa, K.; Hatada, R.; Okuwaki, K.; Mochizuki, Y. Interaction Analysis on the SARS-CoV-2 Spike Protein Receptor Binding Domain Using Visualization of the Interfacial Electrostatic Complementarity. J. Phys. Chem. Lett. 2021, 12, 11267–11272. [Google Scholar] [CrossRef]
- Heifetz, A.; Trani, G.; Aldeghi, M.; MacKinnon, C.H.; McEwan, P.A.; Brookfield, F.A.; Chudyk, E.I.; Bodkin, M.; Pei, Z.; Burch, J.D.; et al. Fragment Molecular Orbital Method Applied to Lead Optimization of Novel Interleukin-2 Inducible T-Cell Kinase (ITK) Inhibitors. J. Med. Chem. 2016, 59, 4352–4363. [Google Scholar] [CrossRef]
- Heifetz, A.; James, T.; Southey, M.; Bodkin, M.J.; Bromidge, S. Guiding Medicinal Chemistry with Fragment Molecular Orbital (FMO) Method. Methods Mol. Biol. 2020, 2114, 37–48. [Google Scholar] [CrossRef]
- Hengphasatporn, K.; Harada, R.; Wilasluck, P.; Deetanya, P.; Sukandar, E.R.; Chavasiri, W.; Suroengrit, A.; Boonyasuppayakorn, S.; Rungrotmongkol, T.; Wangkanont, K.; et al. Promising SARS-CoV-2 Main Protease Inhibitor Ligand-binding Modes Evaluated using LB-PaCS-MD/FMO. Sci. Rep. 2022, 12, 17984. [Google Scholar] [CrossRef]
- Nakamura, S.; Saito, R.; Yamamoto, S.; Terauchi, Y.; Kittaka, A.; Takimoto-Kamimura, M.; Kurita, N. Proposal of Novel Inhibitors for Vitamin-D Receptor: Molecular Docking, Molecular Mechanics and Ab initio Molecular Orbital Simulations. Biophys. Chem. 2021, 270, 106540. [Google Scholar] [CrossRef]
- Lim, H.; Hong, H.; Hwang, S.; Kim, S.J.; Seo, S.Y.; No, K.T. Identification of Novel Natural Product Inhibitors against Matrix Metalloproteinase 9 Using Quantum Mechanical Fragment Molecular Orbital-based Virtual Screening Methods. Int. J. Mol. Sci. 2022, 23, 4438. [Google Scholar] [CrossRef]
- Choi, J.; Kim, H.-J.; Jin, X.; Lim, H.; Kim, S.; Roh, I.-S.; Kang, H.-E.; No, K.T.; Sohn, H.-J. Application of the Fragment Molecular Orbital Method to Discover Novel Natural Products for Prion Disease. Sci. Rep. 2018, 8, 13063. [Google Scholar] [CrossRef]
- Hengphasatporn, K.; Wilasluck, P.; Deetanya, P.; Wangkanont, K.; Chavasiri, W.; Visitchanakun, P.; Leelahavanichkul, A.; Paunrat, W.; Boonyasuppayakorn, S.; Rungrotmongkol, T.; et al. Halogenated Baicalein As a Promising Antiviral Agent toward SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2022, 62, 1498–1509. [Google Scholar] [CrossRef]
- Deb, I.; Wong, H.; Tacubao, C.; Frank, A.T. Quantum Mechanics Helps Uncover Atypical Recognition Features in the Flavin Mononucleotide Riboswitch. J. Phys. Chem. B 2021, 125, 8342–8350. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Fedorov, D.G. Solvent Screening in Zwitterions Analyzed with the Fragment Molecular Orbital Method. J. Chem. Theory Comput. 2019, 15, 5404–5416. [Google Scholar] [CrossRef]
- Fedorov, D.G. Analysis of solute-solvent interactions using the solvation model density combined with the fragment molecular orbital method. Chem. Phys. Lett. 2018, 702, 111–116. [Google Scholar] [CrossRef]
- Akaki, T.; Nakamura, S.; Nishiwaki, K.; Nakanishi, I. Fragment Molecular Orbital Based Affinity Prediction Toward Pyruvate Dehydrogenase Kinases: Insights into the Charge Transfer in Hydrogen Bond Networks. Chem. Pharm. Bull. 2023, 71, 299–306. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Brekhov, A.; Mironov, V.; Alexeev, Y. Molecular Electrostatic Potential and Electron Density of Large Systems in Solution Computed with the Fragment Molecular Orbital Method. J. Phys. Chem. A 2019, 123, 6281–6290. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K.; Li, H.; Jensen, J.H.; Gordon, M.S. The Polarizable Continuum Model (PCM) Interfaced with the Fragment Molecular Orbital Method (FMO). J. Comput. Chem. 2006, 27, 976–985. [Google Scholar] [CrossRef]
- Li, H.; Fedorov, D.G.; Nagata, T.; Kitaura, K.; Jensen, J.H.; Gordon, M.S. Energy gradients in combined fragment molecular orbital and polarizable continuum model (FMO/PCM) calculation. J. Comput. Chem. 2010, 31, 778–790. [Google Scholar] [CrossRef]
- Albino, S.L.; da Silva Moura, W.C.; Reis, M.M.; Sousa, G.L.; da Silva, P.R.; de Oliveira, M.G.; Borges, T.K.; Albuquerque, L.F.; de Almeida, S.M.; de Lima, M.D.; et al. ACW-02 an Acridine Triazolidine Derivative Presents Antileishmanial Activity Mediated by DNA Interaction and Immunomodulation. Pharmaceuticals 2023, 16, 204. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Rifai, E.A.; van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1–Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Okiyama, Y.; Watanabe, C.; Fukuzawa, K.; Mochizuki, Y.; Nakano, T.; Tanaka, S. Fragment Molecular Orbital Calculations with Implicit Solvent Based on the Poisson-Boltzmann Equation: II. Protein and Its Ligand-Binding System Studies. J. Phys. Chem. B 2019, 123, 957–973. [Google Scholar] [CrossRef]
- Watanabe, C.; Watanabe, H.; Fukuzawa, K.; Parker, L.J.; Okiyama, Y.; Yuki, H.; Yokoyama, S.; Nakano, H.; Tanaka, S.; Honma, T. Theoretical Analysis of Activity Cliffs among Benzofuranone-Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach. J. Chem. Inf. Model. 2017, 57, 2996–3010. [Google Scholar] [CrossRef]
- Nakanishi, I.; Fedorov, D.G.; Kitaura, K. Molecular recognition mechanism of FK506 binding protein: An all-electron fragment molecular orbital study. Proteins Struct. Funct. Bioinform. 2007, 68, 145–158. [Google Scholar] [CrossRef]
- Kříž, K.; Řezáč, J. Benchmarking of Semiempirical Quantum-Mechanical Methods on Systems Relevant to Computer-Aided Drug Design. J. Chem. Inf. Model. 2020, 60, 1453–1460. [Google Scholar] [CrossRef]
- Schindler, C.E.; Baumann, H.; Blum, A.; Böse, D.; Buchstaller, H.-P.; Burgdorf, L.; Cappel, D.; Chekler, E.; Czodrowski, P.; Dorsch, D. Large-scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60, 5457–5474. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Subsystem Analysis for the Fragment Molecular Orbital Method and Its Application to Protein-Ligand Binding in Solution. J. Phys. Chem. A 2016, 120, 2218–2231. [Google Scholar] [CrossRef]
- Kříž, K.; Řezáč, J. Reparametrization of the COSMO Solvent Model for Semiempirical Methods PM6 and PM7. J. Chem. Inf. Model. 2019, 59, 229–235. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Hui, C.; Pawson, T. SH2-Containing Phosphotyrosine Phosphatase as a Target of Protein-Tyrosine Kinases. Science 1993, 259, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Somani, R.R.; Madan, D.P.; Rai, P.R. Protein Tyrosine Phosphatase SHP-2 as Drug Target. Mini Rev. Org. Chem. 2016, 13, 410–420. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic Mutations in PTPN11 in Juvenile Myelomonocytic Leukemia, Myelodysplastic Syndromes and Acute Myeloid Leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The Tyrosine Phosphatase Shp2 (PTPN11) in Cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef]
- Song, Z.; Wang, M.; Ge, Y.; Chen, X.; Xu, Z.; Sun, Y.; Xiong, X. Tyrosine Phosphatase SHP2 Inhibitors in Tumor-targeted Therapies. Acta Pharm. Sin. B 2021, 11, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.P.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, M.; Zhang, H.; Yu, B. Double-edged Roles of Protein Tyrosine Phosphatase SHP2 in Cancer and its Inhibitors in Clinical Trials. Pharmacol. Ther. 2022, 230, 107966. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Wang, M.; Yang, C.-Y.; Wang, S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J. Med. Chem. 2020, 63, 7510–7528. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Pei, Y.; Song, N.; Xu, L.; Feng, B.; Wang, H.; Luo, X.; Hu, X.; Qiu, X. Discovery of Thalidomide-based PROTAC Small Molecules as the Highly Efficient SHP2 Degraders. Eur. J. Med. Chem. 2021, 218, 113341. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Wu, C.; Yang, K.; Wang, Q.; Zhou, Y.; Chen, L.; Li, H. Novel PROTACs for Degradation of SHP2 Protein. Bioorg. Chem. 2021, 110, 104788. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. The COSMO and COSMO-RS solvation models. Wires. Comput. Mol. Sci. 2018, 8, e1338. [Google Scholar] [CrossRef]
- Söderhjelm, P.; Kongsted, J.; Ryde, U. Ligand Affinities Estimated by Quantum Chemical Calculations. J. Chem. Theory Comput. 2010, 6, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, N.; Otsuka, T.; Hirano, Y.; Taiji, M. Use of the Multilayer Fragment Molecular Orbital Method to Predict the Rank Order of Protein–Ligand Binding Affinities: A Case Study Using Tankyrase 2 Inhibitors. ACS Omega 2018, 3, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Gu, Y.; Liang, J.; Ning, M.; Yang, J.; Zhang, Y.; Qu, H.; Yang, Y.; Leng, Y.; Zhou, B. Discovery of Highly Potent and Selective Thyroid Hormone Receptor β Agonists for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2023, 66, 3284–3300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship Between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 per cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Second order Møller-Plesset Perturbation Theory Based upon the Fragment Molecular Orbital Method. J. Chem. Phys. 2004, 121, 2483–2490. [Google Scholar] [CrossRef]
- Otsuka, T.; Okimoto, N.; Taiji, M. Assessment and acceleration of binding energy calculations for protein–ligand complexes by the fragment molecular orbital method. J. Comput. Chem. 2015, 36, 2209–2218. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. The Importance of Three-body Terms in the Fragment Molecular Orbital Method. J. Chem. Phys. 2004, 120, 6832–6840. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Pair Interaction Energy Decomposition Analysis. J. Comput. Chem. 2007, 28, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC2016; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016. [Google Scholar]
- O’Boyle, N.M.; Vandermeersch, T.; Flynn, C.J.; Maguire, A.R.; Hutchison, G.R. Confab-Systematic Generation of Diverse Low-energy Conformers. J. Cheminform. 2011, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-binding Quantum Chemistry Methods. Wires. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Lu, T. Molclus Grogram Version 1.9.9.2. Available online: http://www.keinsci.com/research/molclus.html (accessed on 28 December 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.N.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Witte, J.; Goldey, M.; Neaton, J.B.; Head-Gordon, M. Beyond Energies: Geometries of Nonbonded Molecular Complexes as Metrics for Assessing Electronic Structure Approaches. J. Chem. Theory Comput. 2015, 11, 1481–1492. [Google Scholar] [CrossRef]
- Černý, J.; Kabeláč, M.; Hobza, P. Double-Helical → Ladder Structural Transition in the B-DNA is Induced by a Loss of Dispersion Energy. J. Am. Chem. Soc. 2008, 130, 16055–16059. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, D.G.; Gilson, M.K.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Linear Fit Form | Performance Evaluation Metrics | ||||
|---|---|---|---|---|---|---|
| R a | ρ a | τ a | MUE ↓ | RMSE | ||
| 1 | FMO | 0. (0.0%) | 0. (0.0%) | 0. (0.0%) | 0. (0.0%) | 1. (0.0%) |
| 2 | FMO_PBSA | 0. (30.6%↑) | 0. (26.5%↑) | 0. (29.7%↑) | 0. (−29.6%↓) | 0. (−27.1%↓) |
| 3 | FMO_GBSA | 0. (32.2%↑) | 0. (26.5%↑) | 0. (31.9%↑) | 0. (−30.7%↓) | 0. (−28.9%↓) |
| 4 | FMO_COSMO | 0. (37.1%↑) | 0. (34.3%↑) | 0. (42.5%↑) | 0. (−38.4%↓) | 0. (−35.0%↓) |
| 5 | FMO_PCM | 0. (−30.6%↓) | 0. (6.2%↑) | 0. (8.5%↑) | 1. (12.0%↑) | 1. (19.2%↑) |
| 6 | FMO_SMD | 0. (17.7%↑) | 0. (17.1%↑) | 0. (19.1%↑) | 0. (−15.3%↓) | 0. (−13.1%↓) |
| 7 | FMO_COSMO_SE | 0. (40.3%↑) | 0. (35.9%↑) | 0. (46.8%↑) | 0. (−41.7%↓) | 0. (−37.7%↓) |
| 8 | FMO_COSMO_SE_IE | 0. (40.3%↑) | 0. (35.9%↑) | 0. (44.6%↑) | 0. (−41.7%↓) | 0. (−37.7%↓) |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Part A | Part B | SHP-2 IC50(μM) a | ΔGexp b | FMOScore (kcal/mol) | |||
| ΔGpred c | ΔEint | Δ | ΔGsol | |||||
| SHP099 |  | 0.092 ± 0.001 | −9.593 | −7.964 | −141.73 | 7.55 | 196.22 | |
| 1 |  |  | 1.494 ± 0.024 | −7.943 | −7.677 | −146.17 | 9.58 | 190.18 |
| 2 |  |  | 15.610 ± 1.490 | −6.553 | −6.544 | −111.51 | 9.72 | 189.51 |
| 3 |  |  | 5.174 ± 1.080 | −7.207 | −7.129 | −118.01 | 7.74 | 190.95 |
| 4 |  |  | 23.490 ± 2.860 | −6.311 | −6.066 | −120.11 | 13.06 | 173.71 |
| 5 |  |  | 15.110 ± 0.800 | −6.573 | −6.812 | −124.42 | 9.76 | 173.78 |
| 6 |  |  | 10.650 ± 0.920 | −6.780 | −6.840 | −125.27 | 10.25 | 184.50 |
| 7 |  |  | 11.250 ± 1.190 | −6.747 | −7.049 | −138.37 | 11.70 | 191.28 |
| 8 |  |  | 2.290 ± 0.660 | −7.690 | −7.685 | −159.38 | 12.18 | 198.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Chen, X.; Fan, S.; Chang, L.; Chu, L.; Zhang, Y.; Wang, J.; Li, S.; Xie, J.; Hu, J.; et al. Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. Int. J. Mol. Sci. 2024, 25, 671. https://doi.org/10.3390/ijms25010671
Yuan Z, Chen X, Fan S, Chang L, Chu L, Zhang Y, Wang J, Li S, Xie J, Hu J, et al. Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. International Journal of Molecular Sciences. 2024; 25(1):671. https://doi.org/10.3390/ijms25010671
Chicago/Turabian StyleYuan, Zhen, Xingyu Chen, Sisi Fan, Longfeng Chang, Linna Chu, Ying Zhang, Jie Wang, Shuang Li, Jinxin Xie, Jianguo Hu, and et al. 2024. "Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors" International Journal of Molecular Sciences 25, no. 1: 671. https://doi.org/10.3390/ijms25010671
APA StyleYuan, Z., Chen, X., Fan, S., Chang, L., Chu, L., Zhang, Y., Wang, J., Li, S., Xie, J., Hu, J., Miao, R., Zhu, L., Zhao, Z., Li, H., & Li, S. (2024). Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. International Journal of Molecular Sciences, 25(1), 671. https://doi.org/10.3390/ijms25010671