

Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2)

Abstract
1. Introduction
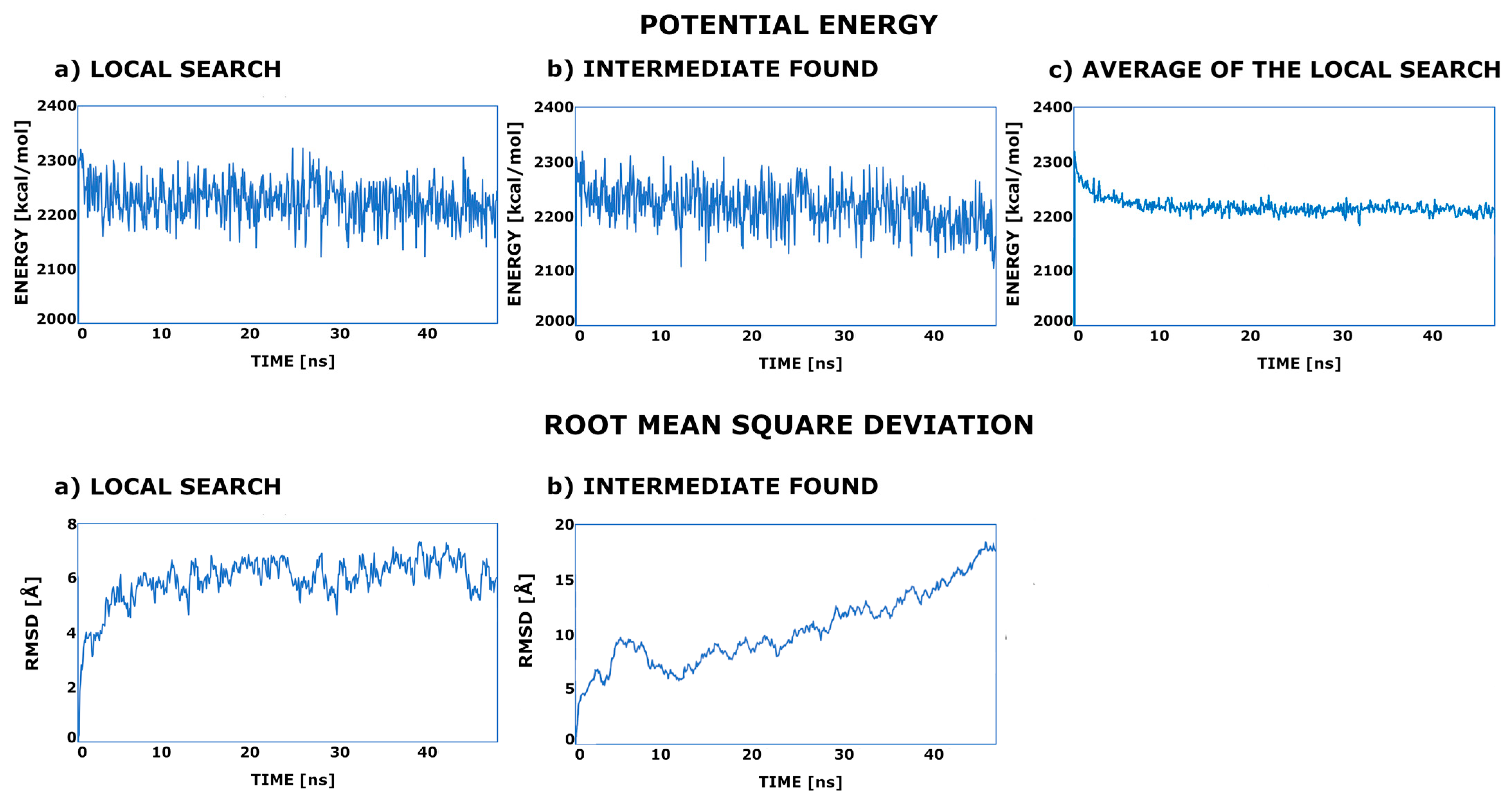
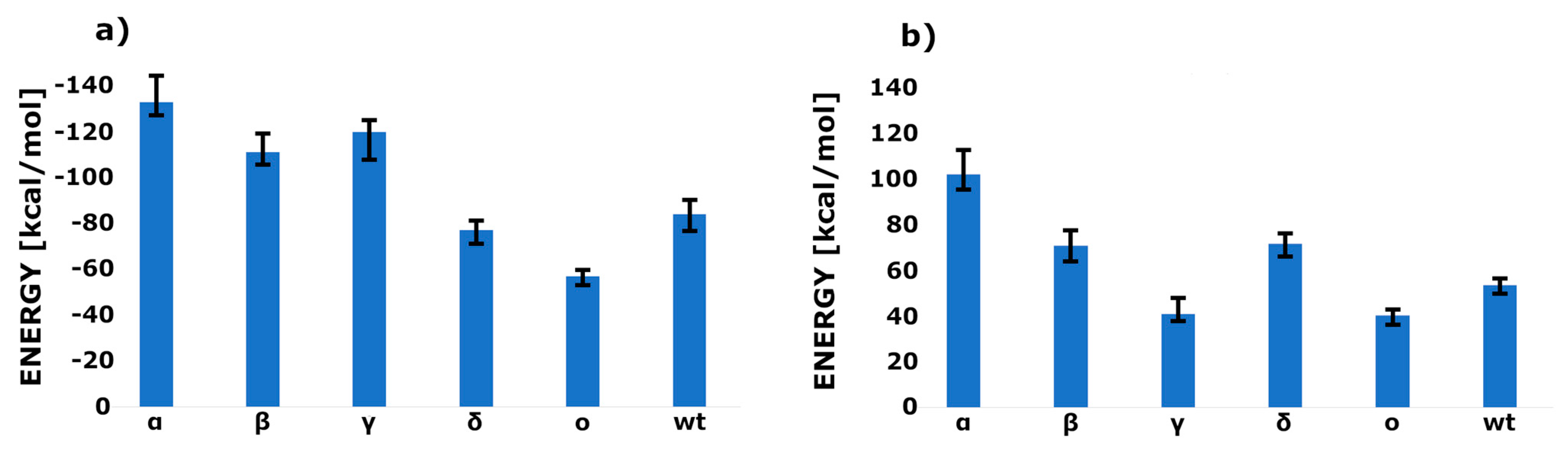
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, F.; Deng, Y.; Li, W. Coronavirus Disease 2019: What We Know? J. Med. Virol. 2020, 92, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Umakanthan, S.; Sahu, P.; Ranade, A.V.; Bukelo, M.M.; Rao, J.S.; Abrahao-Machado, L.F.; Dahal, S.; Kumar, H.; Kv, D. Origin, Transmission, Diagnosis and Management of Coronavirus Disease 2019 (COVID-19). Postgrad. Med. J. 2020, 96, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Eom, J.K. Systematic Literature Review on Impacts of COVID-19 Pandemic and Corresponding Measures on Mobility. Transportation 2023. [Google Scholar] [CrossRef] [PubMed]
- Torge, D.; Bernardi, S.; Arcangeli, M.; Bianchi, S. Histopathological Features of SARS-CoV-2 in Extrapulmonary Organ Infection: A Systematic Review of Literature. Pathogens 2022, 11, 867. [Google Scholar] [CrossRef] [PubMed]
- Bartas, M.; Volná, A.; Beaudoin, C.A.; Poulsen, E.T.; Červeň, J.; Brázda, V.; Špunda, V.; Blundell, T.L.; Pečinka, P. Unheeded SARS-CoV-2 Proteins? A Deep Look into Negative-Sense RNA. Brief. Bioinf. 2022, 23, bbac045. [Google Scholar] [CrossRef] [PubMed]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and Activity of Human TMPRSS2 Protease Implicated in SARS-CoV-2 Activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Casalino, L.; Dommer, A.C.; Gaieb, Z.; Barros, E.P.; Sztain, T.; Ahn, S.-H.; Trifan, A.; Brace, A.; Bogetti, A.T.; Clyde, A.; et al. AI-Driven Multiscale Simulations Illuminate Mechanisms of SARS-CoV-2 Spike Dynamics. Int. J. High Perform. Comput. Appl. 2021, 35, 432–451. [Google Scholar] [CrossRef]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A Multiscale Coarse-Grained Model of the SARS-CoV-2 Virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, Z.; Li, Y.; Lin, X.; Gu, N. Evaluation of Interactions between SARS-CoV-2 RBD and Full-Length ACE2 with Coarse-Grained Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 936–944. [Google Scholar] [CrossRef]
- Pak, A.J.; Yu, A.; Ke, Z.; Briggs, J.A.G.; Voth, G.A. Cooperative Multivalent Receptor Binding Promotes Exposure of the SARS-CoV-2 Fusion Machinery Core. Nat. Commun. 2022, 13, 1002. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor Binding and Priming of the Spike Protein of SARS-CoV-2 for Membrane Fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM Structures of MERS-CoV and SARS-CoV Spike Glycoproteins Reveal the Dynamic Receptor Binding Domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef] [PubMed]
- Sztain, T.; Ahn, S.-H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A Glycan Gate Controls Opening of the SARS-CoV-2 Spike Protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Luisetto, M.; Khaled, E.; Mashori, G.; Yesvi, A.; Latyschev, O. OPEN and CLOSED State of SPIKE SARS-CoV-2: Relationship with Some Integrin Binding. A Biological Molecular Approach to Better Understand the Coagulant Effect. Arch. Biotechnol. Biomed. 2021, 5, 049–056. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant Residues Modulate Conformational Opening in SARS-CoV-2 Spike Protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef]
- Brotzakis, Z.F.; Löhr, T.; Vendruscolo, M. Determination of Intermediate State Structures in the Opening Pathway of SARS-CoV-2 Spike Using Cryo-Electron Microscopy. Chem. Sci. 2021, 12, 9168–9175. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-Electron Microscopy Structures of the SARS-CoV Spike Glycoprotein Reveal a Prerequisite Conformational State for Receptor Binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational Dynamics of SARS-CoV-2 Trimeric Spike Glycoprotein in Complex with Receptor ACE2 Revealed by Cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.-P.; Pfenning, A.R.; Zhao, H.; et al. Broad Host Range of SARS-CoV-2 Predicted by Comparative and Structural Analysis of ACE2 in Vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, X.; Zhou, X.; Chen, P.; Liang, H.; Li, X.; Zhong, W.; Hao, P. Molecular Simulation of SARS-CoV-2 Spike Protein Binding to Pangolin ACE2 or Human ACE2 Natural Variants Reveals Altered Susceptibility to Infection. J. Gen. Virol. 2020, 101, 921–924. [Google Scholar] [CrossRef]
- Zhai, X.; Sun, J.; Yan, Z.; Zhang, J.; Zhao, J.; Zhao, Z.; Gao, Q.; He, W.-T.; Veit, M.; Su, S. Comparison of Severe Acute Respiratory Syndrome Coronavirus 2 Spike Protein Binding to ACE2 Receptors from Human, Pets, Farm Animals, and Putative Intermediate Hosts. J. Virol. 2020, 94, e00831-20. [Google Scholar] [CrossRef]
- Day, C.J.; Bailly, B.; Guillon, P.; Dirr, L.; Jen, F.E.-C.; Spillings, B.L.; Mak, J.; Von Itzstein, M.; Haselhorst, T.; Jennings, M.P. Multidisciplinary Approaches Identify Compounds That Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2–ACE2 Receptor Interactions. mBio 2021, 12, e03681-20. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Biskupek, I.; Sieradzan, A.; Czaplewski, C.; Liwo, A.; Lesner, A.; Giełdoń, A. Theoretical Investigation of the Coronavirus SARS-CoV-2 (COVID-19) Infection Mechanism and Selectivity. Molecules 2022, 27, 2080. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y.; et al. Molecular Insights into Receptor Binding of Recent Emerging SARS-CoV-2 Variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, V.; Chrysovergis, A.; Ragos, V.; Tsiambas, E.; Katsinis, S.; Manoli, A.; Papouliakos, S.; Roukas, D.; Mastronikolis, S.; Peschos, D.; et al. From Delta to Omicron: S1-RBD/S2 Mutation/Deletion Equilibrium in SARS-CoV-2 Defined Variants. Gene 2022, 814, 146134. [Google Scholar] [CrossRef] [PubMed]
- Andre, M.; Lau, L.-S.; Pokharel, M.D.; Ramelow, J.; Owens, F.; Souchak, J.; Akkaoui, J.; Ales, E.; Brown, H.; Shil, R.; et al. From Alpha to Omicron: How Different Variants of Concern of the SARS-Coronavirus-2 Impacted the World. Biology 2023, 12, 1267. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Mandal, N.; Padhi, A.K.; Rath, S.L. Molecular Insights into the Differential Dynamics of SARS-CoV-2 Variants of Concern. J. Mol. Graph. Model. 2022, 114, 108194. [Google Scholar] [CrossRef]
- Zahmatkesh, S.; Sillanpaa, M.; Rezakhani, Y.; Wang, C. Review of Concerned SARS-CoV-2 Variants like Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529), as Well as Novel Methods for Reducing and Inactivating SARS-CoV-2 Mutants in Wastewater Treatment Facilities. J. Hazard. Mater. Adv. 2022, 7, 100140. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The Penultimate Rotamer Library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Karczyńska, A.S.; Ziȩba, K.; Uciechowska, U.; Mozolewska, M.A.; Krupa, P.; Lubecka, E.A.; Lipska, A.G.; Sikorska, C.; Samsonov, S.A.; Sieradzan, A.K.; et al. Improved Consensus-Fragment Selection in Template-Assisted Prediction of Protein Structures with the UNRES Force Field in CASP13. J. Chem. Inf. Model. 2020, 60, 1844–1864. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Lee, J.; Ripoll, D.R.; Pillardy, J.; Scheraga, H.A. Protein Structure Prediction by Global Optimization of a Potential Energy Function. Proc. Natl. Acad. Sci. USA 1999, 96, 5482–5485. [Google Scholar] [CrossRef] [PubMed]
- Khalili, M.; Liwo, A.; Jagielska, A.; Scheraga, H.A. Molecular Dynamics with the United-Residue Model of Polypeptide Chains. II. Langevin and Berendsen-Bath Dynamics and Tests on Model α-Helical Systems. J. Phys. Chem. B 2005, 109, 13798–13810. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, C.; Karczyńska, A.; Sieradzan, A.K.; Liwo, A. UNRES Server for Physics-Based Coarse-Grained Simulations and Prediction of Protein Structure, Dynamics and Thermodynamics. Nucleic Acids Res. 2018, 46, W304–W309. [Google Scholar] [CrossRef] [PubMed]
- Rotkiewicz, P.; Skolnick, J. Fast Procedure for Reconstruction of Full-atom Protein Models from Reduced Representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Voelz, V.A.; Bowman, G.R.; Beauchamp, K.; Pande, V.S. Molecular Simulation of Ab Initio Protein Folding for a Millisecond Folder NTL9(1−39). J. Am. Chem. Soc. 2010, 132, 1526–1528. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Haran, G.; Zhou, H.-X. Fundamental Aspects of Protein−Protein Association Kinetics. Chem. Rev. 2009, 109, 839–860. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| wt | α | β | γ | δ | o | |
|---|---|---|---|---|---|---|
| initial | 70% | 73% | 68% | 70% | 63% | 60% |
| intermediate | 20% | 25% | 30% | 28% | 30% | 25% |
| Mutations Identified in the RBD | Residues Located on the RBD–ACE2 Interface in the Intermediate State | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | o | wt-6LZG | wt | α | β | γ | δ | o |
| 339, G→D | |||||||||||
| 371, S→L | |||||||||||
| 373, S→P | |||||||||||
| 375, S→F | |||||||||||
| R403 | R403 | R403 | R403 | R403 | R403 | ||||||
| D405 | D405 | D405 | |||||||||
| E406 | |||||||||||
| R408 | |||||||||||
| Q409 | |||||||||||
| Q414 | |||||||||||
| T415 | |||||||||||
| 417, K→N | 417, K→T | 417, K→N | K417 | K417 | K417 | N417 | T417 | K417 | N417 | ||
| I418 | I418 | ||||||||||
| Y421 | |||||||||||
| 440, N→K | |||||||||||
| 446, G→S | |||||||||||
| N478 | N448 | N448 | N448 | N448 | |||||||
| 452, L→R | |||||||||||
| Y453 | Y453 | Y453 | |||||||||
| L455 | L455 | L455 | L455 | L455 | L455 | L455 | |||||
| F456 | F456 | F456 | |||||||||
| R457 | R457 | R457 | R457 | ||||||||
| T470 | T470 | ||||||||||
| Y473 | Y473 | ||||||||||
| 477, S→N | S477 | ||||||||||
| 478, T→K | T478 | ||||||||||
| N481 | N481 | N481 | |||||||||
| V483 | V483 | V483 | V483 | ||||||||
| 484, E→K | 484, E→K | 484, E→A | E484 | E484 | E484 | K484 | K484 | E484 | A484 | ||
| F486 | F486 | F486 | F486 | F486 | F486 | F486 | |||||
| 487, T→K | N487 | N487 | N487 | N487 | |||||||
| Y489 | Y489 | Y489 | Y489 | Y489 | Y489 | Y489 | |||||
| F490 | F490 | F490 | F490 | ||||||||
| P491 | |||||||||||
| L492 | L492 | L492 | L492 | ||||||||
| 493, Q→R | Q493 | Q493 | R493 | ||||||||
| S494 | S494 | S494 | |||||||||
| Y495 | Y495 | Y495 | Y495 | Y495 | Y495 | ||||||
| 496, G→S | |||||||||||
| F497 | |||||||||||
| 498, Q→R | Q498 | Q498 | Q498 | Q498 | R498 | ||||||
| T500 | T500 | ||||||||||
| 501, N→Y | 501, N→Y | 501, N→Y | 501, N→Y | N501 | Y501 | Y501 | Y501 | Y501 | Y501 | ||
| V503 | |||||||||||
| 505, Y→H | Y505 | Y505 | Y505 | Y505 | Y505 | Y505 | H505 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biskupek, I.; Gieldon, A. Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2). Int. J. Mol. Sci. 2024, 25, 679. https://doi.org/10.3390/ijms25010679
Biskupek I, Gieldon A. Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2). International Journal of Molecular Sciences. 2024; 25(1):679. https://doi.org/10.3390/ijms25010679
Chicago/Turabian StyleBiskupek, Iga, and Artur Gieldon. 2024. "Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2)" International Journal of Molecular Sciences 25, no. 1: 679. https://doi.org/10.3390/ijms25010679
APA StyleBiskupek, I., & Gieldon, A. (2024). Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2). International Journal of Molecular Sciences, 25(1), 679. https://doi.org/10.3390/ijms25010679